
Abstract
Hippo effectors YAP and TAZ control cell fate and survival through various mechanisms, including transcriptional regulation of key genes. However, much of this research has been marked by conflicting results, as well as controversy over whether YAP and TAZ are redundant. A substantial portion of the discordance stems from their contradictory roles in stem cell self-renewal vs. differentiation and cancer cell survival vs. apoptosis. In this review, we present an overview of the multiple context-dependent functions of YAP and TAZ in regulating cell fate decisions in stem cells and organoids, as well as their mechanisms of controlling programmed cell death pathways in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
YAP and TAZ are effectors of the Hippo pathway, functioning as transcriptional co-regulators that play roles in various cellular contexts. YAP was initially recognized for its roles in inducing hepatomegaly and apoptosis resistance in mice and for its ortholog Yorkie (Yki) positively regulating organ growth in Drosophila [1]. Since YAP and TAZ lack DNA-binding domains, they partner with transcription factors (TFs) (e.g., TEAD1-4, AP-1) to modulate the expression of their target genes [2, 3]. YAP and/or TAZ in complex with TEAD typically associate with distal enhancers and occasionally with promoters, and their functions include controlling transcriptional pause release, H3K27 acetylation, and nucleosome occupancy to influence target gene expression [4,5,6]. This complex modulates genes involved in cell fate specification, including lineage markers, self-renewal factors, and apoptosis-related genes, which results in the maintenance or loss of cell identity depending on the physiological context.
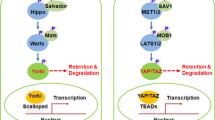
YAP and TAZ are primarily controlled by the Hippo pathway, which is composed of a series of kinases that activate one another and negatively regulate YAP and TAZ’s protein stability and nuclear localization [7]. These kinases were initially identified as tumor suppressors in Drosophila that negatively regulated the transcriptional activity of Yki, which is equivalent to mammalian YAP, and the TF scalloped, equivalent to TEAD [8, 9]. These kinases are in turn influenced by various upstream signals including G protein-coupled receptors (GPCRs), Wnt proteins, cell–cell contact, and mechanical force [10]. GPCR ligands activate YAP and TAZ by inhibiting upstream kinases such as LATS1/2 and stimulating actin polymerization, which tends to promote YAP activity [11,12,13]. Since its discovery, Hippo signaling has been shown to regulate a myriad of cellular processes, including embryogenesis, tumorigenesis, chromatin remodeling, metabolism, and the inflammatory response [8, 10, 14, 15]. We have presented a brief summary of Hippo signaling in Fig. 1.

A graphical summary of Hippo signaling. a When Hippo signaling is on, MST1/2 kinases phosphorylate LATS1/2 kinases, activating them to phosphorylate YAP and TAZ at multiple sites. These phosphorylation events are sufficient for sequestration in the cytoplasm via 14-3-3. Additional phosphorylation events lead to polyubiquitination and proteasome-mediated degradation. In the nucleus, VGLL competes for binding to TEAD, further reducing YAP and TAZ activity. b When Hippo is off, frequently by activation of Rho kinase or inactivation of Hippo kinases, YAP and TAZ translocate to the nucleus to regulate target genes in concert with TEAD factors and other TFs
YAP and TAZ are notable in their ability to change both cell fate and survival. Depending on the context, they can either maintain a stem or progenitor state or stimulate differentiation and morphogenesis. We consider cell fate and survival to be linked, as morphological and biochemical similarities between apoptosis and differentiation have caused some to call them “twins separated at birth” [16]. Numerous pathways influence cell survival, but we will focus on the intrinsic apoptotic pathway, driven by activation of caspase-9 [17,18,19]. The rate of caspase-9 activation is impacted by the relative abundance of Bcl-2 family proteins, which influence mitochondrial homeostasis; although many exist, anti-apoptotic BCL-2 and pro-apoptotic BAX are often transcriptionally regulated by YAP [20]. Though many studies cite that YAP regulates apoptosis, there have been few if any systematic analyses comparing the mechanisms in different contexts.
Contextual dissection of YAP and TAZ (both as individual proteins and comparing them as paralogs) is key, because prior literature has shown many conflicting roles for these proteins in cell fate changes, both in self-renewal vs. differentiation and survival vs. cell death [21], whereas other processes like cell motility and regulation by the cytoskeleton seem to show more consensus. It also remains controversial whether they are functionally redundant or fulfill unique roles [22]. Stem cells and cancer are relevant systems for this undertaking, as they share capacity for differentiation and self-renewal as well as similar signaling pathways [23,24,25]. Thus, this review will expound on how YAP and TAZ regulate cell fate in stem cells and apoptosis in cancer, and what confounding variables have contributed to contradictions within the literature.
Functions of YAP in embryogenesis, stem cell fate and survival, and organoids
In vivo roles of YAP in the developing mouse and human embryo
Phenotypes of Yap1 insufficiency in the mouse embryo
The controversy surrounding Yap1 in development typically involves its roles in early embryogenesis. Yap1−/− embryos undergo developmental arrest around E8.5; nevertheless, the precise mechanisms underlying the development defects remain unclear [26]. Despite typically undergoing successful gastrulation, these embryos show a consistently short and wide body axis, caudal dysgenesis, lack of chorioallantoic fusion, and yolk sac vasculature defects. On the other hand, Wwtr1−/− (encoding Taz) embryos are viable, although they develop multicystic kidney disease as early as E15.5, and the majority of them do not survive to adulthood [27,28,29]. This stark difference in phenotype strongly implies non-redundancy between YAP and TAZ in development. During the earliest stages of embryogenesis, loss of either gene individually does not cause a pre-implantation defect; however, dual knockout (KO) Yap1−/−;Wwtr1−/− embryos die before the 32 cell stage, implying that they are essential but redundant for morula formation [30]. While more sophisticated conditional KOs have shown that Yap1 plays critical roles in the cranial neural crest, female reproductive tract, and heart valves, these phenotypes tended to be late or post-natal, and it would be fruitful to apply conditional KO in the early embryo or placenta to revisit the null phenotypes [31,32,33].
Roles of Yap1 in mouse embryogenesis
One possible mechanism for these null phenotypes may be cell–cell competition regulated by Yap1 in the pre-implantation mouse embryo [34]. Beginning at E3.75, Yap1 starts to translocate from the cytoplasm to the nucleus of some cells in the inner cell mass (ICM), resulting in variable Tead activity that controls expression of Myc and core pluripotency factors. By E4.0, ICM cells that still have primarily cytoplasmic Yap1 undergo apoptosis, and this process ensures a high-quality epiblast. Therefore, it is conceivable that embryos entirely lacking Yap1 undergo developmental errors due to lack of quality control, leading to body axis disruption and other phenotypes, as quality control is particularly crucial at this early stage of development [35]. However, that does not explain the high penetrance of these phenotypes, as a defect in quality control should not affect all embryos.
Given the recent revelation that many embryonic phenotypes can in fact be traced to placental abnormalities [36] and the fact that the placenta originates from the trophectoderm (TE) of the blastocyst, it is worth considering whether Yap1 or Taz null phenotypes originate from TE lineage defects. Indeed, the Yap1–Tead4 complex has been shown to be critical for TE specification. In the outer cells of the blastocyst, simultaneous lack of Hippo signaling [37] and the presence of Notch signaling [38] allows for nuclear localization of Yap1. This in turn allows the Yap1–Tead4 complex to activate expression of Cdx2 and Gata3, specifying the TE. Consequently, it was initially thought that Tead4 is absolutely required for TE formation. However, Kaneko & DePamphilis rebutted this view by showing that Tead4 is dispensable for TE specification when two-cell and eight-cell embryos are cultured in conditions that relieve oxidative stress [39]. Despite strong evidence that Yap1 and Tead4 are key for TE development under physiological conditions, it still remains unknown whether the embryonic phenotypes of null embryos are caused by defects in extraembryonic tissues or are merely concurrent with them.
YAP in the human vs. the mouse embryo and placenta
Like in the mouse, YAP is essential for the development of the human TE and placenta, and its expression is negatively correlated with pregnancy disorders like preeclampsia [40, 41]. The YAP–TEAD4 transcriptional program was recently shown to maintain human trophoblast stemness in primary cytotrophoblasts and repress cell fusion [40]. This was confirmed in another study using both a mouse model and human trophoblast stem cells (TSCs) derived from patients who had experienced pregnancy loss [42]. However, the precise roles of YAP in the human ICM are contested. Yap1 is still inactive and cytoplasmically sequestered in the ICM of the mouse blastocyst by E3.5, whereas it is nuclear in the TE. This is partially established by the Par-aPKC system, which polarizes the blastocyst and thus suppresses Hippo signaling in the outer cells but activates it in the ICM [43]. In contrast to mice, in the human blastocyst, YAP undergoes nuclear accumulation in both the ICM and TE 5–6 days post-fertilization [44]. Whether this may be due to a mismatch in the staging of the human and mouse blastocyst remains undetermined. Nevertheless, YAP has also been shown to sustain pluripotency in human embryonic stem cells (hESCs), and YAP overexpression promotes acquisition of naïve pluripotency, which does not occur in mouse embryonic stem cells (mESCs) [44, 45]. If it were merely an embryo staging issue, YAP would not promote naïve pluripotency in one species but drive differentiation in another. These concerns about the contrasting roles of YAP in mouse vs. human have prompted investigation using in vitro methods that model the blastocyst, namely ESCs and induced pluripotent stem cells (iPSCs).
In vitro roles of YAP in stem cell self-renewal and differentiation
Controversies about mouse Yap1 in self-renewal vs. differentiation
In mESCs, it was initially argued that Yap1 is essential in the nucleus for maintaining self-renewal downstream of LIF by sustaining the expression of core pluripotency factors [46, 47]. This was unusual, because in the mouse ICM, Yap1 is known to be cytoplasmic as mentioned earlier. Further studies using multiple CRISPR KOs have shown that mESCs lacking Yap1 are functionally indistinguishable from WT in regards to proliferation and maintenance of stemness [45]. Furthermore, like in the mouse ICM, Yap1 was found to be cytoplasmic and only translocated to the nucleus upon differentiation. Overexpression of Yap1 also leads to flattened colonies and premature upregulation of lineage markers [45]. These findings are more consistent with in vivo observations, because during post-gastrulation, around E7.5, Yap1 is critical for expression of the early endoderm regulon [48]. Finally, reducing nuclear YAP by inhibiting RHOA GTPase in mouse E3.5 blastocysts leads to the downregulation of TE markers and upregulation of ICM markers, showing that Yap1 may actually transcriptionally antagonize ESC core factors in mouse [49].
However, there continues to be a debate, as KO of upstream Hippo kinases leads to a differentiation defect during teratoma formation that is rescued when Yap1 is knocked down, implying that Yap1 represses lineage specification in teratomas [50]. The contradictory nature of these reports may be due in part to the use of different mESC lines, methods, or failure to test non-cell-autonomous effects of Yap1. Surprisingly, it has been recently shown that during reprogramming, cells that overexpress Yap1 promote the reprogramming of nearby somatic cells that are not overexpressing Yap1; indeed, cell-autonomous expression of Yap1 actually inhibits the acquisition of pluripotency [51]. Additionally, constitutive Yap1 expression promotes astrogenesis in neighboring cells during late embryonic development by upregulating Cntf and represses astrogenesis in a cell-autonomous manner [52]. Thus, whether a cell expresses Yap1 may not be as important as whether its neighbor expresses Yap1. Collectively, these studies show that Yap1 is likely unimportant for maintenance of mESC self-renewal, but it does participate in lineage specification, and can influence cell fate in a non-cell-autonomous manner.
YAP promotes self-renewal and naïve pluripotency in hESCs
Meanwhile, for hESCs, YAP generally inhibits differentiation and promotes self-renewal. Although this may appear to be the reverse of its roles in mESCs, one may initially hypothesize that this is likely due in part to the distinct developmental stages that mouse and hESCs represent on the pluripotency spectrum. After all, both hESCs and iPSCs in standard culture conditions represent a primed pluripotent state, which is more similar to the post-implantation E4.5–5.5 epiblast. Contrarily, in standard mESC culture, cells exhibit naïve pluripotency reminiscent of the E3.5 pre-implantation ICM [53]. The primed state is marked by X chromosome inactivation and highly inefficient germline contribution, whereas both X chromosomes are active in naïve ESCs and pre-implantation chimeras are readily formed. However, primed vs. naïve pluripotency cannot explain the differential impacts of YAP in mouse vs human, as overexpression of Yap1 in mESCs causes premature upregulation of lineage markers, whereas overexpression of YAP in hESCs causes them to enter a naïve state of pluripotency [44, 45]. We propose that it is more likely that differences in binding partners, genomic targets, sequence, isoform, or even the degree of overexpression may contribute to these opposite results rather than developmental staging alone. To our knowledge, depletion or ablation of YAP in naïve hESCs has not yet been performed; thus, it is unclear whether YAP is essential for maintaining human naïve pluripotency or merely acquiring it.
