
Abstract
In recent years, cellular senescence has become the focus of attention in multiple areas of biomedical research. Typically defined as an irreversible cell cycle arrest accompanied by increased cellular growth, metabolic activity and by a characteristic messaging secretome, cellular senescence can impact on multiple physiological and pathological processes such as wound healing, fibrosis, cancer and ageing. These unjustly called ‘zombie cells’ are indeed a rich source of opportunities for innovative therapeutic development. In this review, we collate the current understanding of the process of cellular senescence and its two-faced nature, i.e. beneficial/detrimental, and reason this duality is linked to contextual aspects. We propose the senescence programme as an endogenous pro-resolving mechanism that may lead to sustained inflammation and damage when dysregulated or when senescent cells are not cleared efficiently. This pro-resolving model reconciles the paradoxical two faces of senescence by emphasising that it is the unsuccessful completion of the programme, and not senescence itself, what leads to pathology. Thus, pro-senescence therapies under the right context, may favour inflammation resolution. We also review the evidence for the multiple therapeutic approaches under development based on senescence, including its induction, prevention, clearance and the use of senolytic and senomorphic drugs. In particular, we highlight the importance of the immune system in the favourable outcome of senescence and the implications of an inefficient immune surveillance in completion of the senescent cycle. Finally, we identify and discuss a number of challenges and existing gaps to encourage and stimulate further research in this exciting and unravelled field, with the hope of promoting and accelerating the clinical success of senescence-based therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction, definitions and history
Senescence, /sɪˈnɛs(ə)ns/, noun—the condition or process of deterioration with age
Dating back to the seventeenth century, the term senescence was used to refer to the general and gradual accumulation of changes over time leading to the decline of organismal function, essentially aging. In this context, this process is now referred to as organismal senescence. Senescence also applies to plants, appreciable during autumn when chlorophyll is recycled revealing the bright red leaf colour of carotenoids, and to unicellular organisms like bacteria when they exhibit decreased growth rate [1]. However, the type of senescence, subject of this review, that has recently captured the attention of the scientific community, pharmaceutical industry and even the general public is known as cellular senescence, typically defined as the irreversible or stable cell cycle arrest even in the presence of mitogenic stimuli [2, 3]. This rather simple definition encompasses a very complex and highly heterogeneous phenomenon, in which the stable cell cycle arrest can be driven by multiple mechanisms and associated with diverse phenotypes and functions. Together with loss of proliferative potential, a key feature that distinguishes senescence from other arrested cellular states, such as quiescence or terminal differentiation, is the acquisition of the ‘senescence-associated secretory phenotype’ (SASP) [4], which has strong implications on the outcome of senescence and in the development of therapeutic interventions.
The discovery of cellular senescence in 1961 by Hayflick and Moorhead [5] represented a ground-breaking finding as it was the first demonstration that normal cells are not immortal, as believed at the time, and provided the basis for a scientific explanation of the evolutionary theory of ageing initiated by Peter B. Medawar in 1952. In his lecture ‘An unresolved Problem of Biology’ delivered at University College London, he stated that the force of natural selection declines with age as the probability of an organism to exist declines (e.g. due to diseases, accidents, predators) in parallel to its reproductive value, a concept known as selection shadow. Hence, gene variants that are beneficial in early life are favoured over genes beneficial late in life, in perfect agreement with Darwinian theories. George C. Williams built on this model and introduced in 1957 the concept of antagonistic pleiotropy [6], based on genes having multiple functions: harmful late-acting genes can remain and accumulate in the population if they have a beneficial effect early in life. Hence, the biological and inevitable mortality of cells discovered by Hayflick and Moorhead fitted with Medawar and Williams model of ageing: cellular senescence is evolutionary conserved, because it confers survival advantage early in life, even if, past reproduction age, manifests deleterious effects. The cost–benefit in evolutionary terms is favourable.
Forty years on, our view of cellular senescence has been expanded, shaken, refined and now apparently settled on the consensus that the actual process of senescence is necessary and desirable to avoid propagation of cellular damage, but it is the failure of timely clearing of these cells from tissues that leads to persistent damage, a process where the (aged) immune system might play a major role, as will be discussed later. Worth mentioning at this point is the concept of immunosenescence which broadly speaking refers to the general and progressive decline of immune function during ageing. Senescence in T cells refers to the loss of proliferative potential generally resulting from excessive telomere erosion and associated with increased cytokine release [7]. However, it is not clear what immunosenescence refers to in terminally differentiated, hence non-proliferative, immune cells like, for example, neutrophils. Pears and apples are being mixed under the same label. Beyond a perhaps irrelevant discrepancy on definitions, an immediate consequence is that markers normally used to identify and study cellular senescence may be unsuitable to study immunosenescence, like proliferation arrest markers. Even in T cells, where senescence fits with the accepted definition of cellular senescence, it is not clear whether the typical markers p16 or β-galactosidase are involved [7]. Hence, a consensus of what constitutes immunosenescence and specific definitions applicable to proliferative and terminally differentiated immune cells are still needed.
In this review, we present senescence in the context of the resolution of inflammation and propose that a failure of this endogenous protective mechanism leads to persistent damage and inflammation. We also highlight the heterogeneity of senescence, how this diversity impacts research and opportunities and we bring together different views, contradictory only in appearance, on the role of senescence in health and disease and why consideration of ‘context’ is determining. In addition, we discuss the influence of the immune system in the successful completion of the senescence programme and how senescence can be harnessed therapeutically from multiple angles. Finally, we raise existing knowledge gaps and current challenges that need to be addressed to advance the therapeutic exploitation of senescence, hoping to stimulate and encourage the field to transform patient care based on strategies targeting senescence in multiple therapeutic areas such as ageing, tissue repair and cancer.
The endogenous defensive role of cellular senescence
General pathways and mission of senescence
Cellular senescence is a state of permanent cell cycle arrest induced in proliferating cells by multiple stressors and conditions. The most common mechanism involves the activation by p53 of cyclin-dependent kinase (CDK) inhibitors, such as p16 (CDKN2A), p15 (CDKN2B), p21 (CDKN1A) or p27 (CDKN1B). These inhibitors block the actions of CDK/cyclins complexes, preventing the phosphorylation of the retinoblastoma protein, Rb [3]. Hypophosphorylated Rb remains bound to the transcription factor E2F, preventing the transition of the cell cycle from G1 to S phase. This loss of proliferative potential occurs even in the presence of serum or growth factors, a key difference from quiescence, where cells, whilst non-proliferative, retain the potential to proliferate again when mitogenic stimuli become available [8]. Quiescence is an exit from the cell cycle into G0, while senescence is an arrest in G1. This implies another key distinctive feature of senescent cells: although proliferation is halted, growth is still ongoing, reflected in the increase in cell size, lysosomes and metabolic activity, all hallmarks of cellular senescence [2, 9, 10]. Quiescence is a stand-by mode, while senescence is a flight forward.
The increased activity of senescent cells results in a characteristic messaging secretome, the SASP, that confers new functions to the senescent cells. This is an important difference with apoptosis. Although both processes seem to share the mission of eliminating damaged cells and limit the propagation of the damage to daughter cells, it is not completely clear what determines one fate or another. Some redundancy exists, as there is evidence that one can compensate, to some extent, for the absence of the other [11]. Apoptosis is per se a cell death mechanism, but senescent cells remain alive for long periods of time [12], as if they could still offer a last service to the host before ultimately being eliminated. This last service is offered by the SASP [13, 14], a combination of factors that initiate the tissue repair programme and attract the immune system to induce their own clearance. The SASP also amplifies the senescence process by inducing secondary senescence to neighbouring cells either via soluble factors [15, 16] or via extracellular vesicles [17] and contributes to their increased survival. Moreover, the SASP can also promote a pro-regenerative response by favouring cell plasticity and stem cell activity [18] and induce vascular remodelling [19].
In summary, both arms of senescence, proliferation arrest and secretome, play specific homeostatic and defensive roles in physiological and pathological situations. This is discussed next.
Cancer, tissue repair and development
The first type of senescence discovered by Hayflick and Moorhead [5], now referred to replicative senescence, is caused by the progressive shortening of telomeres after a limited number of cell divisions resulting in the activation of the DNA damage response (DDR) once a critical telomere length has been reached. This response leads to senescence, preventing proliferation of aberrant cells and inhibiting tumour formation [20]. Similarly, the DDR can be triggered by the acquisition of activating or inactivating mutations in oncogenes or tumour suppressor genes, respectively, leading to the activation of mainly p53 and p16 or p21, initiating the senescence programme [3]. This oncogene-induced senescence was first discovered for the gene HRAS [21] and later on for other genes such as PTEN, MYC, BRAF, TP53, RAC1 and many others [22], and represents a potent endogenous anti-cancer mechanism acting at very early stages of tumorigenesis. Melanocytic nevi on skin surface are a visible example of oncogene-induced senescence, in which BRAF is mutated in ~ 80% of the cases [23] typically acquiring the activating mutation BRAFV600E. This illustrates the essential protective role of senescence under the constant environmental oncogenic pressure exerted by UV radiation. Indeed, germline mutations in CDKN2A, permitting senescence bypass, greatly increase melanoma susceptibility [24]. Inactivating mutations in other pro-senescence mediators like CDKN1A, associated with bladder cancer [25], or RB1, linked to retinoblastoma, provide further evidence of the anti-tumour role of senescence. In mice, mutations in CDKN2A gene, affecting either or both encoded proteins p16 and p19, or in CDKN1A, result in increased tumour development susceptibility [26, 27].