YAP promotes pluripotency in hESCs through a variety of mechanisms. It is known that SMADs sustain the expression of NANOG in the absence of WNT3, but when WNT3 is present, SMADs cooperate with β-catenin to induce mesendoderm marker expression [54]. YAP impairs the recruitment of SMADs and reduces occupancy of RNA polymerase II (RNAPII) at the WNT3 locus, preventing expression of WNT3 [55]. YAP expression appears to be highly correlated with the self-renewing state: growing hESCs in 3D culture increases both YAP and core factor expression, and verteporfin-mediated inhibition of YAP strongly reduces expression of both core and naïve markers [56]. Intriguingly, YAP also maintains self-renewal of the human TE by binding to the promoters of CCNA and CDK6 in cytotrophoblasts [40]. This would imply that in the human blastocyst, both the ICM and the TE identities are sustained by YAP, compared to only the TE in the mouse. We have summarized the effects of YAP in cell fate changes in Fig. 2, although this covers not just its effects in stem cells, but in all other major cell fate decisions covered in this review as well.

An overview of YAP-mediated cell fate changes. a Yap1 promotes differentiation of mESCs via upregulation of lineage markers, particularly trophoblast. b YAP promotes naive pluripotency of hESCs via upregulation of core factors and suppression of β-catenin activity. c In tandem with TEAD4, endogenous YAP promotes self-renewal of TSCs in both human and mouse by activating stemness markers. d Ectopic expression of YAP allows dedifferentiation of somatic cells like neurons to a more stem-like state. e YAP is essential for regeneration in some organs (lung, liver, retina, and intestines) by facilitating temporary cell fate switch to a progenitor-like state after organ injury. f In some organoids (lung, kidney, and esophageal), YAP expression promotes branching and morphogenesis. g MSCs are pushed to the osteogenic fate on stiff substrate when YAP is active. h YAP can either prevent apoptosis in cancer cells by binding to TEAD, or promote apoptosis by binding to p73
Early lineage specification and differentiation in both human and mouse
Further research confirms the importance of Yap1 in mediating the development of all three germ layers, particularly the neuroectoderm in mouse as well as repressing the mesendoderm lineage in human. During differentiation, Rassf1a promotes the phosphorylation of Yap1, which causes it to switch binding partners from Tead2 and β-catenin to p73 and initiate lineage-specific transcriptional programs [57]. However, not all three germ layers are equally favored, and it seems that in mice, Yap1 favors the ectoderm lineage [58]. Mechanistically, activation of Yap1 (for example, via KO of upstream Hippo kinases Mst1/2) leads to the formation of novel phase-separated Med1-positive super-enhancers that promote the expression of ectoderm markers [55] while downregulating mesendoderm markers [58]. In humans, YAP appears to inhibit the cardiac fate as culturing YAP-/- hESCs in Activin A results in the spontaneous derivation of beating cardiomyocytes. Therefore, YAP’s complexity and its associated signaling crosstalk seem to depend on species.
Yap1 seems to regulate apoptosis in ESCs, as well. Apoptosis occurs naturally during differentiation in part to cull mESCs that fail to exit self-renewal efficiently, and Bax−/−Bak−/− double KO cells, which cannot undergo apoptosis, display marked defects in embryoid body and teratoma formation [59]. Apoptosis during differentiation occurs not only during LIF withdrawal but also during other modes of differentiation, such as epiblast [60]. Differentiating mESCs that lack Yap1, regardless of their ultimate fate, undergo 2–3-fold higher rates of apoptosis due to defective expression of Bcl-2 and Mcl1 [61]. Yap1−/− mitochondria are more sensitive to apoptotic stress, leading to hyperactivation of caspase-9, and this can be rescued by overexpression of Bcl-2 or Bcl-xL. Intriguingly, double knockdown of YAP and TAZ in hESCs results in cell death that can also be rescued by BCL-xL overexpression, implying that they redundantly guard against apoptosis during primed pluripotency [62]. Although it has not yet been tested whether YAP protects against apoptosis during hESC differentiation, YAP appears to fill a dual role in stem cell survival and identity that varies depending on species and lineage.
YAP and somatic stem cells
Reacquisition of a stem-like state via YAP overexpression
Though not as potent as ESCs, many lineage-restricted progenitor/stem cells exist in post-natal tissues, and their cell fate is often guided by YAP. It has recently been appreciated that activation of YAP helps mediate the reversion of differentiated cells to a more primitive, stem-like state. Organ regeneration after injury is mediated by the expansion and differentiation of tissue-specific somatic (sometimes referred to as adult) stem cells. Unfortunately, in humans, somatic stem cells are often scarce, limiting regeneration. Typical approaches for stem cell therapy involve reprogramming of patient-derived fibroblasts into iPSCs; however, iPSCs can form tumors when introduced into adult tissues if not completely differentiated [63]. Thus, the derivation and transplantation of non-tumorigenic somatic stem cells have been proposed as an alternative to iPSCs.
Fascinatingly, brief overexpression of YAP or TAZ for 5–15 days can convert luminal mammary cells, neurons, and pancreatic acinar cells into their respective lineage-restricted somatic stem cells [64, 65]. For example, it is possible to reconstitute a cleared mammary gland with a YAP-induced mammary stem cell-derived organoid, and the resultant regenerated gland can differentiate into clusters of milk-secreting alveoli in a pregnant mouse [65]. In the adult mouse heart, constitutively activating YAP partially reprograms cardiomyocytes into a fetal-like, proliferative state via chromatin remodeling [66]. Furthermore, overexpression of YAP rejuvenates senescent mesenchymal stem cells and alleviates symptoms of osteoarthritis in mice, and conditional KO of YAP in astrocytes inhibits regeneration of axons after spinal cord injury [67,68,69,70]. These findings open tantalizing possibilities for safer stem cell therapy.
Endogenous YAP and regeneration
Aside from intentional and ectopic overexpression of YAP to generate somatic stem cells, reverse senescence, or promote regeneration, it has become increasingly clear that normal organ repair after injury involves transient YAP activation. For example, colonic regeneration in a mouse model of colitis and restoration of the adult heart post-myocardial infarction involves YAP and Lgr5+ stem cells in intestinal tissues, and organoids undergo expansion after injury in a YAP-dependent manner [71,72,73,74,75]. Also, YAP is critical for regeneration in the lung after damage to the alveolar epithelium, in the liver for hepatocytes to reprogram into biliary progenitors after the injury, and in the retina for Müller glia to proliferate after eye injury [76,77,78,79,80,81]. In several of the aforementioned examples, the mechanism by which YAP promotes regeneration is not elucidated. However, at least in pancreatic and mammary contexts, dedifferentiation caused by YAP or TAZ activation appears to be dependent on increased autophagy [82].
This has been observed in other species as well. In the mouse liver, acute inactivation of YAP efficiently dedifferentiates hepatocytes into self-renewing liver progenitor-like cells [83]. Also, caudal fin regeneration in zebrafish occurs through Yap1 promoting osteoblast differentiation, which is reminiscent of how Yap1 and Taz promote osteoblast activity in mice [84, 85]. These studies strongly evince that YAP serves as a common mechanism for differentiated cells to revert to more primitive states in response to tissue damage and participate in regeneration, and this capacity of YAP presents an invaluable opportunity for patient-specific stem cell therapy without needing to generate iPSCs.
YAP functions in organoid cultures
Contextual roles of YAP in promoting morphogenesis in organoids
Organoids have been proposed as an in vitro method to explore how pluripotent or somatic stem cells form tissues and organs in 3D [86]. They circumvent some of the ethical and financial concerns of in vivo models while also allowing for straightforward genetic manipulation and patient-specific genotypes [87]. Intriguingly, multiple organoid models have shown diverse roles of YAP in either promoting or blocking differentiation and morphogenesis. For example, an organoid model of severe liver damage reveals that the activity of YAP is essential for TET1, a DNA demethylase implicated in reprogramming, to epigenetically reset ductal cells, allowing them to differentiate into cholangiocytes and hepatocytes [88]. In lung organoids, nuclear YAP is found at invasive tubular structures, and knockdown of YAP disrupts the formation of these structures [89]. Similarly, in kidney organoids, nuclear YAP is observed at the tips of elongating ureteric buds, which further supports its roles in cell migration and tissue morphogenesis [90]. Conversely, inhibition of the retinoic acid pathway prevents alveolar epithelial differentiation by augmenting YAP nuclear translocation [91], whereas YAP promotes epithelial differentiation in mouse esophageal organoids [92]. Additionally, retinoic acid inhibition promotes a regenerative state in intestinal organoids and blocks differentiation by maintaining active nuclear YAP [93]. Thus, organoid models also suggest context-dependent roles of YAP in driving or suppressing differentiation.
Organoids have also allowed insights into mechanical and paracrine regulation of YAP. In intestinal organoids, high matrix stiffness increases YAP activity and promotes intestinal stem cell expansion [94]. When mouse intestinal organoids are co-cultured with fibroblasts, the fibroblasts secrete prostaglandin E2, promoting the expansion of the stem cell population via Yap1 [95]. Furthermore, bile acids promote intestinal organoid growth, self-renewal, and fate specification, likely through YAP [96]. Similarly, matrix stiffness promotes liver organoid formation via focal adhesion kinase and YAP [97]. In brain organoids cultured in hyaluronic acid, a common component of the extracellular matrix (ECM), YAP is indispensable for forebrain patterning [98]. Given that embryogenesis is also known to be influenced by the extracellular matrix and paracrine signaling [99], it would be intriguing to investigate how both the matrix and paracrine factors influence YAP functions in vivo and in vitro.
Using organoids to study roles of YAP in early embryogenesis
As ethical concerns make it difficult to molecularly dissect the roles and regulation of YAP in the human embryo, researchers have started using 2D micropatterning and 3D models such as blastoids, peri-implantation organoids, and gastruloids to understand germ layer specification, pre-gastrulation development, and body plan specification [100]. Despite the existence of these models, they have not been used to elucidate embryogenesis-related functions of YAP in greater depth. Recently, hESCs have been used to generate a trophoblastic spheroid that has been proposed as a blastocyst surrogate; YAP expression is more intense and nuclear in these spheroids, and inhibition of YAP leads to defects in attachment and outgrowth [101]. This is promising, but follow-up studies are needed. Gastruloids in particular represent an exciting avenue for elucidating the regulation of YAP during embryogenesis in mouse vs. human. Placental organoids should also be pursued due to the essential roles of YAP and TEAD4 in TE specification in both species [42].
Functions of TAZ and canonical downstream targets in cell fate
Biochemical differences between YAP and TAZ
Differences in protein stability
Many researchers in the field of Hippo signaling consider YAP and TAZ to have very similar functions, even considering them redundant to the point where they are referred to interchangeably as YAP/TAZ [64, 102]. As paralogs with high (~ 50%) sequence identity, these two proteins possess both redundant (i.e., they can genetically compensate for one another) and non-redundant roles [2]. They have a similar target gene profile, and they are redundant in the oviduct, heart, urinary tract, and other organs [31, 103,104,105]. However, they have numerous context-specific differences and fulfill distinct roles during embryogenesis and cell fate specification.
One of the starkest differences between YAP and TAZ is the presence of an additional N-terminal phosphodegron in TAZ, leading to a much shorter half-life [106]. In mouse embryonic fibroblasts, the half-life of TAZ is a mere 2 h, whereas YAP protein levels remain stable for 6 h [107]. YAP and TAZ are frequently regulated by altering their protein stability, although they are sometimes impacted differently. For example, nonreceptor tyrosine kinase PYK2 enhances tyrosine phosphorylation of TAZ but not YAP in triple-negative breast cancer, increasing its stability, and their expression is positively correlated in primary breast tumors [108]. Furthermore, in both mouse and human, YAP promotes the degradation of TAZ via GSK3 and HSP90, whereas TAZ does not affect the protein abundance of YAP [109]. This is controversial as in human corneal fibroblasts, TAZ knockdown leads to elevated YAP levels but not vice versa [110]. Having both paralogs—one relatively stable, the other rapidly fluctuating, and both possibly engaging in negative feedback—may be advantageous for mammalian cell homeostasis.