The senescence programme also orchestrates tissue remodelling associated with development as well as wound healing. Senescence occurs in the embryo in multiple structures like the endolymphatic sac, the inner ear, the apical ectodermal ridge and neural tube, and although some redundancy exists with apoptosis, impaired senescence in certain structures can result in morphological defects [3, 11, 28]. Senescence is also involved in the regulation of placental formation and function, and a deficient senescence programme has been associated with pregnancy complications like intrauterine growth restriction [29].
The role of senescence in tissue remodelling in the adult is also well established. Importantly, the SASP released by senescent cells influences the microenvironment orchestrating the process of wound healing. Proliferating myofibroblasts at early stages of wound healing, are driven into senescence by matricellular proteins like CCN1 during the remodelling phase [30]. Much evidence suggests that senescent cells get locked in a pro-remodelling state characterized by a reduction in collagen deposition and increase in the expression of remodelling enzymes like matrix metalloproteases. This characteristic pro-remodelling signature has been shown in transcriptomic analysis of dermal senescent fibroblasts [31], in mouse models of cutaneous wound healing [30, 32], in hepatic stellate cells [33] and in synovial fibroblasts from rheumatoid arthritis [34]. Interestingly, these examples represent very distinct types of senescence, including replicative, chemically induced and MC1 agonist-mediated senescence, suggesting that the pro-remodelling phenotype may be a common feature of senescence. Furthermore, repair can be impaired by the active elimination of senescent cells, using either the p16-3MR mouse model [32] or administration of senolytics [34]. Physiological pre-programmed cellular senescence in the adult also occurs during thymic involution [35] and megakaryocyte maturation [36].
Senescence as a pro-resolving mechanism
The termination of the inflammatory response is achieved by a number of endogenous mediators that promote the active and safe completion of the inflammatory response leading to restoration of homeostasis. The realization of the active, rather than passive nature of this process, gave birth to a new field of research, the resolution of inflammation [37], and to a new strategy to target inflammatory conditions, resolution pharmacology [38], based on promoting and reinforcing those endogenous pathways and mediators. Another important insight was the realisation that chronic inflammation can derive from the actual failure of these endogenous pathways. Conditions such as rheumatoid arthritis [39], atherosclerosis [40] and inflammatory bowel disease [41] are now seen as a failure of resolution and among the pro-resolving pathways and mediators under pre- and clinical investigation are melanocortins [42], formyl-peptide receptors [43] and lipid resolvins [44].
A common feature of non-resolving inflammation is the dysregulated persistence of certain cells after the inflammatory insult has been neutralized, cells that keep dancing when the music has already stopped. Persistence of myofibroblasts that refuse to die by evading apoptosis impairs wound resolution by promoting fibrosis [45]. Persistence of activated neutrophils with increased lifespan or impaired clearance can delay resolution by causing excessive tissue damage [46]. Similarly, persistence of senescent cells prevents resolution by the influence of damaging SASP. Persistent myofibroblasts can be targeted with pro-apoptotic drugs like ABT-263 [47], similar to persistent neutrophils which can be eliminated with CDK inhibitors like R-roscovitine [46]. Hence, persistent senescent cells can similarly be targeted for elimination to induce resolution.
One of the major processes that drives resolution is the effective and timely elimination of apoptotic immune cells once they have completed the function for which they were recruited to the tissue. Consequently, the process of efferocytosis [48], or phagocytosis of apoptotic cells, is commonly used as a functional assay to assess the potential of new pro-resolving candidate molecules. Efferocytosis is crucial to achieve resolution because non-cleared apoptotic cells will turn necrotic, releasing damaging contents which further promote inflammation [48, 49] (Fig. 1). Similarly, senescence seems to follow a cycle of sensing a harmful stimulus followed by containment and elimination, a catch-it-bin-it-kill-it model (Fig. 2). For many years, it has been debated whether in certain circumstances, like in ageing, the process of senescence was per se detrimental, quite in line with the antagonistic pleiotropy model [6], suggesting the existence of late-acting harmful genes. There is now substantial evidence stating that senescence may always favour a protective role but it is the inefficiency of the immune system what reveals the dark face of senescence [50, 51]. An inefficient coupling of the immune system to complete the senescence programme and close the cycle allows the persistence of senescent cells in tissues for long periods of time while releasing pro-inflammatory factors that promote chronic inflammation and damage (Fig. 1). The reasons why this clearance may fail will be discussed in detail.

Cell clearance and the resolution of inflammation. a The resolution of inflammation requires the efficient and timely clearance of apoptotic cells from tissues by efferocytosis. A defective clearance may lead to secondary necrosis further promoting inflammation. b Similarly, the clearance of senescent cells, efficient and timely, is also required to complete the senescence programme successfully. An impaired clearance will lead to persistent action of SASP components leading to a failure of resolution

The purpose of senescence. Cellular senescence is initiated upon detection of cellular damage or stress. The damage is contained by preventing spread to daughter cells and finally cleared by eliminating damaged cells and inducing tissue repair
We propose here that the process of cellular senescence represents another endogenous protective pro-resolving mechanism that when dysregulated, it leads to persistent inflammation and sustained damage, and similarly to other pro-resolving mechanisms, it can be targeted to favour resolution. We recently demonstrated that the activation of the pro-resolving melanocortin receptor 1 (MC1) induced senescence in hyperactivated synovial fibroblasts from rheumatoid arthritis (RA) patients and favoured resolution of inflammatory arthritis in a serum-transfer mouse model [34]. RA is characterized by persistent inflammation in synovial joints, in which fibroblasts play a crucial role in the chronicity of the process [39]. Synovial fibroblasts present features of tumour-like cells including excessive proliferation and invasiveness of surrounding tissues. Although non-malignant, somatic mutations in tumour suppressor genes can be found, which have been shown to be epigenetically transformed into a permanent state of activation [52]. This imprinted aggressive behaviour not only sustains the pro-inflammatory microenvironment, but it is also responsible for the destruction of cartilage and bone within the joints. Thus, induction of senescence in these cells by activating the pro-resolving receptor MC1 can stop the cycle of reciprocal activation and favour resolution of inflammation [34].
This pro-resolving model of senescence provides a framework that solves the paradoxical co-existence of physiological and pathological consequences of senescence, and provides a much clearer view on why, when and how, senescent cells should be targeted. A failure to complete the programme that brings tissues back to homeostasis after a previous insult is what determines the detrimental effects of senescence, something largely determined by the context, as explained below.
The bright and dark sides of senescence: reconciling opposite views
Senescence in context
Scientific literature contains as much evidence for the protective actions of senescence as for its detrimental role (Fig. 3). In addition to cancer prevention and treatment, promotion of wound healing and embryogenesis, beneficial effects of senescence have been observed in liver [33, 53], renal [54] and skin [30] fibrosis, myocardial infarct [55], pulmonary hypertension [56], nerve regeneration [57], metabolic dysfunction[58], atherosclerosis, and rheumatoid arthritis [34, 59]. On the other hand, senescence may worsen intervertebral disc regeneration [60], fibrotic pulmonary disease [61], hepatic steatosis [62], Parkinson’s disease [63], allograft survival [63], muscle regeneration [64], brain function [65], osteoarthritis[66], type 2 diabetes [67] and radiotherapy-induced bone loss [68]. Paradoxically, detrimental effects of senescence have also been reported in myocardial infarct [69] and dysfunction [70], cancer progression [71], metabolic dysfunction [72] and atherosclerosis [73, 74].

Physiological and pathological consequences of senescence. Beneficial and detrimental effects of senescence have been reported, suggesting the implementation of both pro- and anti-senescence approaches depending on the context
However, evolutionary speaking, such a conserved mechanism would not have evolved if it were detrimental to the host. Amassing the existing literature on the subject, it is clear that multiple factors are involved in defining the impact of cellular senescence. We propose the consideration of four main contextual aspects (cell type, cellular state, SASP type and ageing) that largely determine the protective or adverse outcome of senescence (Fig. 4). First, the cell type affected by senescence can define the result. For example, senescence occurring in hepatocytes, the parenchymal cells found in the liver, results in dysregulated fat deposition, leading to hepatic steatosis [62]. However, when senescence is induced in myofibroblasts or in hepatic stellate cells, which are mesenchymal cells involved in healing by acquiring a fibroblast-like phenotype upon injury, the effect can be beneficial by limiting fibrosis [33, 53]. In myocardial infarct, senescence happening in the functional parenchymal cells of the heart, i.e. cardiomyocytes, increases mortality in mice [69], while if senescence occurs in cardiac fibroblasts, it promotes tissue repair [55].

Contextual aspects determining senescence outcome. Whether the results of senescence are beneficial or pathological, this is largely determined by multiple aspects, commonly overlapping. The most important ones include: cell type, cell state, type of SASP released and age, which has a strong influence on the surveillance capacity of the immune system
The cellular state is another major determinant of the outcome of senescence. In rheumatoid arthritis, synovial fibroblasts are epigenetically locked in a hyperactivated state, sustaining immune cell recruitment and activation within the joints that further promotes inflammation and damage. This vicious cycle can be broken by inducing senescence in those fibroblasts, favouring the resolution of inflammation [34]. A different scenario takes place in osteoarthritis, in which senescence occurring in cartilage producing chondrocytes (parenchymal, indeed), is associated with cartilage degeneration [66].
Induction of senescence in a transformed cell is unequivocally beneficial to prevent cancer at an early stage. However, the type of SASP -third aspect of context- released in each particular case can greatly influence the outcome at later stages of tumorigenesis. Cancer cells can evade the immune system by various mechanisms and cancer immunotherapy is based on boosting the immune system to help find and/or destroy cancer cells. Then, the ability of the SASP to attract the immune system can contribute to the clearance of senescent tumour cells [75]. For example, CCL-5 secreted by senescent melanoma cells promotes the recruitment of tumour-infiltrating leukocytes leading to a tumour suppressive environment [76]. Conversely, in other forms of cancer, SASP contributes to cancer progression by the release of factors like IL-6 and IL-1α [71] as well as by other mechanisms including induction of epithelial–mesenchymal transition [77], mTOR activation[78], recruitment of immature myeloid cells [79], or induction of the checkpoint molecule PD-L1 [80]. Thus, the type of SASP plays an important role in favouring resolution and avoiding further tissue damage.
Beyond cancer, the frequent pro-inflammatory nature of the SASP is believed to be responsible for the toxic effects that accumulating senescent cells exert in aged organisms [81], which links to the fourth aspect of context, ageing. This was elegantly shown by Velarde et al. applying in young and old mice the model of mitochondrial dysfunction based on superoxide dismutase 2 (Sod2) deficiency, which leads to cellular senescence. They demonstrated that Sod2 deficiency, and hence increased senescence, promotes wound healing and epidermal reepithelization in young mice, while the opposite was observed in old mice [82]. The pathogenic effect exerted by ageing on the outcome of senescence is largely determined by factors like stem cell exhaustion [83] and by the fitness of the immune system, as discussed next.
The crucial role of the immune system
The completion of the senescent programme, the kill-it step (Fig. 2), requires the effective and timely cooperation of the immune system, and possibly, this is the most important factor for determining a successful resolution (Fig. 1). Immune surveillance of senescent cells is mainly carried out by natural killer (NK) cells, macrophages and T cells. NK cells target senescent cells via the receptor NKG2D binding to the ligands MICA and ULBP2 on the surface of senescent cells, and mediate their killing by the release of perforin and granzymes [84, 85]. CD8+ and CD4+ T cells are also involved in the elimination of senescent cells [86, 87]. The completion of the clearance to favour resolution requires the involvement of phagocytic cells like macrophages. Enrichment of F4/80+ cells was detected around areas of senescent cells in postpartum uterus, and their depletion caused enlargement of those areas [88]. In addition, it was found that monocytes/macrophages were required to complete the clearance of senescent hepatocytes after CD4+ T cells activity [86]. Macrophage plasticity may also be important and can be influenced by the SASP. Senescent stellate cells were shown to induce macrophage class-switch towards M1 phenotype favouring their clearance, while proliferating cells induced an M2 phenotype creating a tumour-promoting environment [89]. This co-existence of non-senescent cancer cells seems to be essential in the final outcome of senescence as it has also been shown that non-senescent hepatocellular carcinoma cells block the maturation of CCR2+ myeloid precursors preventing the clearance of senescent cells, favouring tumour growth [79].
The impact that organismal ageing exerts on the efficiency of the immune system and its surveillance capacity seems to drive the persistence of senescent cells within tissues and consequently their pathogenic effects. In a bleomycin model, it has been shown that senescent cell turnover can take from days in young mice to weeks in old animals [50], and that the reduced CD8+ T cell cytotoxic activity associated with age accelerates the accumulation of senescent cells [51]. However, inefficient clearance is not always due to an intrinsic defect in immune cells. For the immune system to find senescent cells, these have to be discoverable. Senescent cells can play ‘hide and seek’ and evade the immune system by the expression or release of certain mediators. The mechanisms for clearance of apoptotic cells are well understood and require the release of find-me signals (e.g. S1P, CX3CL1), with a balanced expression of eat-me (e.g. phosphatidylserine, calreticulin) and do not-eat-me signals (e.g. CD47, CD31, PAI-1) [90, 91]. These signals have been extensively investigated and exploited to promote resolution of inflammation [92,93,94]. Although these signals are not so well understood for senescent cells, several do not-eat-me signals have been reported, like expression of checkpoint molecule HLA-E, which blocks NKG2D-mediated cytotoxic response by NK and CD8+ T cells [87], or expression of decoy receptor 2, Dcr2, which inhibits the activation of death receptors 4 and 5 [85]. Other mechanisms may exist that render senescent cells immunologically cold and appropriate immune recognition signals are necessary to generate a response by immune cells. Moreover, the expression of these signals may depend on the influence of context-specific microenvironments. On the other hand, post-translationally modified vimentin has been proposed as an eat-me signal for senescent cells [95]. Little is known about whether additional eat-me signals exist in senescent cells, or if the senescent programme evolved to be coupled to subsequent apoptosis, relying then on the typical apoptosis eat-me signals to complete clearance.
Not all senescent cells are created equal
To produce a unified classification of senescence has proven difficult as multiple criteria could be applied: replicative or premature, presence or absence of DNA damage, intrinsically or extrinsically induced, damage-induced or developmentally programmed, and more. It is striking how a rather simple phenomenon, cell cycle arrest, can generate a vast diversity of cellular states. Multiple stimuli can result in senescence via multiple mechanisms, in multiple cell types, resulting in multiple SASP types, and multiple outcomes. Senescence heterogeneity occurs at many levels with implications in disease phenotypes, potential therapeutic approaches, in the way we conduct research and in the interpretation of results.
Several and diverse types of stimuli can induce senescence, including telomere attrition [5], oncogenic transformation [21], PTEN loss [96], mitochondrial dysfunction [97], cytosolic DNA [98], chemicals and genotoxic drugs [99, 100], disturbed blood flow [101], ultraviolet radiation [102], cell fusion [103], proteasome inhibition [104], or by melanocortin receptor 1 agonism [34]. The mechanisms by which senescence is executed also differ. While p16 (CDKN2A) plays a prominent role in oncogene-induced senescence [21], it is mainly p21 (CDKN1A) that mediates senescence during telomere loss [105] and embryonic development [11]. In hepatic tumour cells, however, it was shown that cells can evade senescence even when p16, p19, p21 and p53 are all expressed [106]. Moreover, the pro-senescence effect of p16 in human cells is more prominent than in mouse cells [107]. The DNA damage response mediates senescence as a result of telomere loss [105, 108] and in certain types of oncogene senescence [3], but not when senescence is elicited by mitochondrial dysfunction [97] or by developmental cues [11].
Heterogeneity in the SASP composition is also well documented. Although in general terms, the SASP is typically described as pro-inflammatory, there are many examples in which this is not the case. Senescence induced by mitochondrial dysfunction in human fibroblasts lacks IL-1-related cytokines [97], and senescence induced by ectopic expression of p16 does not increase typical SASP cytokines like IL-6, IL-8 or CXCL-1 [109]. Senescence induced by proteasome inhibition in lung fibroblasts also leads to a predominantly non-inflammatory secretome [110]. In addition, atypical non-inflammatory SASP is also associated with senescence induction by certain drugs, like treatment with doxorubicin on endothelial cells [111] and MC1-selective ligands on synovial fibroblasts [34]. It has been suggested that persistent DNA damage mediates cytokine secretion by senescent cells [112] via the activation of NF-κB [113]. It might then be plausible that in those types of senescence processes lacking a DDR response, the resulting SASP will not be pro-inflammatory. It remains to be known which scenario will be more favourable to achieve resolution: a pro-inflammatory SASP able to call the immune system to induce clearance, or a silent SASP that does not cause damage to the surrounding tissues but may delay clearance.
The sensitivity of cells to senolytics is also heterogeneous. Senolytics are drugs that induce lysis of senescent cells and are under investigation as potential therapies for cancer, ageing and other conditions. However, depending on the type of senescence, some senolytics may be more effective than others. The senolytic Bcl-2 inhibitor ABT-263, navitoclax, was effective in inducing apoptosis in murine embryonic fibroblasts [114], but ineffective in murine sarcoma cells overexpressing p16 [115]. Navitoclax was also ineffective in inducing cell death in melanoma cells, an effect attributed to increased levels of anti-apoptotic MCL-1. Interestingly, co-administration with the MCL-1 inhibitor S63845 markedly sensitized the cells to death by navitoclax [116].