TEAD complex formation and dimerization
Aside from these differences in half-life, YAP and TAZ display different binding modes to TEAD as well as distinct, isoform-specific capacities for dimerization and phase separation. Whereas human and mouse YAP binds to TEAD in a 1:1 heterodimeric complex, the crystal structure of the mouse Taz–Tead4 is a heterotetrameric complex where two molecules of Taz bind to two molecules of Tead4, likely due to the absence of a PXXΦP motif in the Taz linker sequence [111, 112]. Furthermore, unlike YAP, TAZ overexpression can induce phase separation, allowing it to compartmentalize partner TFs and promote transcription, and TAZ can homodimerize via its coiled-coil domain, whereas YAP cannot [113, 114]. Thus, organisms may require both YAP and TAZ due to their regulation being so distinct; YAP levels may remain stable over relatively long periods, whereas TAZ levels can rapidly increase, homodimerize, and phase separate, decreasing quickly when it is no longer needed. However, their comparative abilities to dimerize and phase separate remain controversial. A long isoform of YAP known as YAP2L can homodimerize both in vitro and in vivo, while shorter isoforms cannot [115]. Also, YAP does have an intrinsically disordered region, and upon hyperosmotic stress, YAP forms liquid-like condensates around super-enhancers [116]. Future research focusing on isoform-specific functions of YAP and TAZ should be carried out to explain these inconsistencies and determine in what physiological contexts they are most relevant.
Domain architecture, binding partners, and unique targets
These biochemical differences between YAP and TAZ as well as differences in primary sequence contribute to differences in binding partners and transcriptional targets. Aside from an additional phosphodegron in TAZ, these two proteins have slightly different domain architecture. In YAP, from N- to C-terminus, this includes a proline-rich domain, a TEAD-binding domain, 1–2 WW domains that interact with PPXY motifs on proteins like LATS, p73, and many others, an SH3-binding domain, a coiled-coil domain, a transactivation domain, and a PDZ-binding domain [117,118,119,120,121,122,123]. TAZ only has one WW domain and lacks a proline-rich domain or SH3-binding domain. We have detailed the domain structure of YAP and TAZ and indicated several of their associated binding partners in Fig. 3.

YAP and TAZ domain architecture. Overview of YAP and TAZ domains and phosphodegrons as well as key binding partners. YAP’s PXXΦP motif as well as regulatory serines and a tyrosine that are known to be phosphorylated are also indicated. BD binding domain
As a result of differences in sequence and domain architecture, TAZ, but not YAP, binds to Smad4 in mesenchymal stem cells, facilitating its nuclear localization, and enhancing osteogenesis in partnership with RUNX2 [124]. TAZ also promotes osteogenesis in bone marrow mesenchymal stem cells and periodontal ligament cells [125, 126]. YAP also increases osteogenesis, yet it works with a different set of binding partners, and in tandem, they regulate osteoblast and osteoclast activity [85, 127]. TAZ blocks adipogenesis by acting as a co-repressor of PPARγ via direct interaction and repression of its targets [128]. Intriguingly, parafibromin, a nuclear scaffold, interacts with YAP after parafibromin is tyrosine phosphorylated, but after dephosphorylation, it interacts with TAZ instead [129]. Finally, these distinct binding partners result in some unique transcriptional targets. The integrin ITGAV is a target of TAZ, but not YAP, in hepatocellular carcinoma, and it is associated with poor clinical outcome, indicating unique roles of TAZ in oncogenesis [130]. In ovarian cancer, ANGPTL4 is a direct target of TAZ but not YAP [131]. Intriguingly, in non-small cell lung cancer, their overall transcriptional programs have little overlap: YAP activates genes involved in cell cycle progression whereas TAZ activates genes critical for migration, resulting in differential sensitivity to chemotherapeutics [132]. Therefore, their unique transcriptional targets can have biologically significant consequences. Future research should combine structural and in vivo approaches to elucidate the functions of more of their targets and, most urgently, how TAZ and YAP can regulate different sets of genes despite their similarities.
TAZ in cell fate specification
In development and stem cells
As noted earlier, YAP KO is embryonic lethal after E8.5 due to gross body axis defects, yolk sac vascular defects, and failure to undergo chorioallantoic fusion, whereas TAZ KO leads to kidney and lung defects as well as post-natal lethality. They also have unique roles in maintaining primed pluripotency. Cytoplasmic TAZ promotes the self-renewal of both hESCs and mouse epiblast stem cells (mEpiSCs) via sequestration of β-catenin, whereas nuclear translocation of TAZ results in mEpiSC differentiation [133]. However, in that study, YAP did not associate with β-catenin in either hESCs or mEpiSCs. Thus, the two are not functionally redundant in early embryogenesis or in primed pluripotency, though it is unknown whether TAZ also participates in the primed-to-naïve transition as YAP does.
In somatic cells
Despite dramatic disparities in embryonic phenotype which have not yet been explored in mechanistic detail, most of the research comparing YAP and TAZ in differentiation has focused on adult tissues. TAZ is much less studied than YAP, and they are rarely directly compared, limiting our ability to assess their redundancy. However, it is known that YAP and TAZ exert complementary time-sensitive roles in the lungs, whereas in fibroblasts, T cells, and myoblasts, YAP and TAZ play somewhat opposing roles. Volckaert et al. in 2019 found that cytoplasmic Yap1 suppresses Fgf10 by promoting the degradation of β-catenin during early lung development, enabling alveolar epithelial differentiation, whereas Taz promotes the differentiation of alveolar type 1 cells [134]. Furthermore, TAZ, but not YAP, is essential for the differentiation of alveolar type 2 to alveolar type 1 cells, and conditional deletion of TAZ reduces lung regeneration and increases fibrosis [135].
Meanwhile, in corneal fibroblasts, their roles are contrasting rather than complementary. Knockdown of YAP in these cells leads to reduced CTGF and Smad2/3/4 expression, whereas knockdown of TAZ upregulates CTGF and αSMA, and simultaneous depletion causes cell death much like in hESCs [110]. TAZ is required for pro-inflammatory TH17 helper T-cell differentiation and prevents differentiation toward immunomodulatory Treg cells [136]. Meanwhile, YAP inhibits naïve T-cell differentiation regardless of ultimate fate, and the deletion of YAP enhances differentiation to TH1, TH17, TH2, and Treg fates under polarizing conditions [137]. Whereas both Taz and Yap1 promote myoblast proliferation, only Taz binds to Tead4 in myoblasts to enhance myogenic differentiation [138]. In sum, despite observations that YAP and TAZ are similar enough to be redundant in many circumstances, their biochemical and regulatory differences are sufficient to result in distinct consequences for cell fate specification, as summarized in Table 1. Future studies should address the physiological implications and structural underpinnings of their non-redundancy.
YAP/TAZ and apoptosis in cancer
YAP as an oncogene or a tumor suppressor via apoptosis regulation
Cancer types where YAP reduces apoptosis
Due to metabolic and signaling similarities between cancer and stem cells, it is unsurprising that YAP regulates similar processes in the two cell types [24, 139]. YAP orchestrates many processes during oncogenesis, including metastasis, chemoresistance, and cancer stemness. Focusing on apoptosis, in most cancer types, ranging from gastric to colon to renal, YAP inhibits apoptosis, as seen in mESCs, to help drive tumor progression. Furthermore, YAP expression is generally associated with poor prognosis and heightened tumor aggressiveness in cancers like oral squamous cell carcinoma and cholangiocarcinoma [140, 141]. However, much like the disagreement about whether YAP promotes self-renewal vs. differentiation in ESCs, there have been reports where YAP has functioned as a tumor suppressor and promoted apoptosis rather than preventing it [142].
In gastric cancer, YAP reduces apoptosis by promoting mitophagy downstream of SIRT1 activity, MFN2 expression, and upregulation of growth factors, whereas the targeting of YAP increases apoptosis [143,144,145]. In clear cell renal cell carcinoma [146] and rhabdomyosarcoma [147], YAP is upregulated, and YAP depletion increases apoptosis and decreases proliferation, even in murine xenografts in vivo. Inhibition of YAP via the chemotherapeutic norcantharidin enhances apoptosis in non-small cell lung carcinoma [148]. Similarly, targeting YAP in liver cancer [149, 150] and breast cancer [151, 152] induces apoptosis, reduces proliferation, and increases chemosensitivity. Thus, it is evident that in many different cancer types, YAP impedes apoptosis, and its depletion is sufficient to cause apoptosis in cancer, which establishes YAP as a valuable therapeutic target in the clinic.
Mechanisms of apoptosis regulation via YAP
Like during mESC differentiation, YAP primarily regulates apoptosis in cancer via transcriptional regulation of anti- and pro-apoptotic genes, most commonly BCL-2 and BAX. In colon adenocarcinoma, overexpression of YAP or TAZ reduces apoptosis via the upregulation of BCL-2 and downregulation of BAX; reducing YAP levels is sufficient to trigger apoptosis [153, 154]. BAX is repressed by YAP in prostate cancer [155,156,157], and YAP, TEAD, and SLUG collaborate to directly repress pro-apoptotic BMF in dormant non-small cell lung cancer [158]. In senescent tumor cells, YAP sustains expression of the anti-apoptotic factor survivin [159]. YAP activates expression of BCL-xL in bladder cancer, and depletion of YAP increases apoptosis [160]. Using a hybrid spheroid model of cell competition, Liu et al. showed that glioblastoma cells express YAP heterogeneously, and YAP-low cells experience more apoptosis, reminiscent of the E4.5 mouse epiblast, though it is not specified what downstream targets are involved [161]. Thus, many of the mechanisms by which YAP influences ESC survival are shared in cancer.
YAP as a tumor suppressor
Although YAP has often been described as an oncogene, it can act as pro-apoptotic in select contexts. YAP’s capacity for triggering apoptosis by binding p73 and upregulating BAX has been known for nearly 2 decades [142, 162,163,164]. YAP switches binding partners from TEAD factors to p73 after phosphorylation by c-Abl on Y357 in response to DNA damage [165]. More recent research has shown that YAP can increase apoptosis in breast cancer [166, 167], lung cancer [168], and liver cancer [169] cells, typically downstream of or alongside apoptotic stimuli like chemotherapy. This phenomenon has even been shown in non-cancer contexts, such as damaged mouse livers where Yap1high hepatocytes undergo apoptosis [170]. Intriguingly, in Huntington’s disease, YAP in conjunction with TEAD can induce endoplasmic reticulum swelling and necrosis in primary neurons [171]. It still remains unknown whether targets of the YAP-p73 complex other than BAX are relevant to promoting apoptosis as well as whether other TFs can switch YAP to a pro-apoptotic transcriptional program.
Regulation of YAP in cancer
Regulation of YAP protein levels in cancer
YAP itself is positively regulated by various genes that tend to be highly expressed in cancer. Many of these oncogenes reduce apoptosis in cancer cells by increasing YAP protein levels or its stability. Overexpression of E3 ubiquitin ligase FBXW7 increases apoptosis in hepatocellular carcinoma by targeting YAP for ubiquitination and degradation, and the expression of these two proteins is inversely correlated in tumors [172]. Ankyrin repeat-containing protein Kank1, when overexpressed, reduces the proliferation and increases the apoptosis of oral squamous cell carcinoma by reducing Yap1 and Taz protein expression [141]. The knockdown of ubiquitin-specific protease USP22 causes gastric cancer cell apoptosis via a reduction in YAP protein levels [173]. Therefore, altered post-translational regulation of YAP may be a common strategy for tumors to resist apoptotic stimuli.
Noncoding RNAs and YAP
In addition to post-translational regulation of YAP itself, various noncoding RNAs are overexpressed in cancer and suppress apoptosis by targeting Hippo kinases to indirectly raise YAP levels. miR-25, which is overexpressed in non-small cell lung cancer and suppresses apoptosis, increases YAP1 levels by targeting LATS2 [174]. miR-224-3p also represses LATS2, and its suppression leads to apoptosis [175]. The lncRNA MALAT, which is highly expressed in pancreatic cancer tissues, also targets LATS1, and knockdown of MALAT also induces apoptosis in pancreatic cancer cells [176]. Upstream of LATS kinases is KIBRA, which is targeted by miR-21 expressed in lung adenocarcinoma tissues [177]. miR-4319, a tumor suppressor that is lost in breast and non-small cell lung cancer, increases apoptosis by targeting LIN28, leading to destabilization of RFX5, a positive regulator of YAP mRNA expression [178]. In summary, tumors manipulate both post-translational regulation of YAP as well as post-transcriptional regulation of its upstream kinases via noncoding RNAs to further their own survival. Future studies should examine whether the suppression of these noncoding RNAs can facilitate cancer treatment.