One of the major implications of this multi-level heterogeneity is the lack of universal markers of senescence, which impacts scientific research in particular when comparing different studies. Multiple markers have been described, some of which are more common than others among the different types of senescence and are typically used in senescence research (Fig. 5). The most frequently used include the overexpression of cyclin inhibitors, detection of β-galactosidase reflecting lysosomal expansion, decrease of proliferation rate, detection of heterochromatin foci or markers of DNA damage and release of pro-inflammatory cytokines. However, as previously discussed, these features are not always present. Other markers have also been described in specific types of senescence like lamin B1 loss in HCA2 fibroblasts [117], MGST1 in COPD-derived fibroblasts [118], β3 integrin in primary fibroblasts [119] and WNT16B during replicative senescence in fibroblasts [120]. If we accept the definition of cellular senescence as cell cycle arrest with maintained cellular growth, as a minimum, measurements or markers indicative of decreased proliferation (cell number, proliferation marker Ki67, cell cycle phases, etc.), accompanied with markers indicative of increased growth or metabolic activity that distinguishes senescence from other cell arrested cellular states (e.g. increased cell size, lysosomal expansion reflected in β-galactosidase or lipofuscin content, etc.) should be used to determine the presence of senescence (Fig. 5). Other common hallmarks of senescence, however, may require the analysis of specific biomarkers, like particular SASP components [4, 113, 121], presence of DNA damage or involvement of specific CDK inhibitors. This may help to define the specific type of senescence as they tend to be context-dependent. This approach, while feasible during in vitro studies, presents difficulties in detecting senescence in vivo. Typically, detection of cell cycle inhibitors, β-galactosidase in tissue sections or whole organs, lipofuscin staining or reporter mice like the p16-3MR [32], INK-ATAAC [122], humanised p16-luc [123] and p21-p-luc mice [124] are currently available.

Hallmarks of senescence and biomarkers used in research. Cell cycle arrest ([122, 166]) increased cell size ([10, 166, 177]), lysosomal compartment expansion ([178,179,180]), DNA damage ([117, 181,182,183,184]), SASP components ([4]), anti-apoptotic pathways ([185]). Abbreviations: Immunofluorescence (IF), flow cytometry (FC), western blot (WB), colorimetric (C), histochemistry (HC), DNA damage response (DDR)
As universal markers may not exist, the creation of a catalogue compiling all types of senescence, with features and markers characterizing each one, would represent a very helpful initiative. Such a resource might include sections like cell type affected, physiological/pathological context, inducing stimulus, DNA damage involvement, participating cyclin inhibitors or SASP components. Recently, the literature-based resource SeneQuest (https://senequest.net) have been made available to search for genes related to senescence.
Senescence in the pipeline
Opportunities for therapeutic exploitation of senescence
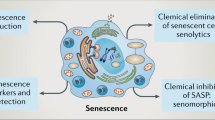
Cellular senescence offers an arsenal of potential therapeutic uses: for some, its intrinsic anti-cancer properties can be further exploited, while for others, senescence holds the secrets of youth. However, the bi-directional nature of these options, that is promotion or elimination depending on the target disease, presents important challenges: what is beneficial for one purpose may potentially be detrimental for another. The therapeutic potential of promoting, preventing, killing, modulating and clearing senescence will now be discussed (Fig. 6).

Potential therapeutic strategies based on senescence. Senescence can be targeted from multiple angles and has the potential to benefit multiple therapeutic indications. Abbreviations: activator/agonist ( ⊕), inhibitor/antagonist ( ⊖), overexpression (↑)
Several pro-senescence drugs are currently approved for the treatment of cancer, including methotrexate, etoposide, tamoxifen, palbociclib, doxorubicin or cisplatin, among many others, and generally induce senescence by a DNA-damage-mediated mechanism or interference with cell cycle components, among other mechanisms [99, 100, 125, 126]. Side effects derived from persistent SASP or from the pro-senescence action in off-target tissues limit their use [126, 127]. However, the induction of senescence also represents an opportunity to generate new vulnerabilities in cancer cells. Wang et al. demonstrated that p53 mutant liver cancer cells made senescent by treatment with CDC7 kinase inhibitors, become susceptible to senolysis by the antidepressant sertraline [128]. Interestingly, it has also been reported that p53 mutant breast cancers are resistant to chemotherapy with doxorubicin when co-treatment with hormone therapy is not used, because cells bypass apoptosis and become senescent. However, these p53 mutant cells rendered senescent by doxorubicin became sensitive to tamoxifen [129]. Hence, this double-hit approach suggests that induction of senescence followed by senolysis can enhance therapeutic efficacy in cancer. Induction of senescence to favour wound healing and induce resolution of fibrosis or chronic inflammatory conditions like rheumatoid arthritis may require less toxic approaches like the recently discovered pro-senescence activity of MC1-selective agonists [34], acting via the activation of a highly druggable GPCR which ligands have been shown to be safe in clinical trials. Other pro-senescence approaches reported include the inhibition of aurora kinases [130], Shp2 inhibitors [131], suicide gene therapy [115], MDM2/p53 interaction inhibitors [132], treatment with CCN1 protein [30] or interleukin 22 [133]. Interestingly, it was also shown that reprogramming approaches by ectopic expression of OSKM or Yamanaka factors (OCT4, SOX2, KLF4, MYC) to induce pluripotent stem cells (iPS) is strongly associated with induction of senescence where senescent cells create a tissue context involving IL-6 that favours OSKM-driven reprogramming in surrounding cells [134].
Complete suppression of senescence will invariably promote cancer development. However, there might be ways to prevent certain types of stress-related senescence by reducing the relevant stress signals. This approach is based on the information theory of ageing, suggesting that the accumulation of damage at the cellular level decreases cellular repair mechanisms, leading to decreased functionality of the whole organism. Ageing produces accumulation of DNA instability leading to re-distribution of the epigenetic regulators sirtuins to engage into repair mechanisms as a survival circuit, which is accompanied by disengagement of other cellular activities. This epigenetic noise (or information loss) caused by exhaustion of the survival mechanism, leads to loss of cellular identity terminating in senescence. Although this theory, so far, has only been fully demonstrated in yeast [135, 136], substantial evidence suggests a mayor role of sirtuins in eukaryotic cells senescence too. Sirt1 prevents telomere shortening in mice [137], while Sirt6 overexpression prevented replicative senescence in human cells [138]. Caloric restriction, by inducing sirtuins and enhancing DNA repair mechanisms, can reduce senescence burden in ageing and extend lifespan [139]. Caloric restriction can be mimicked with sirtuin-activating compounds (STACs) such as resveratrol, fisetin, or NAD+ precursors, which extend the lifespans of yeast, mice and humans [140,141,142]. Consistently, it has been shown that the inhibition of Sirt1 promotes premature senescence in mouse fibroblasts [143]. However, sirtuin inhibitors like sirtinol [144] present anti-senescence activity, suggesting that further research is needed to fully elucidate the role of sirtuins, or their targeting, in senescence. Indeed, it has been suggested that the dual outcome of sirtuin inhibition might depend on p53 status [145]. STACs act by activating AMPK which in turn inhibits mTOR. Hence, AMPK activators like metformin and mTOR inhibitors like rapamycin also prevent senescence and ageing [142, 146]. Inhibition of SIRT1 was also achieved by expression of the microRNA miR-34a, reducing senescence in human adipose tissue-derived mesenchymal stem cells [147].
The elimination of senescent cells using senolytics is being intensively investigated for applications in cancer, ageing and facilitation of resolution in fibrosis. Hence, under the right context, senolytics may also act as pro-resolving drugs. Senolytics aim to induce death in senescent cells without affecting proliferating or quiescent cells, by targeting a wide range of vulnerabilities existing specifically in senescent cells. These include the increased expression of BCL anti-apoptotic proteins (navitoclax and other BH3-mimetics), the PI3K/AKT pathway (quercetin), ephrin receptors (dasatinib), lysosomal V-ATPase (bafilomycin A1, concanamycin A), Na + /K + ATPase (cardiac glycosides), MDM2/p53 interaction (UBX0101), mTOR pathway (sertraline), oxidation resistance 1 (piperlongumine), USP7 protease (P22007), urokinase-type plasminogen activator receptor (CAR-T cells) and bromodomain and extra-terminal (BET) family proteins [66, 128, 148,149,150,151,152,153,154]. General cytotoxic drugs like the antibiotic duocarmycin can also be converted into senolytics by addition of a galactose motif, creating a pro-drug that is preferentially cleaved under high β-galactosidase expression, producing a targeted-delivery system for senescent cells [155]. Other antibiotics like azithromycin and roxithromycin have shown preferential cytotoxic activity in senescent fibroblasts [156], although the exact mechanism has not been reported. As previously discussed, different types of senescence display different sensitivities to senolytics.