TAZ vs. YAP in cancer
TAZ as an oncogene
Much like YAP, TAZ is generally considered an oncogene whose expression is correlated with poor cancer prognosis [179, 180]. Depletion or inhibition of TAZ in prostate cancer [22], breast cancer [181], or glioma [93] induces apoptosis. Additionally, there are miRNAs that repress TAZ that tend to be downregulated in cancer. For example, miR-550-1 targets TAZ in acute myeloid leukemia [112] and miR-455-3p targets TAZ in pancreatic cancer [182]. Notably, unlike YAP, TAZ has a natural antisense transcript known as WWTR1-AS1; knockdown of this transcript in head–neck squamous cell carcinoma reduces TAZ expression and increases apoptosis [183]. Though YAP and TAZ are rarely compared in parallel in any given cancer study, it has been shown that TAZ, but not YAP, is upregulated in alveolar rhabdomyosarcoma, and its depletion induces apoptosis and reduces xenograft growth [184]. Importantly, their unique roles in cancer have clinical significance. YAPhigh cancer cell lines are sensitive to the chemotherapeutic paclitaxel, but TAZhigh cell lines are not, and many other cancer drugs seem to affect the two groups differently [132]. Given these findings, future studies on YAP or TAZ in cancer should strive to consider both paralogs and avoid treating them as functionally redundant.
TAZ as a tumor suppressor
Even though TAZ does not interact with p73 like YAP does to promote apoptosis, TAZ can act as a tumor suppressor via both apoptotic and non-apoptotic mechanisms. In multiple myeloma, the WWTR1 promoter is hypermethylated and thus silenced; re-expression of TAZ leads to apoptosis via repression of MYC and its targets [185]. TAZ was recently discovered to promote ferroptosis, an iron-dependent form of non-apoptotic programmed cell death, in ovarian cancer [131]. Intriguingly, TAZ, but not YAP, sensitizes renal cell carcinoma cell lines to ferroptosis via regulation of EMP1, whereas YAP promotes ferroptosis in colon cancer via different targets such as ACSL4 [186, 187]. However, compared to the research on their roles in tumorigenesis, much less is known about how and why these proteins promote cell death in certain situations. Thus, additional research should be performed to determine whether it is possible to take advantage of tumor suppressive YAP or TAZ in chemotherapy.
Future perspectives
YAP and TAZ are essential for proper development, but their precise roles can differ dramatically depending on the cellular context. They can promote regeneration and reprogramming in some tissues while driving differentiation and morphogenesis in others, and their classical categorization as oncogenes is an oversimplification given their surprising ability to act as tumor suppressors in some cancer contexts. Furthermore, YAP and TAZ crosstalk extensively with other pathways, especially canonical Wnt signaling and β-catenin, but again the nature and implications of these interactions depend strongly on tissue type. In sum, YAP and TAZ display profoundly multifaceted capabilities in regulating cell fate specification and cell survival. The controversies surrounding their roles in self-renewal and apoptosis serve as a cautionary tale for extrapolating findings concerning YAP and TAZ outside of their original biological contexts.
Great strides have been made in recent years concerning the functions and regulation of YAP and TAZ, and yet much remains unknown. Most notably, YAP’s role along the pluripotency spectrum—from naïve to primed—has not been fully elucidated, despite the availability of organoid models for embryogenesis and increasingly nuanced views of how each stage of pluripotency is acquired and maintained. Additionally, it is unknown why YAP behaves as an oncogene in some contexts, but a tumor suppressor in others, and the consequences of how programmed cell death and cell fate specification interact. Furthermore, aside from differences between YAP and TAZ themselves, functional differences between the poorly characterized isoforms of YAP or TAZ may be biologically significant. Finally, it is tempting to determine whether it is possible to harness the pro-regenerative potential of YAP-mediated reprogramming without risking tumorigenesis, and whether these YAP-mediated processes proceed via distinct or shared genomic targets and binding partners. In recent years, various small molecules, peptides, and even a genetically encoded inhibitor have been shown to antagonize or promote YAP and TAZ activity, which should facilitate future research on these proteins [188,189,190,191,192]. However, we also speculate that increasing awareness of non-redundant roles between YAP and TAZ will motivate the development of paralog- or even isoform-specific inhibitors.
Aside from these specific inquiries, future research on YAP and TAZ should focus on incorporating single-cell and single-molecule approaches, revisiting KO phenotypes and conflicting roles in stem cells using organoid models of the early embryo, and investigating non-cell-autonomous functions of YAP. Furthermore, we contend that differences between in vitro culture systems in different labs and seeding density also influence YAP localization and activity, and such confounding factors should be taken into more careful consideration during both experimental design and execution.
References
Huang J, Wu S, Barrera J, Matthews K, Pan D (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the <em>Drosophila</em> Homolog of YAP. Cell 122(3):421–434. https://doi.org/10.1016/j.cell.2005.06.007
Varelas X (2014) The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141(8):1614. https://doi.org/10.1242/dev.102376
Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015) Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17(9):1218–1227. https://doi.org/10.1038/ncb3216
Galli GG, Carrara M, Yuan W-C, Valdes-Quezada C, Gurung B, Pepe-Mooney B, Zhang T, Geeven G, Gray NS, de Laat W, Calogero RA, Camargo FD (2015) YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell 60(2):328–337. https://doi.org/10.1016/j.molcel.2015.09.001
Kim M, Kim T, Johnson Randy L, Lim D-S (2015) Transcriptional co-repressor function of the Hippo pathway transducers YAP and TAZ. Cell Rep 11(2):270–282. https://doi.org/10.1016/j.celrep.2015.03.015
Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schübeler D, Bauer A (2015) YAP1 exerts its transcriptional control via TEAD-mediated activation of enhancers. PLoS Genet 11(8):e1005465–e1005465. https://doi.org/10.1371/journal.pgen.1005465
Seo J, Kim J (2018) Regulation of Hippo signaling by actin remodeling. BMB Rep 51(3):151–156. https://doi.org/10.5483/bmbrep.2018.51.3.012
Misra JR, Irvine KD (2018) The hippo signaling network and its biological functions. Annu Rev Genet 52:65–87. https://doi.org/10.1146/annurev-genet-120417-031621
Pan D (2010) The hippo signaling pathway in development and cancer. Dev Cell 19(4):491–505. https://doi.org/10.1016/j.devcel.2010.09.011
Park JH, Shin JE, Park HW (2018) The role of hippo pathway in cancer stem sell biology. Mol Cells 41(2):83–92. https://doi.org/10.14348/molcells.2018.2242
Feng X, Liu P, Zhou X, Li M-T, Li F-L, Wang Z, Meng Z, Sun Y-P, Yu Y, Xiong Y, Yuan H-X, Guan K-L (2016) Thromboxane A2 activates YAP/TAZ protein to induce vascular smooth muscle cell proliferation and migration. J Biol Chem 291(36):18947–18958. https://doi.org/10.1074/jbc.M116.739722
Miller E, Yang J, DeRan M, Wu C, Su Andrew I, Bonamy Ghislain MC, Liu J, Peters Eric C, Wu X (2012) Identification of serum-derived Sphingosine-1-Phosphate as a small molecule regulator of YAP. Chem Biol 19(8):955–962. https://doi.org/10.1016/j.chembiol.2012.07.005
Yu F-X, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu X-D, Mills GB, Guan K-L (2012) Regulation of the hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150(4):780–791. https://doi.org/10.1016/j.cell.2012.06.037
Totaro A, Panciera T, Piccolo S (2018) YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20(8):888–899. https://doi.org/10.1038/s41556-018-0142-z
Zhang Q, Han X, Chen J, Xie X, Xu J, Zhao Y, Shen J, Hu L, Xu P, Song H, Zhang L, Zhao B, Wang Y-J, Xia Z (2018) Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) mediate cell density-dependent proinflammatory responses. J Biol Chem 293(47):18071–18085. https://doi.org/10.1074/jbc.RA118.004251
Bell RAV, Megeney LA (2017) Evolution of caspase-mediated cell death and differentiation: twins separated at birth. Cell Death Differ 24(8):1359–1368. https://doi.org/10.1038/cdd.2017.37
Hu Q, Wu D, Chen W, Yan Z, Shi Y (2013) Proteolytic processing of the caspase-9 zymogen is required for apoptosome-mediated activation of caspase-9. J Biol Chem 288(21):15142–15147. https://doi.org/10.1074/jbc.M112.441568
Li Y, Zhou M, Hu Q, Bai X-C, Huang W, Scheres SHW, Shi Y (2017) Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci U S A 114(7):1542–1547. https://doi.org/10.1073/pnas.1620626114
Yuan S, Akey CW (2013) Apoptosome structure, assembly, and procaspase activation. Structure (London, England : 1993) 21(4):501–515. https://doi.org/10.1016/j.str.2013.02.024
Kale J, Osterlund EJ, Andrews DW (2018) BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25(1):65–80. https://doi.org/10.1038/cdd.2017.186
Zhang X, Abdelrahman A, Vollmar B, Zechner D (2018) The ambivalent function of YAP in apoptosis and cancer. Int J Mol Sci 19(12):3770. https://doi.org/10.3390/ijms19123770
Reggiani F, Gobbi G, Ciarrocchi A, Sancisi V (2020) YAP and TAZ are not identical twins. Trends Biochem Sci. https://doi.org/10.1016/j.tibs.2020.08.012
Chae YC, Kim JH (2018) Cancer stem cell metabolism: target for cancer therapy. BMB Rep 51(7):319–326. https://doi.org/10.5483/bmbrep.2018.51.7.112
Ratajczak MZ, Bujko K, Mack A, Kucia M, Ratajczak J (2018) Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms. Leukemia 32(12):2519–2526. https://doi.org/10.1038/s41375-018-0294-7
Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434(7035):843–850. https://doi.org/10.1038/nature03319
Morin-Kensicki EM, Boone BN, Howell M, Stonebraker JR, Teed J, Alb JG, Magnuson TR, O’Neal W, Milgram SL (2006) Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol Cell Biol 26(1):77–87. https://doi.org/10.1128/MCB.26.1.77-87.2006
Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K, Qi Z, Ponniah S, Hong W, Hunziker W (2007) Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci U S A 104(5):1631–1636. https://doi.org/10.1073/pnas.0605266104
Makita R, Uchijima Y, Nishiyama K, Amano T, Chen Q, Takeuchi T, Mitani A, Nagase T, Yatomi Y, Aburatani H, Nakagawa O, Small EV, Cobo-Stark P, Igarashi P, Murakami M, Tominaga J, Sato T, Asano T, Kurihara Y, Kurihara H (2008) Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am J Phys Renal Phys 294(3):F542–F553. https://doi.org/10.1152/ajprenal.00201.2007
Tian Y, Kolb R, Hong J-H, Carroll J, Li D, You J, Bronson R, Yaffe MB, Zhou J, Benjamin T (2007) TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex. Mol Cell Biol 27(18):6383–6395. https://doi.org/10.1128/MCB.00254-07