A fourth type of approach to targeting senescence consists of using drugs that modulate the composition of the SASP, commonly termed senomorphics. The search for senomorphics has been focused on the discovery of drugs that reduce the pro-inflammatory activity of the SASP. For example, NF-κB inhibitors like apigenin or glucocorticoids can reduce the pro-inflammatory nature of the SASP [157, 158], as well as p38 inhibitors and JAK inhibitors, like SB203580 molecule and ruxolitinib, respectively [159, 160]. The natural compound rapamycin, via inhibition of mTOR, also modulates the composition of the SASP towards a less damaging secretome, and has shown to increase lifespan in mice [161, 162]. A senomorphic therapy is aimed at reducing the detrimental effects of a persistent SASP without the need of eliminating the senescent cells, which might be more advantageous than a senolytic as it may allow senescent cells to perform their physiological and reparative actions, without fuelling further inflammatory processes; however, this view needs to be corroborated. Therapeutic areas for potential application of senomorphics include the prevention of ageing associated pathologies, reduction of inflammatory pro-tumour microenvironment in cancer and reduction of damaging SASP during fibrosis.
Clearance of senescent cells can also be promoted therapeutically to facilitate their removal from tissues. The pro-tumorigenic effect of senescent cells derives from their persistence in tissues after containing the oncogenic damage. Thus, strategies that promote the surveillance capacity of immune cells may contribute to tumour regression. Ruscetti et al. demonstrated that treatment with palbociclib induced senescence in pancreatic cancer with a pro-angiogenic SASP, which favoured the recruitment of CD8+ T cells. These recruited cells showed signs of exhaustion but the combination of pro-senescence therapy with blockade of PD-1 checkpoint triggered T cell anti-tumour activity [19]. Hence, therapies to enhance clearance by immune system require not only the recruitment of appropriate cells, but also to ensure that their cytotoxic activity is intact. In addition, as discussed above, senescent cells need to be visible to the immune system to trigger their phagocytosis. Enhancement of apoptotic cell clearance can be achieved for example, by targeting the eat-me signal phosphatidylserine with a modified annexin A5 that interacts with αvβ3 receptors on the surface of macrophages [92]. This approach not only favoured resolution but it also induced the release of anti-inflammatory IL-10 by the phagocyte. This switch from pro- to anti-inflammatory signals during efferocytosis has been referred to as “tolerate me” signals [163]. Whether a similar non-phlogistic mechanism occurs during senescent cell engulfment remains to be studied. Another example of efferocytosis enhancement includes the matricellular protein CCN1, which facilitates the clearance of apoptotic neutrophils by a similar mechanism, bridging the apoptotic cell with the phagocyte [164]. Interestingly, as mentioned earlier, CCN1 induces senescence during physiological wound healing but whether CCN1 could also act as an opsonin for senescent cells has not been determined. On the other hand, blocking the do not-eat-me signal CD47 was demonstrated effective in promoting phagocytosis of apoptotic cells and preventing atherosclerosis [165]. The identification of similar signals that selectively tag senescent cells may lead to similar pro-phagocytic approaches as the one already existing for apoptosis.
Finally, reversal of senescence is typically investigated with the aim to discover factors that mediate the irreversibility of senescence [166]. It was found that telomere-induced senescence may be reversed by p53 or Rb inactivation if p16 is expressed at low levels [167], indicating the requirement of high levels of p16 to sustain the irreversible state. Non-replicative senescence in CD4+ memory T cells showed dependence on the p38 pathway [168]. Hypothetically, the reversal of senescence may benefit ageing-related pathologies by enhancing immunosenescence or by replenishing depleted stem cell pools. However, this approach brings the risk to spread the damage that led to senescence induction in the first place. The protein Rpl1 induces the bypass of replicative senescence but also contributed to transformation in rasVal12 mutant cells [169]. It is also believed that the transformation from nevi (senescent melanocytes) to melanoma occurs when senescent cells regain the ability to proliferate by acquiring additional abnormalities, hence reverting their senescent status [170].
Ongoing approaches in clinical development
Despite all the existing challenges, multiple clinical investigations with senescence-related approaches are currently undergoing. We identified 250 clinical trials registered at the World Health Organization repository with the majority (180) being interventional (Fig. 7, Table 1).

Clinical trials related to senescence. Information retrieved from the WHO International Clinical Trial Registry Platform in July 2020 using the search terms: senescence, senescent, senolytics, senolysis, senomorphic, senotherapy, senotherapeutic. This figure shows the classification of the trials according to different criteria: observational/interventional, whether they are actively recruiting or not, and the trial phase. Of the 180 interventional studies identified, 41 were actively recruiting. Of the 69 observational studies, 27 were actively recruiting. A selection is shown in Table 1
Unity Biotechnology Inc. has completed a phase I placebo-controlled trial where the primary outcome was to address the safety and tolerability of a single intra-articular injection of UBX0101, monitoring the incidence of serious and non-serious adverse events in knee osteoarthritis (NCT03513016). The compound is reported by the company as an inhibitor of the interaction between MDM2/p53, and has displayed senolytic properties in pre-clinical research [171]. A phase II trial with ~ 180 participants is currently ongoing to investigate the efficacy of a single dose on osteoarthritis pain score (NCT04129944). A small pilot study in five participants with Alzheimer’s disease is being conducted to assess brain penetrance of the senolytic compounds dasatinib and quercetin (NCT04063124). Another trial has investigated the efficacy of these two senolytics, given orally, in eliminating senescent cells in chronic kidney disease patients (NCT02848131). Results for this trial, led by Mayo Clinic, have been published [171], reporting significant decreases in senescent cell burden. Oral administration of fisetin, a sirtuin activator but also reported as senolytic, is being trialled in ~ 70 patients with osteoarthritis to address safety (NCT04210986). Observational studies on the role of senescence in several diseases are also being conducted, including COPD, diabetes, wound healing, complicated pregnancies and circulatory shock (Table 1).
Challenges in senescence research and potential therapies
While reviewing the recent advances in senescence research and directions towards potential clinical applications, we identified a number of challenges, gaps and research paths that may help advance the field of senescence and accelerate its successful translation into clinics.
We now know that senescence comes in many flavours, induced by multiple mechanisms leading to different SASPs, that may, or may not, be pro-inflammatory. Senescence is intensively studied in the context of fibrosis where bleomycin is used to induce the disease in animal models. However, bleomycin itself induces senescence by causing direct DNA strand breaks [172], suggesting that the SASP and its actions may greatly differ from the ones produced by endogenously induced senescence during would healing and fibrosis, and as such, results may not be extrapolatable into human disease. Fibrosis models have also been used to test senolytics and generally, when such treatments improve fibrosis, it is believed that senescent cells were the pathogenic cells. However, as Lagares et al. demonstrated, senolytics can also target and kill activated (i.e. non-senescent) myofibroblasts [47], which are major drivers of fibrosis. Therefore, the protective effects of senolytics in fibrosis should not be directly attributed to the elimination of senescent cells without performing more in-depth investigations.
Pro-senescence therapies are currently used to treat several forms of cancer. The major obstacles derive from the induction of senescence in off-target tissues, which may promote their premature ageing. Hence, selective and targeted senescence to cancer cells would be preferred. The group of Bernards et al. is working in that direction by performing screenings of pro-senescence targets in cancer cells. They identified not only molecules that induce senescence specifically in liver cancer cells, but, following a second screening, they also found specific senolytics which favour their elimination [128]. This is an excellent example of the integration of a pro-senescence followed by senolytic approach to ensure completion of the senescence programme, which they call the ‘one-two punch’ approach. Certain types of cancer are actually derived from mutations in genes related to senescence, like CDKN2A, inducing malignancy due to their ability to bypass senescence [24]. A better understanding of alternative routes to induce senescence may help to design novel therapies for those cancers.
Anti-senescence approaches are based on preventing the build-up of informational noise caused by cellular stress that leads to senescence. It has been reported that inhibitors of mTOR like rapamycin have anti-senescence, senolytic and senomorphic activity with potential to reduce age-related pathologies. However, rapamycin may also prevent oncogene-induced senescence by preventing up-regulation of p53, hence impairing our major endogenous anti-cancer mechanism from acting [96]. These potential detrimental actions require further investigation to ensure the safety of such approaches.
Some strategies focus on identifying common targets of senescent cells to develop broad-spectrum senolytics [149, 154, 155]. These can be very useful as research tools and will allow the comparison of different studies if all senescent cells, regardless of their type and context, respond to the same senolytic. However, selective senolytics for each type of senescence, and possible each cell type, may be more beneficial for therapeutic applications than a one-size-fits-all approach. For example, we may need to favour the elimination of senescent cells from a tumour while preserving senescent cells physiologically dealing with wound healing. This may reduce the therapeutic indications available for such drugs, but might provide a more balanced risk/benefit profile. However, this will require the identification of specific vulnerabilities for each type of senescence. In addition, all animal studies with senolytics, as with all studies essentially, are performed under controlled conditions where mice do not suffer from additional co-morbidities, traumas, or wounds. It is then yet unknown how senolytics will perform in more real-life situations. Another challenge related to senolytics is that the elimination of senescent cells may not restore organ or tissue function if not coupled with pro-regeneration approaches [45]. The elimination of senescent chondrocytes in osteoarthritis may help to reduce the sustained release of pro-inflammatory factors, but that alone may not lead to restoration of the chondrocyte population due to the intrinsic limited regenerative capability of this avascularised tissue. The same concept applies to fibrosis, where repopulation with functional parenchymal cells, not only elimination of senescent ones, is required for restoration of tissue function. The effects of senolysis on stem cell exhausted tissues may not restore function.