Nishioka N, Inoue K-i, Adachi K, Kiyonari H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N, Makita R, Kurihara H, Morin-Kensicki EM, Nojima H, Rossant J, Nakao K, Niwa H, Sasaki H (2009) The hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse Trophectoderm from inner cell mass. Dev Cell 16(3):398–410. https://doi.org/10.1016/j.devcel.2009.02.003
Godin P, Tsoi M, Paquet M, Boerboom D (2020) YAP and TAZ are required for the postnatal development and the maintenance of the structural integrity of the oviduct. Reproduction 160(2):307–318. https://doi.org/10.1530/REP-20-0202
Wang J, Xiao Y, Hsu C-W, Martinez-Traverso IM, Zhang M, Bai Y, Ishii M, Maxson RE, Olson EN, Dickinson ME, Wythe JD, Martin JF (2016) Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development (Cambridge, England) 143(3):504–515. https://doi.org/10.1242/dev.126920
Zhang H, von Gise A, Liu Q, Hu T, Tian X, He L, Pu W, Huang X, He L, Cai C-L, Camargo FD, Pu WT, Zhou B (2014) Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J Biol Chem 289(27):18681–18692. https://doi.org/10.1074/jbc.M114.554584
Hashimoto M, Sasaki H (2019) Epiblast formation by TEAD-YAP-dependent expression of Pluripotency factors and competitive elimination of unspecified cells. Dev Cell 50(2):139-154.e135. https://doi.org/10.1016/j.devcel.2019.05.024
Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, Strasser A (2018) Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell 173(5):1217-1230.e1217. https://doi.org/10.1016/j.cell.2018.04.036
Perez-Garcia V, Fineberg E, Wilson R, Murray A, Mazzeo CI, Tudor C, Sienerth A, White JK, Tuck E, Ryder EJ, Gleeson D, Siragher E, Wardle-Jones H, Staudt N, Wali N, Collins J, Geyer S, Busch-Nentwich EM, Galli A, Smith JC, Robertson E, Adams DJ, Weninger WJ, Mohun T, Hemberger M (2018) Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 555(7697):463–468. https://doi.org/10.1038/nature26002
Wicklow E, Blij S, Frum T, Hirate Y, Lang RA, Sasaki H, Ralston A (2014) HIPPO pathway members restrict SOX2 to the inner cell mass where it promotes ICM fates in the mouse blastocyst. PLoS Genet 10(10):e1004618–e1004618. https://doi.org/10.1371/journal.pgen.1004618
Rayon T, Menchero S, Nieto A, Xenopoulos P, Crespo M, Cockburn K, Cañon S, Sasaki H, Hadjantonakis A-K, de la Pompa JL, Rossant J, Manzanares M (2014) Notch and hippo converge on Cdx2 to specify the trophectoderm lineage in the mouse blastocyst. Dev Cell 30(4):410–422. https://doi.org/10.1016/j.devcel.2014.06.019
Kaneko KJ, DePamphilis ML (2013) TEAD4 establishes the energy homeostasis essential for blastocoel formation. Development (Cambridge, England) 140(17):3680–3690. https://doi.org/10.1242/dev.093799
Meinhardt G, Haider S, Kunihs V, Saleh L, Pollheimer J, Fiala C, Hetey S, Feher Z, Szilagyi A, Than NG, Knöfler M (2020) Pivotal role of the transcriptional co-activator YAP in trophoblast stemness of the developing human placenta. Proc Natl Acad Sci 117(24):13562. https://doi.org/10.1073/pnas.2002630117
Liu R, Wei C, Ma Q, Wang W (2020) Hippo-YAP1 signaling pathway and severe preeclampsia (sPE) in the Chinese population. Pregnancy Hypertens 19:1–10. https://doi.org/10.1016/j.preghy.2019.11.002
Saha B, Ganguly A, Home P, Bhattacharya B, Ray S, Ghosh A, Rumi MAK, Marsh C, French VA, Gunewardena S, Paul S (2020) TEAD4 ensures postimplantation development by promoting trophoblast self-renewal: an implication in early human pregnancy loss. Proc Natl Acad Sci U S A 117(30):17864. https://doi.org/10.1073/pnas.2002449117
Hirate Y, Hirahara S, Inoue K-i, Kiyonari H, Niwa H, Sasaki H (2015) Par-aPKC-dependent and -independent mechanisms cooperatively control cell polarity, Hippo signaling, and cell positioning in 16-cell stage mouse embryos. Dev Growth Differ 57(8):544–556. https://doi.org/10.1111/dgd.12235
Qin H, Hejna M, Liu Y, Percharde M, Wossidlo M, Blouin L, Durruthy-Durruthy J, Wong P, Qi Z, Yu J, Qi LS, Sebastiano V, Song JS, Ramalho-Santos M (2016) YAP induces human naive pluripotency. Cell Rep 14(10):2301–2312. https://doi.org/10.1016/j.celrep.2016.02.036
Chung H, Lee B-K, Uprety N, Shen W, Lee J, Kim J (2016) Yap1 is dispensable for self-renewal but required for proper differentiation of mouse embryonic stem (ES) cells. EMBO Rep 17(4):519–529. https://doi.org/10.15252/embr.201540933
Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LSB, Abujarour R, Ding S, Guan K-L (2010) The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev 24(11):1106–1118. https://doi.org/10.1101/gad.1903310
Tamm C, Böwer N, Annerén C (2011) Regulation of mouse embryonic stem cell self-renewal by a Yes–YAP–TEAD2 signaling pathway downstream of LIF. J Cell Sci 124(7):1136. https://doi.org/10.1242/jcs.075796
Peng G, Suo S, Cui G, Yu F, Wang R, Chen J, Chen S, Liu Z, Chen G, Qian Y, Tam PPL, Han J-DJ, Jing N (2019) Molecular architecture of lineage allocation and tissue organization in early mouse embryo. Nature 572(7770):528–532. https://doi.org/10.1038/s41586-019-1469-8
Marikawa Y, Alarcon VB (2019) RHOA activity in expanding blastocysts is essential to regulate HIPPO-YAP signaling and to maintain the trophectoderm-specific gene expression program in a ROCK/actin filament-independent manner. Mol Hum Reprod 25(2):43–60. https://doi.org/10.1093/molehr/gay048
Bae JS, Kim SM, Jeon Y, Sim J, Jang JY, Son J, Hong W, Park MK, Lee H (2019) Loss of Mob1a/b impairs the differentiation of mouse embryonic stem cells into the three germ layer lineages. Exp Mol Med 51(11):1–12. https://doi.org/10.1038/s12276-019-0342-z
Hartman AA, Scalf SM, Zhang J, Hu X, Chen X, Eastman AE, Yang C, Guo S (2020) YAP non-cell-autonomously promotes pluripotency induction in mouse cells. Stem Cell Rep 14(4):730–743. https://doi.org/10.1016/j.stemcr.2020.03.006
Han Z, Yu Y, Cai B, Xu Z, Bao Z, Zhang Y, Bamba D, Ma W, Gao X, Yuan Y, Zhang L, Yu M, Liu S, Yan G, Jin M, Huang Q, Wang X, Hua B, Yang F, Pan Z, Liang H, Liu Y (2020) YAP/TEAD3 signal mediates cardiac lineage commitment of human-induced pluripotent stem cells. J Cell Physiol 235(3):2753–2760. https://doi.org/10.1002/jcp.29179
Morgani S, Nichols J, Hadjantonakis A-K (2017) The many faces of Pluripotency: in vitro adaptations of a continuum of in vivo states. BMC Dev Biol 17(1):7–7. https://doi.org/10.1186/s12861-017-0150-4
Wang L, Xu X, Cao Y, Li Z, Cheng H, Zhu G, Duan F, Na J, Han J-DJ, Chen Y-G (2017) Activin/Smad2-induced histone H3 Lys-27 Trimethylation (H3K27me3) reduction is crucial to initiate Mesendoderm differentiation of human embryonic stem cells. J Biol Chem 292(4):1339–1350. https://doi.org/10.1074/jbc.M116.766949
Estarás C, Hsu H-T, Huang L, Jones KA (2017) YAP repression of the WNT3 gene controls hESC differentiation along the cardiac mesoderm lineage. Genes Dev 31(22):2250–2263
McKee C, Brown C, Chaudhry GR (2019) Self-assembling scaffolds supported long-term growth of human primed embryonic stem cells and upregulated core and naïve Pluripotent markers. Cells 8(12):1650. https://doi.org/10.3390/cells8121650
Papaspyropoulos A, Bradley L, Thapa A, Leung CY, Toskas K, Koennig D, Pefani D-E, Raso C, Grou C, Hamilton G, Vlahov N, Grawenda A, Haider S, Chauhan J, Buti L, Kanapin A, Lu X, Buffa F, Dianov G, von Kriegsheim A, Matallanas D, Samsonova A, Zernicka-Goetz M, O’Neill E (2018) RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nature Communications 9(1):424. https://doi.org/10.1038/s41467-017-02786-5
Sun X, Ren Z, Cun Y, Zhao C, Huang X, Zhou J, Hu R, Su X, Ji L, Li P, Mak King Lun K, Gao F, Yang Y, Xu H, Ding J, Cao N, Li S, Zhang W, Lan P, Sun H, Wang J, Yuan P (2020) Hippo-YAP signaling controls lineage differentiation of mouse embryonic stem cells through modulating the formation of super-enhancers. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa482
Wang ES, Reyes NA, Melton C, Huskey NE, Momcilovic O, Goga A, Blelloch R, Oakes SA (2015) Fas-activated mitochondrial apoptosis culls stalled embryonic stem cells to promote differentiation. Curr Biol 25(23):3110–3118. https://doi.org/10.1016/j.cub.2015.10.020
Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M (2011) Reconstitution of the mouse Germ Cell Specification Pathway in Culture by Pluripotent Stem Cells. Cell 146(4):519–532. https://doi.org/10.1016/j.cell.2011.06.052
LeBlanc L, Lee B-K, Yu AC, Kim M, Kambhampati AV, Dupont SM, Seruggia D, Ryu BU, Orkin SH, Kim J (2018) Yap1 safeguards mouse embryonic stem cells from excessive apoptosis during differentiation. Elife 7:e40167. https://doi.org/10.7554/eLife.40167
Ohgushi M, Minaguchi M, Sasai Y (2015) Rho-signaling-directed YAP/TAZ activity underlies the long-term survival and expansion of human embryonic stem cells. Cell Stem Cell 17(4):448–461. https://doi.org/10.1016/j.stem.2015.07.009
Bar-Nur O, Verheul C, Sommer AG, Brumbaugh J, Schwarz BA, Lipchina I, Huebner AJ, Mostoslavsky G, Hochedlinger K (2015) Lineage conversion induced by pluripotency factors involves transient passage through an iPSC stage. Nat Biotechnol 33(7):761–768. https://doi.org/10.1038/nbt.3247
Panciera T, Azzolin L, Di Biagio D, Totaro A, Cordenonsi M, Piccolo S (2018) De Novo generation of Somatic stem cells by YAP/TAZ. J Vis Exp 135:57462. https://doi.org/10.3791/57462
Panciera T, Azzolin L, Fujimura A, Di Biagio D, Frasson C, Bresolin S, Soligo S, Basso G, Bicciato S, Rosato A, Cordenonsi M, Piccolo S (2016) Induction of expandable tissue-specific stem/progenitor cells through transient expression of YAP/TAZ. Cell Stem Cell 19(6):725–737. https://doi.org/10.1016/j.stem.2016.08.009
Monroe TO, Hill MC, Morikawa Y, Leach JP, Heallen T, Cao S, Krijger PHL, de Laat W, Wehrens XHT, Rodney GG, Martin JF (2019) YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev Cell 48(6):765-779.e767. https://doi.org/10.1016/j.devcel.2019.01.017
Elbediwy A, Vincent-Mistiaen ZI, Spencer-Dene B, Stone RK, Boeing S, Wculek SK, Cordero J, Tan EH, Ridgway R, Brunton VG, Sahai E, Gerhardt H, Behrens A, Malanchi I, Sansom OJ, Thompson BJ (2016) Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development (Cambridge, England) 143(10):1674–1687. https://doi.org/10.1242/dev.133728
Fu L, Hu Y, Song M, Liu Z, Zhang W, Yu F-X, Wu J, Wang S, Izpisua Belmonte JC, Chan P, Qu J, Tang F, Liu G-H (2019) Up-regulation of FOXD1 by YAP alleviates senescence and osteoarthritis. PLoS Biol 17(4):e3000201–e3000201. https://doi.org/10.1371/journal.pbio.3000201
Lee M-J, Byun MR, Furutani-Seiki M, Hong J-H, Jung H-S (2014) YAP and TAZ regulate skin wound healing. J Invest Dermatol 134(2):518–525. https://doi.org/10.1038/jid.2013.339
Xie C, Shen X, Xu X, Liu H, Li F, Lu S, Gao Z, Zhang J, Wu Q, Yang D, Bao X, Zhang F, Wu S, Lv Z, Zhu M, Xu D, Wang P, Cao L, Wang W, Yuan Z, Wang Y, Li Z, Teng H, Huang Z (2020) Astrocytic YAP promotes the formation of Glia scars and neural regeneration after spinal cord injury. J Neurosci 40(13):2644. https://doi.org/10.1523/JNEUROSCI.2229-19.2020
Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, Chan K, Wrana JL, Gregorieff A (2019) Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569(7754):121–125. https://doi.org/10.1038/s41586-019-1154-y
Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL (2015) Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526(7575):715–718. https://doi.org/10.1038/nature15382
Serra D, Mayr U, Boni A, Lukonin I, Rempfler M, Challet Meylan L, Stadler MB, Strnad P, Papasaikas P, Vischi D, Waldt A, Roma G, Liberali P (2019) Self-organization and symmetry breaking in intestinal organoid development. Nature 569(7754):66–72. https://doi.org/10.1038/s41586-019-1146-y
Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN (2013) Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A 110(34):13839–13844. https://doi.org/10.1073/pnas.1313192110
Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, Larsen HL, Guiu J, Alves MRP, Rundsten CF, Johansen JV, Li Y, Madsen CD, Nakamura T, Watanabe M, Nielsen OH, Schweiger PJ, Piccolo S, Jensen KB (2018) YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 22(1):35-49.e37. https://doi.org/10.1016/j.stem.2017.11.001
Fang Y, Liu C, Shu B, Zhai M, Deng C, He C, Luo M, Han T, Zheng W, Zhang J, Liu S (2018) Axis of serotonin -pERK-YAP in liver regeneration. Life Sci 209:490–497. https://doi.org/10.1016/j.lfs.2018.08.047
Hamon A, García-García D, Ail D, Bitard J, Chesneau A, Dalkara D, Locker M, Roger JE, Perron M (2019) Linking YAP to Müller Glia Quiescence exit in the degenerative retina. Cell Rep 27(6):1712-1725.e1716. https://doi.org/10.1016/j.celrep.2019.04.045
Konishi T, Schuster RM, Lentsch AB (2018) Proliferation of hepatic stellate cells, mediated by YAP and TAZ, contributes to liver repair and regeneration after liver ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 314(4):G471–G482. https://doi.org/10.1152/ajpgi.00153.2017
Liu Z, Wu H, Jiang K, Wang Y, Zhang W, Chu Q, Li J, Huang H, Cai T, Ji H, Yang C, Tang N (2016) MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep 16(7):1810–1819. https://doi.org/10.1016/j.celrep.2016.07.020
Pepe-Mooney BJ, Dill MT, Alemany A, Ordovas-Montanes J, Matsushita Y, Rao A, Sen A, Miyazaki M, Anakk S, Dawson PA, Ono N, Shalek AK, van Oudenaarden A, Camargo FD (2019) Single-cell analysis of the liver epithelium reveals dynamic heterogeneity and an essential role for YAP in homeostasis and regeneration. Cell Stem Cell 25(1):23-38.e28. https://doi.org/10.1016/j.stem.2019.04.004
Tharehalli U, Svinarenko M, Kraus JM, Kühlwein SD, Szekely R, Kiesle U, Scheffold A, Barth TFE, Kleger A, Schirmbeck R, Kestler HA, Seufferlein T, Oswald F, Katz S-F, Lechel A (2018) YAP activation drives liver regeneration after cholestatic damage induced by Rbpj deletion. Int J Mol Sci 19(12):3801. https://doi.org/10.3390/ijms19123801
Totaro A, Zhuang Q, Panciera T, Battilana G, Azzolin L, Brumana G, Gandin A, Brusatin G, Cordenonsi M, Piccolo S (2019) Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc Natl Acad Sci U S A 116(36):17848–17857. https://doi.org/10.1073/pnas.1908228116
Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD (2014) Hippo pathway activity influences liver cell fate. Cell 157(6):1324–1338. https://doi.org/10.1016/j.cell.2014.03.060
Brandão AS, Bensimon-Brito A, Lourenço R, Borbinha J, Soares AR, Mateus R, Jacinto A (2019) Yap induces osteoblast differentiation by modulating Bmp signalling during zebrafish caudal fin regeneration. J Cell Sci 132(22):jcs231993. https://doi.org/10.1242/jcs.231993
Kegelman CD, Mason DE, Dawahare JH, Horan DJ, Vigil GD, Howard SS, Robling AG, Bellido TM, Boerckel JD (2018) Skeletal cell YAP and TAZ combinatorially promote bone development. FASEB J 32(5):2706–2721. https://doi.org/10.1096/fj.201700872R
Kim J, Koo B-K, Knoblich JA (2020) Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol 21(10):571–584. https://doi.org/10.1038/s41580-020-0259-3
Rossi G, Manfrin A, Lutolf MP (2018) Progress and potential in organoid research. Nat Rev Genet 19(11):671–687. https://doi.org/10.1038/s41576-018-0051-9
Aloia L, McKie MA, Vernaz G, Cordero-Espinoza L, Aleksieva N, van den Ameele J, Antonica F, Font-Cunill B, Raven A, Aiese Cigliano R, Belenguer G, Mort RL, Brand AH, Zernicka-Goetz M, Forbes SJ, Miska EA, Huch M (2019) Epigenetic remodelling licences adult cholangiocytes for organoid formation and liver regeneration. Nat Cell Biol 21(11):1321–1333. https://doi.org/10.1038/s41556-019-0402-6
Tan Q, Choi KM, Sicard D, Tschumperlin DJ (2017) Human airway organoid engineering as a step toward lung regeneration and disease modeling. Biomaterials 113:118–132. https://doi.org/10.1016/j.biomaterials.2016.10.046
Matsumoto S, Fujii S, Sato A, Ibuka S, Kagawa Y, Ishii M, Kikuchi A (2014) A combination of Wnt and growth factor signaling induces Arl4c expression to form epithelial tubular structures. EMBO J 33(7):702–718. https://doi.org/10.1002/embj.201386942
Ng-Blichfeldt J-P, Schrik A, Kortekaas RK, Noordhoek JA, Heijink IH, Hiemstra PS, Stolk J, Königshoff M, Gosens R (2018) Retinoic acid signaling balances adult distal lung epithelial progenitor cell growth and differentiation. EBioMedicine 36:461–474. https://doi.org/10.1016/j.ebiom.2018.09.002
Bailey DD, Zhang Y, van Soldt BJ, Jiang M, Suresh S, Nakagawa H, Rustgi AK, Aceves SS, Cardoso WV, Que J (2019) Use of hPSC-derived 3D organoids and mouse genetics to define the roles of YAP in the development of the esophagus. Development 146(23):dev178855. https://doi.org/10.1242/dev.178855
Lukonin I, Serra D, Challet Meylan L, Volkmann K, Baaten J, Zhao R, Meeusen S, Colman K, Maurer F, Stadler MB, Jenkins J, Liberali P (2020) Phenotypic landscape of intestinal organoid regeneration. Nature 586(7828):275–280. https://doi.org/10.1038/s41586-020-2776-9
Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordóñez-Morán P, Clevers H, Lutolf MP (2016) Designer matrices for intestinal stem cell and organoid culture. Nature 539(7630):560–564. https://doi.org/10.1038/nature20168
Roulis M, Kaklamanos A, Schernthanner M, Bielecki P, Zhao J, Kaffe E, Frommelt L-S, Qu R, Knapp MS, Henriques A, Chalkidi N, Koliaraki V, Jiao J, Brewer JR, Bacher M, Blackburn HN, Zhao X, Breyer RM, Aidinis V, Jain D, Su B, Herschman HR, Kluger Y, Kollias G, Flavell RA (2020) Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 580(7804):524–529. https://doi.org/10.1038/s41586-020-2166-3
Sorrentino G, Perino A, Yildiz E, El Alam G, Sleiman MB, Gioiello A, Pellicciari R, Schoonjans K (2020) Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology S0016–5085(20):34739–34739
Sorrentino G, Rezakhani S, Yildiz E, Nuciforo S, Heim MH, Lutolf MP, Schoonjans K (2020) Mechano-modulatory synthetic niches for liver organoid derivation. Nature Commun 11(1):3416–3416. https://doi.org/10.1038/s41467-020-17161-0
Bejoy J, Wang Z, Bijonowski B, Yang M, Ma T, Sang Q-X, Li Y (2018) Differential effects of heparin and hyaluronic acid on neural patterning of human induced pluripotent stem cells. ACS Biomater Sci Eng 4(12):4354–4366. https://doi.org/10.1021/acsbiomaterials.8b01142
Dzamba BJ, DeSimone DW (2018) Chapter Seven - Extracellular Matrix (ECM) and the Sculpting of Embryonic Tissues. In: Litscher ES, Wassarman PM (eds) Current Topics in Developmental Biology, vol 130. Academic Press, pp 245–274. doi:https://doi.org/10.1016/bs.ctdb.2018.03.006
Baillie-Benson P, Moris N, Martinez Arias A (2020) Pluripotent stem cell models of early mammalian development. Curr Opin Cell Biol 66:89–96. https://doi.org/10.1016/j.ceb.2020.05.010
Philippe G, Mayra T, Marilène P, Derek B (2020) YAP and TAZ are required for the postnatal development and the maintenance of the structural integrity of the oviduct. Reproduction 160(2):307–318. https://doi.org/10.1530/REP-20-0202
Oku Y, Nishiya N, Shito T, Yamamoto R, Yamamoto Y, Oyama C, Uehara Y (2015) Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 5:542–549. https://doi.org/10.1016/j.fob.2015.06.007
Artap S, Manderfield LJ, Smith CL, Poleshko A, Aghajanian H, See K, Li L, Jain R, Epstein JA (2018) Endocardial Hippo signaling regulates myocardial growth and cardiogenesis. Dev Biol 440(1):22–30. https://doi.org/10.1016/j.ydbio.2018.04.026
Plouffe SW, Lin KC, Moore JL 3rd, Tan FE, Ma S, Ye Z, Qiu Y, Ren B, Guan K-L (2018) The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J Biol Chem 293(28):11230–11240. https://doi.org/10.1074/jbc.RA118.002715
Reginensi A, Hoshi M, Boualia SK, Bouchard M, Jain S, McNeill H (2015) Yap and Taz are required for Ret-dependent urinary tract morphogenesis. Development (Cambridge, England) 142(15):2696–2703. https://doi.org/10.1242/dev.122044
Huang W, Lv X, Liu C, Zha Z, Zhang H, Jiang Y, Xiong Y, Lei Q-Y, Guan K-L (2012) The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFβ-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem 287(31):26245–26253. https://doi.org/10.1074/jbc.M112.382036
Miranda MZ, Bialik JF, Speight P, Dan Q, Yeung T, Szászi K, Pedersen SF, Kapus A (2017) TGF-β1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J Biol Chem 292(36):14902–14920. https://doi.org/10.1074/jbc.M117.780502
Kedan A, Verma N, Saroha A, Shreberk-Shaked M, Müller A-K, Nair NU, Lev S (2018) PYK2 negatively regulates the Hippo pathway in TNBC by stabilizing TAZ protein. Cell Death Dis 9(10):985–985. https://doi.org/10.1038/s41419-018-1005-z
Finch-Edmondson ML, Strauss RP, Passman AM, Sudol M, Yeoh GC, Callus BA (2015) TAZ protein accumulation is negatively regulated by YAP abundance in mammalian cells. J Biol Chem 290(46):27928–27938
Muppala S, Raghunathan VK, Jalilian I, Thomasy S, Murphy CJ (2019) YAP and TAZ are distinct effectors of corneal myofibroblast transformation. Exp Eye Res 180:102–109. https://doi.org/10.1016/j.exer.2018.12.009
Kaan HYK, Chan SW, Tan SKJ, Guo F, Lim CJ, Hong W, Song H (2017) Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Scientific Rep 7(1):2035–2035. https://doi.org/10.1038/s41598-017-02219-9
Chen L, Loh PG, Song H (2010) Structural and functional insights into the TEAD-YAP complex in the Hippo signaling pathway. Protein Cell 1(12):1073–1083. https://doi.org/10.1007/s13238-010-0138-3
Lu Y, Wu T, Gutman O, Lu H, Zhou Q, Henis YI, Luo K (2020) Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat Cell Biol 22(4):453–464. https://doi.org/10.1038/s41556-020-0485-0
Murakami M, Nakagawa M, Olson EN, Nakagawa O (2005) A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc Natl Acad Sci U S A 102(50):18034. https://doi.org/10.1073/pnas.0509109102
Khanal P, Jia Z, Yang X (2018) Cysteine residues are essential for dimerization of Hippo pathway components YAP2L and TAZ. Sci Rep 8(1):3485. https://doi.org/10.1038/s41598-018-21828-6
Cai D, Feliciano D, Dong P, Flores E, Gruebele M, Porat-Shliom N, Sukenik S, Liu Z, Lippincott-Schwartz J (2019) Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat Cell Biol 21(12):1578–1589. https://doi.org/10.1038/s41556-019-0433-z
Zhu C, Li L, Zhao B (2014) The regulation and function of YAP transcription co-activator. Acta Biochim Biophys Sin 47(1):16–28. https://doi.org/10.1093/abbs/gmu110