A further challenge is that senolytics are developed under the assumption that the immune system may recognise dead senescent cells, i.e. apoptotic cells. Speculating as we write, it may be possible that in certain contexts, phagocytes might not recognize senescent or apoptotic cells, rendering senolysis approaches detrimental, as deficient efferocytosis also leads to secondary cell necrosis, which would sustain pro-inflammatory damage and comport failed resolution. Indeed, inefficient efferocytosis is associated with pathologies like systemic lupus erythematosus due to genetic or acquired C1q deficiency [173]. How senolysis will perform in immunosuppressed patients also remains unknown. An interesting aspect of the clearance of senescent cells is that they are eaten alive, a process termed phagoptosis [174]. This may represent an opportunity for the development of strategies aimed at actively modulating the expression of surface molecules that target them for clearance, similar to approaches already under investigation that actively modulate the composition of the SASP. This is possible, because it is known that the SASP can be modulated without affecting the irreversibility of cell cycle arrest, as they seem to be controlled by independent regulatory networks. Cell cycle arrest is largely dependent on p16/Rb or p53/p21 pathways, whilst the SASP is more dependent on NF-κB, C/EBPβ or mTOR pathways [175] and inhibiting the latter ones does not revert cell cycle arrest.
The field of ageing research is also facing the difficulty in running the long clinical trials that will be required to assess if senolytics may indeed prevent age-related diseases. To facilitate research, the inclusion of organismal senescence (essentially ageing) as a disease category in the WHO International Classification of Diseases (ICD) has been proposed [176]: a major challenge, because accepting that ageing is a disease will have important social, political and medical implications. In the latest ICD version (ICD-11, https://www.icd.who.int), ‘old age’, under the code MG2A, is recognised as a symptom, sign or clinical finding, not elsewhere classified. Although age as a disease is difficult to accept by many, others are happy to conduct self-experimentation by taking available senolytics and STACs like quercetin or resveratrol. This kind of citizen science can be a double-edged sword. In the field of melanocortin research, due to self-administration of illegal melanocortin peptides to boost tanning, the development of melanoma on some of these individuals, likely derived from their risky behaviour in their sun exposure, led to the wrong suspicion that melanocortins may cause melanoma, a conclusion disproved in controlled clinical trials.
Summary
Senescence has emerged in recent years as the rising star among the cellular processes to target to develop innovative therapies due to their potential impact on diverse therapeutic areas, such as cancer, fibrosis and ageing. We propose here the process of cellular senescence as an endogenous pro-resolving mechanism. If we consider their clearance as an integral step of the senescence programme, failure to complete this last step is what results in non-resolving inflammation. Furthermore, we reason on how senescence is a highly heterogeneous phenomenon and we suggest that a precise definition of context will benefit our understanding and enable reliable and meaningful comparison across different studies. Senescence can be targeted from multiples angles (promotion, prevention, killing, modulation, clearance) and it is important to consider the context to determine what, when and how we should target senescence. Whether beneficial or detrimental, this cannot be decided without taking the context into account.
Senescence passes the appraisal, with the set objectives of (i) improving detection strategies, (ii) consideration of contextual aspects to decide beneficial/detrimental role, (iii) understanding better how the process of clearing occurs and could be modulated, (iv) creation of repositories compiling features of all types of senescence (cell type, context, pathways, SASP, biomarkers), and (v) development of more targeted approaches for specific pathological situations. Neither zombie cells nor regenerative powers, deciphering the process of senescence has a great potential to improve human health, potential reflected in the prominent presence of senescence approaches currently under clinical development.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
References
Ackermann M, Stearns SC, Jenal U (2003) Senescence in a bacterium with asymmetric division. Science 300(5627):1920. https://doi.org/10.1126/science.1083532
Hernandez-Segura A, Nehme J, Demaria M (2018) Hallmarks of cellular senescence. Trends Cell Biol 28(6):436–453. https://doi.org/10.1016/j.tcb.2018.02.001
Munoz-Espin D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15(7):482–496. https://doi.org/10.1038/nrm3823
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621. https://doi.org/10.1016/0014-4827(61)90192-6
Williams G (1957) Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 11(4):398–411
Akbar AN, Henson SM, Lanna A (2016) Senescence of T lymphocytes: implications for enhancing human immunity. Trends Immunol 37(12):866–876. https://doi.org/10.1016/j.it.2016.09.002
Blagosklonny MV (2011) Cell cycle arrest is not senescence. Aging (Albany NY) 3(2):94–101. https://doi.org/10.18632/aging.100281
Ogrodnik M, Salmonowicz H, Jurk D, Passos JF (2019) Expansion and cell-cycle arrest: common denominators of cellular senescence. Trends Biochem Sci 44(12):996–1008. https://doi.org/10.1016/j.tibs.2019.06.011
Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, Miettinen TP, Vaites LP, Soares LM, Paulo JA, Harper JW, Buratowski S, Manalis S, van Werven FJ, Holt LJ, Amon A (2019) Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell 176(5):1083–1097. https://doi.org/10.1016/j.cell.2019.01.018
Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M (2013) Programmed cell senescence during mammalian embryonic development. Cell 155(5):1104–1118. https://doi.org/10.1016/j.cell.2013.10.019
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G, Nomenclature Committee on Cell D (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16(1):3–11. https://doi.org/10.1038/cdd.2008.150
Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Sharma V, Ferrucci L, Campisi J, Schilling B (2020) A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 18(1):e3000599. https://doi.org/10.1371/journal.pbio.3000599
Fafian-Labora JA, O’Loghlen A (2020) Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. https://doi.org/10.1016/j.tcb.2020.05.003
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15(8):978–990. https://doi.org/10.1038/ncb2784
Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133(6):1006–1018. https://doi.org/10.1016/j.cell.2008.03.038
Fafian-Labora JA, O’Loghlen A (2020) Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol 30(8):628–639. https://doi.org/10.1016/j.tcb.2020.05.003
Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM (2017) The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31(2):172–183. https://doi.org/10.1101/gad.290635.116
Ruscetti M, Morris JPt, Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho YJ, Fennell M, Li J, Norgard RJ, Wilkinson JE, Alonso-Curbelo D, Sridharan R, Heller DA, de Stanchina E, Stanger BZ, Sherr CJ, Lowe SW, (2020) Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181(2):424–441. https://doi.org/10.1016/j.cell.2020.03.008
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130(2):223–233. https://doi.org/10.1016/j.cell.2007.07.003
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88(5):593–602. https://doi.org/10.1016/s0092-8674(00)81902-9
Gorgoulis VG, Halazonetis TD (2010) Oncogene-induced senescence: the bright and dark side of the response. Curr Opin Cell Biol 22(6):816–827. https://doi.org/10.1016/j.ceb.2010.07.013
Damsky WE, Bosenberg M (2017) Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene 36(42):5771–5792. https://doi.org/10.1038/onc.2017.189
Hayward NK (2003) Genetics of melanoma predisposition. Oncogene 22(20):3053–3062. https://doi.org/10.1038/sj.onc.1206445
Cazier JB, Rao SR, McLean CM, Walker AK, Wright BJ, Jaeger EE, Kartsonaki C, Marsden L, Yau C, Camps C, Kaisaki P, Oxford-Illumina WGSC, Taylor J, Catto JW, Tomlinson IP, Kiltie AE, Hamdy FC (2014) Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nat Commun 5:3756. https://doi.org/10.1038/ncomms4756
Serrano M (2000) The INK4a/ARF locus in murine tumorigenesis. Carcinogenesis 21(5):865–869. https://doi.org/10.1093/carcin/21.5.865
Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M (2001) Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res 61(16):6234–6238
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155(5):1119–1130. https://doi.org/10.1016/j.cell.2013.10.041
Gal H, Lysenko M, Stroganov S, Vadai E, Youssef SA, Tzadikevitch-Geffen K, Rotkopf R, Biron-Shental T, de Bruin A, Neeman M, Krizhanovsky V (2019) Molecular pathways of senescence regulate placental structure and function. EMBO J 38(18):e100849. https://doi.org/10.15252/embj.2018100849
Jun JI, Lau LF (2010) The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12(7):676–685. https://doi.org/10.1038/ncb2070
Shelton DN, Chang E, Whittier PS, Choi D, Funk WD (1999) Microarray analysis of replicative senescence. Curr Biol 9(17):939–945. https://doi.org/10.1016/s0960-9822(99)80420-5
Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31(6):722–733. https://doi.org/10.1016/j.devcel.2014.11.012
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134(4):657–667. https://doi.org/10.1016/j.cell.2008.06.049
Montero-Melendez T, Nagano A, Chelala C, Filer A, Buckley CD, Perretti M (2020) Therapeutic senescence via GPCR activation in synovial fibroblasts facilitates resolution of arthritis. Nat Commun 11(1):745. https://doi.org/10.1038/s41467-020-14421-x
Barbouti A, Evangelou K, Pateras IS, Papoudou-Bai A, Patereli A, Stefanaki K, Rontogianni D, Munoz-Espin D, Kanavaros P, Gorgoulis VG (2019) In situ evidence of cellular senescence in Thymic Epithelial Cells (TECs) during human thymic involution. Mech Ageing Dev 177:88–90. https://doi.org/10.1016/j.mad.2018.02.005
Besancenot R, Chaligne R, Tonetti C, Pasquier F, Marty C, Lecluse Y, Vainchenker W, Constantinescu SN, Giraudier S (2010) A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol. https://doi.org/10.1371/journal.pbio.1000476
Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL (2007) Resolution of inflammation: state of the art, definitions and terms. FASEB J 21(2):325–332. https://doi.org/10.1096/fj.06-7227rev
Perretti M, Leroy X, Bland EJ, Montero-Melendez T (2015) Resolution pharmacology: opportunities for therapeutic innovation in inflammation. Trends Pharmacol Sci 36(11):737–755. https://doi.org/10.1016/j.tips.2015.07.007
Buckley CD (2011) Why does chronic inflammation persist: an unexpected role for fibroblasts. Immunol Lett 138(1):12–14. https://doi.org/10.1016/j.imlet.2011.02.010
Fredman G, Tabas I (2017) Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am J Pathol 187(6):1211–1221. https://doi.org/10.1016/j.ajpath.2017.01.018
Ho GT, Cartwright JA, Thompson EJ, Bain CC, Rossi AG (2020) Resolution of inflammation and gut repair in IBD: translational steps towards complete mucosal healing. Inflamm Bowel Dis 26(8):1131–1143. https://doi.org/10.1093/ibd/izaa045
Montero-Melendez T (2015) ACTH: the forgotten therapy. Semin Immunol 27(3):216–226. https://doi.org/10.1016/j.smim.2015.02.003
Perretti M, D’Acquisto F (2009) Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol 9(1):62–70. https://doi.org/10.1038/nri2470
Serhan CN, Levy BD (2018) Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 128(7):2657–2669. https://doi.org/10.1172/JCI97943
Hinz B, Lagares D (2020) Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol 16(1):11–31. https://doi.org/10.1038/s41584-019-0324-5
Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez-Losa M, Walker TR, Duffin R, Gray M, Crescenzi E, Martin MC, Brady HJ, Savill JS, Dransfield I, Haslett C (2006) Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med 12(9):1056–1064. https://doi.org/10.1038/nm1468
Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, Sakai N, Kuehl T, Ryan J, Bhola P, Montero J, Kapoor M, Baron M, Varelas X, Tschumperlin DJ, Letai A, Tager AM (2017) Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aal3765
Doran AC, Yurdagul A Jr, Tabas I (2020) Efferocytosis in health and disease. Nat Rev Immunol 20(4):254–267. https://doi.org/10.1038/s41577-019-0240-6
Arienti S, Barth ND, Dorward DA, Rossi AG, Dransfield I (2019) Regulation of apoptotic cell clearance during resolution of inflammation. Front Pharmacol 10:891. https://doi.org/10.3389/fphar.2019.00891
Karin O, Agrawal A, Porat Z, Krizhanovsky V, Alon U (2019) Senescent cell turnover slows with age providing an explanation for the Gompertz law. Nat Commun 10(1):5495. https://doi.org/10.1038/s41467-019-13192-4
Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira A, Windheim J, Tsoory M, Schirmbeck R, Amit I, Geiger H, Krizhanovsky V (2018) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9(1):5435. https://doi.org/10.1038/s41467-018-07825-3
Bottini N, Firestein GS (2013) Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 9(1):24–33. https://doi.org/10.1038/nrrheum.2012.190
Kim KH, Chen CC, Monzon RI, Lau LF (2013) Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol 33(10):2078–2090. https://doi.org/10.1128/MCB.00049-13
Wolstein JM, Lee DH, Michaud J, Buot V, Stefanchik B, Plotkin MD (2010) INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am J Physiol Renal Physiol 299(6):F1486-1495. https://doi.org/10.1152/ajprenal.00378.2010
Zhu F, Li Y, Zhang J, Piao C, Liu T, Li HH, Du J (2013) Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 8(9):e74535. https://doi.org/10.1371/journal.pone.0074535
Mouraret N, Marcos E, Abid S, Gary-Bobo G, Saker M, Houssaini A, Dubois-Rande JL, Boyer L, Boczkowski J, Derumeaux G, Amsellem V, Adnot S (2013) Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation 127(16):1664–1676. https://doi.org/10.1161/CIRCULATIONAHA.113.002434
Gomez-Sanchez JA, Gomis-Coloma C, Morenilla-Palao C, Peiro G, Serra E, Serrano M, Cabedo H (2013) Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain 136(Pt 7):2262–2278. https://doi.org/10.1093/brain/awt130
Gonzalez-Navarro H, Vinue A, Sanz MJ, Delgado M, Pozo MA, Serrano M, Burks DJ, Andres V (2013) Increased dosage of Ink4/Arf protects against glucose intolerance and insulin resistance associated with aging. Aging Cell 12(1):102–111. https://doi.org/10.1111/acel.12023
Taniguchi K, Kohsaka H, Inoue N, Terada Y, Ito H, Hirokawa K, Miyasaka N (1999) Induction of the p16INK4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nat Med 5(7):760–767. https://doi.org/10.1038/10480
Patil P, Dong Q, Wang D, Chang J, Wiley C, Demaria M, Lee J, Kang J, Niedernhofer LJ, Robbins PD, Sowa G, Campisi J, Zhou D, Vo N (2019) Systemic clearance of p16(INK4a) -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 18(3):e12927. https://doi.org/10.1111/acel.12927
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532. https://doi.org/10.1038/ncomms14532
Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, Grellscheid SN, Hoeijmakers JHJ, Barnhoorn S, Mann DA, Bird TG, Vermeij WP, Kirkland JL, Passos JF, von Zglinicki T, Jurk D (2017) Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 8:15691. https://doi.org/10.1038/ncomms15691
Chinta SJ, Woods G, Demaria M, Rane A, Zou Y, McQuade A, Rajagopalan S, Limbad C, Madden DT, Campisi J, Andersen JK (2018) Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to parkinson’s disease. Cell Rep 22(4):930–940. https://doi.org/10.1016/j.celrep.2017.12.092
Du J, Klein JD, Hassounah F, Zhang J, Zhang C, Wang XH (2014) Aging increases CCN1 expression leading to muscle senescence. Am J Physiol Cell Physiol 306(1):C28-36. https://doi.org/10.1152/ajpcell.00066.2013
Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, Andersen JK (2015) Cellular senescence and the aging brain. Exp Gerontol 68:3–7. https://doi.org/10.1016/j.exger.2014.09.018
Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, Elisseeff JH (2017) Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23(6):775–781. https://doi.org/10.1038/nm.4324
Sone H, Kagawa Y (2005) Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 48(1):58–67. https://doi.org/10.1007/s00125-004-1605-2
Chandra A, Lagnado AB, Farr JN, Monroe DG, Park S, Hachfeld C, Tchkonia T, Kirkland JL, Khosla S, Passos JF, Pignolo RJ (2020) Targeted reduction of senescent cell burden alleviates focal radiotherapy-related bone loss. J Bone Miner Res 35(6):1119–1131. https://doi.org/10.1002/jbmr.3978
Walaszczyk A, Dookun E, Redgrave R, Tual-Chalot S, Victorelli S, Spyridopoulos I, Owens A, Arthur HM, Passos JF, Richardson GD (2019) Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 18(3):e12945. https://doi.org/10.1111/acel.12945
Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D, Proctor C, Correia-Melo C, Victorelli S, Fielder E, Berlinguer-Palmini R, Owens A, Greaves LC, Kolsky KL, Parini A, Douin-Echinard V, LeBrasseur NK, Arthur HM, Tual-Chalot S, Schafer MJ, Roos CM, Miller JD, Robertson N, Mann J, Adams PD, Tchkonia T, Kirkland JL, Mialet-Perez J, Richardson GD, Passos JF (2019) Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. https://doi.org/10.15252/embj.2018100492
Wiley CD, Schaum N, Alimirah F, Lopez-Dominguez JA, Orjalo AV, Scott G, Desprez PY, Benz C, Davalos AR, Campisi J (2018) Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep 8(1):2410. https://doi.org/10.1038/s41598-018-20000-4
Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, Prata LG, van Dijk TH, Verkade E, Casaclang-Verzosa G, Johnson KO, Cubro H, Doornebal EJ, Ogrodnik M, Jurk D, Jensen MD, Chini EN, Miller JD, Matveyenko A, Stout MB, Schafer MJ, White TA, Hickson LJ, Demaria M, Garovic V, Grande J, Arriaga EA, Kuipers F, von Zglinicki T, LeBrasseur NK, Campisi J, Tchkonia T, Kirkland JL (2019) Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18(3):e12950. https://doi.org/10.1111/acel.12950