Verma A, Jing-Song F, Finch-Edmondson ML, Velazquez-Campoy A, Balasegaran S, Sudol M, Sivaraman J (2018) Biophysical studies and NMR structure of YAP2 WW domain - LATS1 PPxY motif complexes reveal the basis of their interaction. Oncotarget 9(8):8068–8080. https://doi.org/10.18632/oncotarget.23909
Zhang X, Sun F, Qiao Y, Zheng W, Liu Y, Chen Y, Wu Q, Liu X, Zhu G, Chen Y, Yu Y, Pan Q, Wang J (2017) TFCP2 is required for YAP-dependent transcription to stimulate liver malignancy. Cell Rep 21(5):1227–1239. https://doi.org/10.1016/j.celrep.2017.10.017
Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S, Li X, Zhou R, Wang J, Zhao B, Liu J, Qin J, Zou J, Feng X-H, Guan K-L, Xu P (2017) Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat Cell Biol 19(4):362–374. https://doi.org/10.1038/ncb3496
Remue E, Meerschaert K, Oka T, Boucherie C, Vandekerckhove J, Sudol M, Gettemans J (2010) TAZ interacts with zonula occludens-1 and -2 proteins in a PDZ-1 dependent manner. FEBS Lett 584(19):4175–4180. https://doi.org/10.1016/j.febslet.2010.09.020
Domínguez-Calderón A, Ávila-Flores A, Ponce A, López-Bayghen E, Calderón-Salinas J-V, Luis Reyes J, Chávez-Munguía B, Segovia J, Angulo C, Ramírez L, Gallego-Gutiérrez H, Alarcón L, Martín-Tapia D, Bautista-García P, González-Mariscal L (2016) ZO-2 silencing induces renal hypertrophy through a cell cycle mechanism and the activation of YAP and the mTOR pathway. Mol Biol Cell 27(10):1581–1595. https://doi.org/10.1091/mbc.E15-08-0598
Kim M-K, Jang J-W, Bae S-C (2018) DNA binding partners of YAP/TAZ. BMB Rep 51(3):126–133. https://doi.org/10.5483/bmbrep.2018.51.3.015
Park JS, Kim M, Song N-J, Kim J-H, Seo D, Lee J-H, Jung SM, Lee JY, Lee J, Lee YS, Park KW, Park SH (2019) A reciprocal role of the Smad4-Taz axis in Osteogenesis and Adipogenesis of mesenchymal stem cells. Stem Cells 37(3):368–381. https://doi.org/10.1002/stem.2949
Wang N, Li Y, Li Z, Ma J, Wu X, Pan R, Wang Y, Gao L, Bao X, Xue P (2019) IRS-1 targets TAZ to inhibit adipogenesis of rat bone marrow mesenchymal stem cells through PI3K-Akt and MEK-ERK pathways. Eur J Pharmacol 849:11–21. https://doi.org/10.1016/j.ejphar.2019.01.064
Wang Y, Hu B, Hu R, Tong X, Zhang M, Xu C, He Z, Zhao Y, Deng H (2020) TAZ contributes to osteogenic differentiation of periodontal ligament cells under tensile stress. J Periodontal Res 55(1):152–160. https://doi.org/10.1111/jre.12698
Pan J-X, Xiong L, Zhao K, Zeng P, Wang B, Tang F-L, Sun D, Guo H-h, Yang X, Cui S, Xia W-F, Mei L, Xiong W-C (2018) YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating β-catenin signaling. Bone Res 6(1):18. https://doi.org/10.1038/s41413-018-0018-7
El Ouarrat D, Isaac R, Lee YS, Oh DY, Wollam J, Lackey D, Riopel M, Bandyopadhyay G, Seo JB, Sampath-Kumar R, Olefsky JM (2020) TAZ is a negative regulator of PPARγ activity in Adipocytes and TAZ deletion improves insulin sensitivity and glucose tolerance. Cell Metab 31(1):162-173.e165. https://doi.org/10.1016/j.cmet.2019.10.003
Tang C, Takahashi-Kanemitsu A, Kikuchi I, Ben C, Hatakeyama M (2018) Transcriptional co-activator functions of YAP and TAZ are inversely regulated by tyrosine phosphorylation status of parafibromin. Science 1:1–15. https://doi.org/10.1016/j.isci.2018.01.003
Weiler SME, Lutz T, Bissinger M, Sticht C, Knaub M, Gretz N, Schirmacher P, Breuhahn K (2020) TAZ target gene ITGAV regulates invasion and feeds back positively on YAP and TAZ in liver cancer cells. Cancer Lett 473:164–175. https://doi.org/10.1016/j.canlet.2019.12.044
Yang W-H, Huang Z, Wu J, Ding C-KC, Murphy SK, Chi J-T (2020) A TAZ-ANGPTL4-NOX2 Axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res 18(1):79–90. https://doi.org/10.1158/1541-7786.MCR-19-0691
Shreberk-Shaked M, Dassa B, Sinha S, Di Agostino S, Azuri I, Mukherjee S, Aylon Y, Blandino G, Ruppin E, Oren M (2020) A division of labor between YAP and TAZ in non-small cell lung cancer. Cancer Res 80(19):4145. https://doi.org/10.1158/0008-5472.CAN-20-0125
Zhou X, Chadarevian JP, Ruiz B, Ying Q-L (2017) Cytoplasmic and nuclear TAZ exert distinct functions in regulating primed pluripotency. Stem Cell Rep 9(3):732–741. https://doi.org/10.1016/j.stemcr.2017.07.019
Volckaert T, Yuan T, Yuan J, Boateng E, Hopkins S, Zhang J-S, Thannickal VJ, Fässler R, De Langhe SP (2019) Hippo signaling promotes lung epithelial lineage commitment by curbing Fgf10 and β-catenin signaling. Development (Cambridge, England) 146(2):dev166454. https://doi.org/10.1242/dev.166454
Sun T, Huang Z, Zhang H, Posner C, Jia G, Ramalingam TR, Xu M, Brightbill H, Egen JG, Dey A, Arron JR (2019) TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insight 4(14):e128674. https://doi.org/10.1172/jci.insight.128674
Geng J, Yu S, Zhao H, Sun X, Li X, Wang P, Xiong X, Hong L, Xie C, Gao J, Shi Y, Peng J, Johnson RL, Xiao N, Lu L, Han J, Zhou D, Chen L (2017) The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat Immunol 18(7):800–812. https://doi.org/10.1038/ni.3748
Stampouloglou E, Cheng N, Federico A, Slaby E, Monti S, Szeto GL, Varelas X (2020) Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol 18(1):e3000591. https://doi.org/10.1371/journal.pbio.3000591
Sun C, De Mello V, Mohamed A, Ortuste Quiroga HP, Garcia-Munoz A, Al Bloshi A, Tremblay AM, von Kriegsheim A, Collie-Duguid E, Vargesson N, Matallanas D, Wackerhage H, Zammit PS (2017) Common and distinctive functions of the hippo effectors Taz and Yap in skeletal muscle stem cell function. Stem Cells 35(8):1958–1972. https://doi.org/10.1002/stem.2652
Toh TB, Lim JJ, Chow EK-H (2017) Epigenetics in cancer stem cells. Mol Cancer 16(1):29–29. https://doi.org/10.1186/s12943-017-0596-9
Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, Sun B, Yin D, Sun J, Song R, Pan S, Sun Y, Jiang H, Zheng T, Liu L (2015) YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 6(19):17206–17220. https://doi.org/10.18632/oncotarget.4043
Fan H, Tian H, Cheng X, Chen Y, Liang S, Zhang Z, Liao Y, Xu P (2020) Aberrant Kank1 expression regulates YAP to promote apoptosis and inhibit proliferation in OSCC. J Cell Physiol 235(2):1850–1865. https://doi.org/10.1002/jcp.29102
Basu S, Totty NF, Irwin MS, Sudol M, Downward J (2003) Akt phosphorylates the yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell 11(1):11–23. https://doi.org/10.1016/S1097-2765(02)00776-1
Yan H, Qiu C, Sun W, Gu M, Xiao F, Zou J, Zhang L (2018) Yap regulates gastric cancer survival and migration via SIRT1/Mfn2/mitophagy. Oncol Rep 39(4):1671–1681. https://doi.org/10.3892/or.2018.6252
Ye C, Wang W, Xia G, Yu C, Yi Y, Hua C, Tu F, Shen L, Chen C, Sun W, Zheng Z (2019) A novel curcumin derivative CL-6 exerts antitumor effect in human gastric cancer cells by inducing apoptosis through Hippo-YAP signaling pathway. Onco Targets Ther 12:2259–2269. https://doi.org/10.2147/OTT.S196914
Zhou Z, Zhu J-S, Gao C-P, Li L-P, Zhou C, Wang H, Liu X-G (2016) siRNA targeting YAP gene inhibits gastric carcinoma growth and tumor metastasis in SCID mice. Oncol Lett 11(4):2806–2814. https://doi.org/10.3892/ol.2016.4319
Cao J-J, Zhao X-M, Wang D-L, Chen K-H, Sheng X, Li W-B, Li M-C, Liu W-J, He J (2014) YAP is overexpressed in clear cell renal cell carcinoma and its knockdown reduces cell proliferation and induces cell cycle arrest and apoptosis. Oncol Rep 32(4):1594–1600
Slemmons KK, Crose LES, Rudzinski E, Bentley RC, Linardic CM (2015) Role of the YAP oncoprotein in priming ras-driven rhabdomyosarcoma. PLoS ONE 10(10):e0140781–e0140781. https://doi.org/10.1371/journal.pone.0140781
Guo J, Wu Y, Yang L, Du J, Gong K, Chen W, Dai J, Li X, Xi S (2017) Repression of YAP by NCTD disrupts NSCLC progression. Oncotarget 8(2):2307–2319. https://doi.org/10.18632/oncotarget.13668
Li M, Chen J, Yu X, Xu S, Li D, Zheng Q, Yin Y (2019) Myricetin suppresses the propagation of hepatocellular carcinoma via down-regulating expression of YAP. Cells 8(4):358. https://doi.org/10.3390/cells8040358
Wang X, Wu B, Zhong Z (2018) Downregulation of YAP inhibits proliferation, invasion and increases cisplatin sensitivity in human hepatocellular carcinoma cells. Oncol Lett 16(1):585–593. https://doi.org/10.3892/ol.2018.8633
Shen Y, Han Z, Liu S, Jiao Y, Li Y, Yuan H (2020) Curcumin inhibits the Tumorigenesis of breast cancer by blocking Tafazzin/yes-associated protein axis. Cancer Manag Res 12:1493–1502. https://doi.org/10.2147/CMAR.S246691
Song Q, Mao B, Cheng J, Gao Y, Jiang K, Chen J, Yuan Z, Meng S (2015) YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. PLoS ONE 10(3):e0120790–e0120790. https://doi.org/10.1371/journal.pone.0120790
Li K, Guo J, Wu Y, Jin D, Jiang H, Liu C, Qin C (2018) Suppression of YAP by DDP disrupts colon tumor progression. Oncol Rep 39(5):2114–2126. https://doi.org/10.3892/or.2018.6297
Li Y, Li L, Zhu M, Ye L, Yang Q (2017) Upregulation of Yes-associated protein and transcriptional co-activator with PDZ-binding motif influences the behavior of LOVO human colon adenocarcinoma cells. Exp Ther Med 14(4):3831–3835. https://doi.org/10.3892/etm.2017.4962
Jin X, Zhao W, Zhou P, Niu T (2018) YAP knockdown inhibits proliferation and induces apoptosis of human prostate cancer DU145 cells. Mol Med Rep 17(3):3783–3788
Sheng X, Li W-B, Wang D-L, Chen K-H, Cao J-J, Luo Z, He J, Li M-C, Liu W-J, Yu C (2015) YAP is closely correlated with castration-resistant prostate cancer, and downregulation of YAP reduces proliferation and induces apoptosis of PC-3 cells. Mol Med Rep 12(4):4867–4876. https://doi.org/10.3892/mmr.2015.4005
Shi Y, Cao T, Sun Y, Xia J, Wang P, Ma J (2019) Nitidine Chloride inhibits cell proliferation and invasion via downregulation of YAP expression in prostate cancer cells. Am J Transl Res 11(2):709–720
Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, Knelson EH, Xie Y, Lim K, Cejas P, Portell A, Lizotte PH, Ficarro SB, Li S, Chen T, Haikala HM, Wang H, Bahcall M, Gao Y, Shalhout S, Boettcher S, Shin BH, Thai T, Wilkens MK, Tillgren ML, Mushajiang M, Xu M, Choi J, Bertram AA, Ebert BL, Beroukhim R, Bandopadhayay P, Awad MM, Gokhale PC, Kirschmeier PT, Marto JA, Camargo FD, Haq R, Paweletz CP, Wong K-K, Barbie DA, Long HW, Gray NS, Jänne PA (2020) Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37(1):104-122.e112. https://doi.org/10.1016/j.ccell.2019.12.006
Ma K, Xu Q, Wang S, Zhang W, Liu M, Liang S, Zhu H, Xu N (2016) Nuclear accumulation of Yes-Associated Protein (YAP) maintains the survival of doxorubicin-induced senescent cells by promoting survivin expression. Cancer Lett 375(1):84–91. https://doi.org/10.1016/j.canlet.2016.02.045
Tan S, Bian X, Wu B, Chen X (2019) RASSF6 is Downregulated in human bladder cancers and regulates Doxorubicin sensitivity and mitochondrial membrane potential via the hippo signaling pathway. Onco Targets Ther 12:9189–9200. https://doi.org/10.2147/OTT.S217041
Liu Z, Yee PP, Wei Y, Liu Z, Kawasawa YI, Li W (2019) Differential YAP expression in glioma cells induces cell competition and promotes tumorigenesis. J Cell Sci 132(5):jcs225714. https://doi.org/10.1242/jcs.225714
Ehsanian R, Brown M, Lu H, Yang XP, Pattatheyil A, Yan B, Duggal P, Chuang R, Doondeea J, Feller S, Sudol M, Chen Z, Van Waes C (2010) YAP dysregulation by phosphorylation or ΔNp63-mediated gene repression promotes proliferation, survival and migration in head and neck cancer subsets. Oncogene 29(46):6160–6171. https://doi.org/10.1038/onc.2010.339
Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, Oren M, Sudol M, Cesareni G, Blandino G (2001) Physical Interaction with yes-associated protein enhances p73 transcriptional activity. J Biol Chem 276(18):15164–15173
Howell M, Borchers C, Milgram SL (2004) Heterogeneous nuclear ribonuclear protein U associates with YAP and regulates its co-activation of Bax transcription. J Biol Chem 279(25):26300–26306
Keshet R, Adler J, Ricardo Lax I, Shanzer M, Porat Z, Reuven N, Shaul Y (2015) c-Abl antagonizes the YAP oncogenic function. Cell Death Differ 22(6):935–945. https://doi.org/10.1038/cdd.2014.182
Deng L-J, Qi M, Peng Q-L, Chen M-F, Qi Q, Zhang J-Y, Yao N, Huang M-H, Li X-B, Peng Y-H, Liu J-S, Fu D-R, Chen J-X, Ye W-C, Zhang D-M (2018) Arenobufagin induces MCF-7 cell apoptosis by promoting JNK-mediated multisite phosphorylation of Yes-associated protein. Cancer Cell Int 18:209–209. https://doi.org/10.1186/s12935-018-0706-9
Cao L, Yao M, Sasano H, Sun P-L, Gao H (2020) YAP increases response to Trastuzumab in HER2-positive Breast Cancer by enhancing P73-induced apoptosis. J Cancer 11(22):6748–6759. https://doi.org/10.7150/jca.48535
Xiao Q, Qian Z, Zhang W, Liu J, Hu E, Zhang J, Li M, Wang J, Kong F, Li Y, Wang R, Tan X, He D, Xiao X (2016) Depletion of CABYR-a/b sensitizes lung cancer cells to TRAIL-induced apoptosis through YAP/p73-mediated DR5 upregulation. Oncotarget 7(8):9513–9524. https://doi.org/10.18632/oncotarget.7069
Simile MM, Latte G, Demartis MI, Brozzetti S, Calvisi DF, Porcu A, Feo CF, Seddaiu MA, Daino L, Berasain C, Tomasi ML, Avila MA, Feo F, Pascale RM (2016) Post-translational deregulation of YAP1 is genetically controlled in rat liver cancer and determines the fate and stem-like behavior of the human disease. Oncotarget 7(31):49194–49216. https://doi.org/10.18632/oncotarget.10246
Miyamura N, Hata S, Itoh T, Tanaka M, Nishio M, Itoh M, Ogawa Y, Terai S, Sakaida I, Suzuki A, Miyajima A, Nishina H (2017) YAP determines the cell fate of injured mouse hepatocytes in vivo. Nat Commun 8:16017–16017. https://doi.org/10.1038/ncomms16017
Mao Y, Chen X, Xu M, Fujita K, Motoki K, Sasabe T, Homma H, Murata M, Tagawa K, Tamura T, Kaye J, Finkbeiner S, Blandino G, Sudol M, Okazawa H (2016) Targeting TEAD/YAP-transcription-dependent necrosis, TRIAD, ameliorates Huntington’s disease pathology. Hum Mol Genet 25(21):4749–4770. https://doi.org/10.1093/hmg/ddw303
Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C, Yao Y, Liu Q (2014) Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer 13:110–110. https://doi.org/10.1186/1476-4598-13-110
Liu H, Liu N, Zhao Y, Zhu X, Wang C, Liu Q, Gao C, Zhao X, Li J (2019) Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging (Albany NY) 11(21):9643–9660. https://doi.org/10.18632/aging.102410
Wu T, Hu H, Zhang T, Jiang L, Li X, Liu S, Zheng C, Yan G, Chen W, Ning Y, Li Y, Lu Z (2019) miR-25 Promotes cell proliferation, migration, and invasion of non-small-cell lung cancer by targeting the LATS2/YAP signaling pathway. Oxid Med Cell Longev 2019:9719723–9719723. https://doi.org/10.1155/2019/9719723
Song L, Huang Y, Zhang X, Han S, Hou M, Li H (2020) Downregulation of microRNA-224-3p hampers retinoblastoma progression via activation of the hippo-YAP signaling pathway by increasing LATS2. Invest Ophthalmol Vis Sci 61(3):32–32. https://doi.org/10.1167/iovs.61.3.32
Zhou Y, Shan T, Ding W, Hua Z, Shen Y, Lu Z, Chen B, Dai T (2018) Study on mechanism about long noncoding RNA MALAT1 affecting pancreatic cancer by regulating Hippo-YAP signaling. J Cell Physiol 233(8):5805–5814. https://doi.org/10.1002/jcp.26357
An Y, Zhang Q, Li X, Wang Z, Li Y, Tang X (2018) Upregulated microRNA miR-21 promotes the progression of lung adenocarcinoma through inhibition of KIBRA and the Hippo signaling pathway. Biomed Pharmacother 108:1845–1855. https://doi.org/10.1016/j.biopha.2018.09.125
Yang Y, Li H, Liu Y, Chi C, Ni J, Lin X (2019) MiR-4319 hinders YAP expression to restrain non-small cell lung cancer growth through regulation of LIN28-mediated RFX5 stability. Biomed Pharmacother 115:108956. https://doi.org/10.1016/j.biopha.2019.108956
Escoll M, Lastra D, Pajares M, Robledinos-Antón N, Rojo AI, Fernández-Ginés R, Mendiola M, Martínez-Marín V, Esteban I, López-Larrubia P, Gargini R, Cuadrado A (2020) Transcription factor NRF2 uses the Hippo pathway effector TAZ to induce tumorigenesis in glioblastomas. Redox Biol 30:101425–101425. https://doi.org/10.1016/j.redox.2019.101425
Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, Sorrentino G, Chianese G, Taglialatela-Scafati O, Trisciuoglio D, Del Bufalo D, Di Martile M, Di Napoli A, Ruco L, Costantini S, Jakopin Z, Budillon A, Melino G, Del Sal G, Ciliberto G, Mancini R (2017) Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 36(32):4573–4584. https://doi.org/10.1038/onc.2017.75
Ding Y, He J, Huang J, Yu T, Shi X, Zhang T, Yan G, Chen S, Peng C (2019) Harmine induces anticancer activity in breast cancer cells via targeting TAZ. Int J Oncol 54(6):1995–2004. https://doi.org/10.3892/ijo.2019.4777
Zhan T, Zhu Q, Han Z, Tan J, Liu M, Liu W, Chen W, Chen X, Chen X, Deng J, Tian X, Huang X (2020) miR-455-3p functions as a tumor suppressor by restraining Wnt/β-catenin signaling via TAZ in pancreatic cancer. Cancer Manag Res 12:1483–1492. https://doi.org/10.2147/CMAR.S235794
Li J, Li Z, Wu Y, Diao P, Zhang W, Wang Y, Yang J, Cheng J (2019) Overexpression of lncRNA WWTR1-AS1 associates with tumor aggressiveness and unfavorable survival in head-neck squamous cell carcinoma. J Cell Biochem 120(10):18266–18277. https://doi.org/10.1002/jcb.29132
Deel MD, Slemmons KK, Hinson AR, Genadry KC, Burgess BA, Crose LES, Kuprasertkul N, Oristian KM, Bentley RC, Linardic CM (2018) The transcriptional coactivator TAZ is a potent mediator of alveolar rhabdomyosarcoma tumorigenesis. Clin Cancer Res 24(11):2616–2630. https://doi.org/10.1158/1078-0432.CCR-17-1207
Grieve S, Wajnberg G, Lees M, Chacko S, Weir J, Crapoulet N, Reiman T (2019) TAZ functions as a tumor suppressor in multiple myeloma by downregulating MYC. Blood Adv 3(22):3613–3625. https://doi.org/10.1182/bloodadvances.2019000374
Yang W-H, Ding C-KC, Sun T, Rupprecht G, Lin C-C, Hsu D, Chi J-T (2019) The Hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep 28(10):2501-2508.e2504. https://doi.org/10.1016/j.celrep.2019.07.107
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen Z-N, Jiang X (2019) Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572(7769):402–406. https://doi.org/10.1038/s41586-019-1426-6
Furet P, Salem B, Mesrouze Y, Schmelzle T, Lewis I, Kallen J, Chène P (2019) Structure-based design of potent linear peptide inhibitors of the YAP-TEAD protein-protein interaction derived from the YAP omega-loop sequence. Bioorg Med Chem Lett 29(16):2316–2319. https://doi.org/10.1016/j.bmcl.2019.06.022
Nouri K, Azad T, Ling M, van Rensburg HJJ, Pipchuk A, Shen H, Hao Y, Zhang J, Yang X (2019) Identification of celastrol as a novel YAP-TEAD inhibitor for cancer therapy by high throughput screening with ultrasensitive YAP/TAZ-TEAD biosensors. Cancers (Basel) 11(10):1596. https://doi.org/10.3390/cancers11101596
Smith SA, Sessions RB, Shoemark DK, Williams C, Ebrahimighaei R, McNeill MC, Crump MP, McKay TR, Harris G, Newby AC, Bond M (2019) Antiproliferative and antimigratory effects of a novel YAP-TEAD interaction inhibitor identified using in silico molecular docking. J Med Chem 62(3):1291–1305. https://doi.org/10.1021/acs.jmedchem.8b01402
Yuan Y, Park J, Feng A, Awasthi P, Wang Z, Chen Q, Iglesias-Bartolome R (2020) YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat Commun 11(1):1472–1472. https://doi.org/10.1038/s41467-020-15301-0
Liu Y, Lu T, Zhang C, Xue Z, Xu J, Busuttil RW, Xia Q, Xu N, Kupiec-Weglinski JW, Ji H (2019) Pituitary adenylate cyclase-activating polypeptides prevent Hepatocyte damage by promoting Yes-associated protein in liver ischemia-reperfusion injury. Transplantation 103(8):1639–1648. https://doi.org/10.1097/TP.0000000000002742
Chen Y-A, Lu C-Y, Cheng T-Y, Pan S-H, Chen H-F, Chang N-S (2019) WW domain-containing proteins YAP and TAZ in the hippo pathway as key regulators in stemness maintenance, tissue homeostasis, and tumorigenesis. Front Oncol 9:60–60. https://doi.org/10.3389/fonc.2019.00060
Vigneron AM, Ludwig RL, Vousden KH (2010) Cytoplasmic ASPP1 inhibits apoptosis through the control of YAP. Genes Dev 24(21):2430–2439. https://doi.org/10.1101/gad.1954310
Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan K-L, Xu Y (2010) Structural insights into the YAP and TEAD complex. Genes Dev 24(3):235–240. https://doi.org/10.1101/gad.1865810
Beyer Tobias A, Weiss A, Khomchuk Y, Huang K, Ogunjimi Abiodun A, Varelas X, Wrana Jeffrey L (2013) Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep 5(6):1611–1624. https://doi.org/10.1016/j.celrep.2013.11.021
Reuven N, Adler J, Meltser V, Shaul Y (2013) The Hippo pathway kinase Lats2 prevents DNA damage-induced apoptosis through inhibition of the tyrosine kinase c-Abl. Cell Death Differ 20(10):1330–1340. https://doi.org/10.1038/cdd.2013.83
Xu Z, Chen J, Shao L, Ma W, Xu D (2015) Promyelocytic leukemia protein enhances apoptosis of gastric cancer cells through Yes-associated protein. Tumor Biology 36(10):8047–8054. https://doi.org/10.1007/s13277-015-3539-3
Acknowledgements
We thank Dr. Bum-Kyu Lee and Mijeong Kim for critical reading of the manuscript. L.L. is supported by funding from the National Science Foundation Graduate Research Fellowship Program (NSF GRFP) and the UT Austin Graduate School Continuing Fellowship. J.K. is supported by awards R01GM112722 from the National Institute of General Medical Sciences (NIH/NIGMS) and Preterm Birth Research Grant from the Burroughs Wellcome Fund (BWF).
Funding
L.L. is supported by funding from the National Science Foundation Graduate Research Fellowship Program (NSF GRFP) and the UT Austin Graduate School Continuing Fellowship. J.K. is supported by awards R01GM112722 from the National Institute of General Medical Sciences (NIH/NIGMS) and the Preterm Birth Research Grant from the Burroughs Wellcome Fund (BWF).
Author information
Authors and Affiliations
Contributions
L.L. and N.R. performed the literature search. L.L. prepared all figures and tables. L.L., N.R., and J.K. wrote and critically revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
LeBlanc, L., Ramirez, N. & Kim, J. Context-dependent roles of YAP/TAZ in stem cell fates and cancer. Cell. Mol. Life Sci. 78, 4201–4219 (2021). https://doi.org/10.1007/s00018-021-03781-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03781-2