Sun XM, Patel DD, Acosta JC, Gil J, Soutar AK (2011) Premature senescence in cells from patients with autosomal recessive hypercholesterolemia (ARH): evidence for a role for ARH in mitosis. Arterioscler Thromb Vasc Biol 31(10):2270–2277. https://doi.org/10.1161/ATVBAHA.111.232223
Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM (2016) Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354(6311):472–477. https://doi.org/10.1126/science.aaf6659
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445(7128):656–660. https://doi.org/10.1038/nature05529
Vilgelm AE, Johnson CA, Prasad N, Yang J, Chen SC, Ayers GD, Pawlikowski JS, Raman D, Sosman JA, Kelley M, Ecsedy JA, Shyr Y, Levy SE, Richmond A (2016) Connecting the dots: therapy-induced senescence and a tumor-suppressive immune microenvironment. J Natl Cancer Inst 108(6):djv406. https://doi.org/10.1093/jnci/djv406
Laberge RM, Awad P, Campisi J, Desprez PY (2012) Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron 5(1):39–44. https://doi.org/10.1007/s12307-011-0069-4
Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, Hubbard GB, Ikeno Y, Javors M, Desprez PY, Benz CC, Kapahi P, Nelson PS, Campisi J (2015) MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17(8):1049–1061. https://doi.org/10.1038/ncb3195
Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, Heikenwalder M, Wang XW, Zender L, Greten TF (2016) Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30(4):533–547. https://doi.org/10.1016/j.ccell.2016.09.003
Xu Q, Long Q, Zhu D, Fu D, Zhang B, Han L, Qian M, Guo J, Xu J, Cao L, Chin YE, Coppe JP, Lam EW, Campisi J, Sun Y (2019) Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell 18(6):e13027. https://doi.org/10.1111/acel.13027
Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM (2017) Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov 16(10):718–735. https://doi.org/10.1038/nrd.2017.116
Velarde MC, Demaria M, Melov S, Campisi J (2015) Pleiotropic age-dependent effects of mitochondrial dysfunction on epidermal stem cells. Proc Natl Acad Sci U S A 112(33):10407–10412. https://doi.org/10.1073/pnas.1505675112
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT (2006) Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443(7110):421–426. https://doi.org/10.1038/nature05159
Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, Golani O, Polic B, Krizhanovsky V (2016) NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany NY) 8(2):328–344. https://doi.org/10.18632/aging.100897
Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V (2013) Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 32(15):1971–1977. https://doi.org/10.1038/onc.2012.206
Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L (2011) Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479(7374):547–551. https://doi.org/10.1038/nature10599
Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, Virasami A, Sebire NJ, Kinsler V, Valdovinos A, LeSaux CJ, Passos JF, Antoniou A, Rustin MHA, Campisi J, Akbar AN (2019) Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat Commun 10(1):2387. https://doi.org/10.1038/s41467-019-10335-5
Egashira M, Hirota Y, Shimizu-Hirota R, Saito-Fujita T, Haraguchi H, Matsumoto L, Matsuo M, Hiraoka T, Tanaka T, Akaeda S, Takehisa C, Saito-Kanatani M, Maeda KI, Fujii T, Osuga Y (2017) F4/80+ macrophages contribute to clearance of senescent cells in the mouse postpartum uterus. Endocrinology 158(7):2344–2353. https://doi.org/10.1210/en.2016-1886
Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, Lowe SW (2013) Non-cell-autonomous tumor suppression by p53. Cell 153(2):449–460. https://doi.org/10.1016/j.cell.2013.03.020
Park SY, Kim IS (2017) Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp Mol Med 49(5):e331. https://doi.org/10.1038/emm.2017.52
Park YJ, Liu G, Lorne EF, Zhao X, Wang J, Tsuruta Y, Zmijewski J, Abraham E (2008) PAI-1 inhibits neutrophil efferocytosis. Proc Natl Acad Sci U S A 105(33):11784–11789. https://doi.org/10.1073/pnas.0801394105
Schutters K, Kusters DH, Chatrou ML, Montero-Melendez T, Donners M, Deckers NM, Krysko DV, Vandenabeele P, Perretti M, Schurgers LJ, Reutelingsperger CP (2013) Cell surface-expressed phosphatidylserine as therapeutic target to enhance phagocytosis of apoptotic cells. Cell Death Differ 20(1):49–56. https://doi.org/10.1038/cdd.2012.107
Gerlach BD, Marinello M, Heinz J, Rymut N, Sansbury BE, Riley CO, Sadhu S, Hosseini Z, Kojima Y, Tang DD, Leeper NJ, Spite M, Barroso M, Rayner KJ, Fredman G (2020) Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ 27(2):525–539. https://doi.org/10.1038/s41418-019-0370-1
Osman R, Tacnet-Delorme P, Kleman JP, Millet A, Frachet P (2017) Calreticulin release at an early stage of death modulates the clearance by macrophages of apoptotic cells. Front Immunol 8:1034. https://doi.org/10.3389/fimmu.2017.01034
Frescas D, Roux CM, Aygun-Sunar S, Gleiberman AS, Krasnov P, Kurnasov OV, Strom E, Virtuoso LP, Wrobel M, Osterman AL, Antoch MP, Mett V, Chernova OB, Gudkov AV (2017) Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc Natl Acad Sci U S A 114(9):E1668–E1677. https://doi.org/10.1073/pnas.1614661114
Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, Cheng K, Varmeh S, Kozma SC, Thomas G, Rosivatz E, Woscholski R, Cognetti F, Scher HI, Pandolfi PP (2010) A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest 120(3):681–693. https://doi.org/10.1172/JCI40535
Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23(2):303–314. https://doi.org/10.1016/j.cmet.2015.11.011
Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L (2018) Cytosolic DNA sensing in organismal tumor control. Cancer Cell 34(3):361–378. https://doi.org/10.1016/j.ccell.2018.05.013
Lozano-Torres B, Estepa-Fernández A, Rovira M, Orzáez M, Serrano M, Martínez-Máñez R, Sancenón F (2019) The chemistry of senescence. Nat Rev Chem 3(7):426–441. https://doi.org/10.1038/s41570-019-0108-0
Petrova NV, Velichko AK, Razin SV, Kantidze OL (2016) Small molecule compounds that induce cellular senescence. Aging Cell 15(6):999–1017. https://doi.org/10.1111/acel.12518
Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, White A, Biswas S, Khamis R, Chong CK, Cheung WM, Sherwin SJ, Bennett MR, Gil J, Mason JC, Haskard DO, Evans PC (2014) Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol 34(5):985–995. https://doi.org/10.1161/ATVBAHA.114.303415
Shin J, Kim JH, Kim EK (2012) Repeated exposure of human fibroblasts to UVR induces secretion of stem cell factor and senescence. J Eur Acad Dermatol Venereol 26(12):1577–1580. https://doi.org/10.1111/j.1468-3083.2011.04223.x
Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V (2013) Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 27(21):2356–2366. https://doi.org/10.1101/gad.227512.113
Torres C, Lewis L, Cristofalo VJ (2006) Proteasome inhibitors shorten replicative life span and induce a senescent-like phenotype of human fibroblasts. J Cell Physiol 207(3):845–853. https://doi.org/10.1002/jcp.20630
Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 14(4):501–513. https://doi.org/10.1016/s1097-2765(04)00256-4
Obata M, Imamura E, Yoshida Y, Goto J, Kishibe K, Yasuda A, Ogawa K (2001) Resistance of primary cultured mouse hepatic tumor cells to cellular senescence despite expression of p16(Ink4a), p19(Arf), p53, and p21(Waf1/Cip1). Mol Carcinog 32(1):9–18. https://doi.org/10.1002/mc.1059
Evan GI, d’Adda di Fagagna F (2009) Cellular senescence: hot or what? Curr Opin Genet Dev 19(1):25–31. https://doi.org/10.1016/j.gde.2008.11.009
d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426(6963):194–198. https://doi.org/10.1038/nature02118
Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J (2011) Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 286(42):36396–36403. https://doi.org/10.1074/jbc.M111.257071
Maciel-Baron LA, Morales-Rosales SL, Aquino-Cruz AA, Triana-Martinez F, Galvan-Arzate S, Luna-Lopez A, Gonzalez-Puertos VY, Lopez-Diazguerrero NE, Torres C, Konigsberg M (2016) Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age (Dordr) 38(1):26. https://doi.org/10.1007/s11357-016-9886-1
Bent EH, Gilbert LA, Hemann MT (2016) A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev 30(16):1811–1821. https://doi.org/10.1101/gad.284851.116
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11(8):973–979. https://doi.org/10.1038/ncb1909
Freund A, Orjalo AV, Desprez PY, Campisi J (2010) Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16(5):238–246. https://doi.org/10.1016/j.molmed.2010.03.003
Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, Wren JD, Niedernhofer LJ, Robbins PD, Kirkland JL (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15(3):428–435. https://doi.org/10.1111/acel.12445
Kohli J, Campisi J, Demaria M (2018) A novel suicide gene therapy for the treatment of p16(Ink4a)-overexpressing tumors. Oncotarget 9(7):7274–7281. https://doi.org/10.18632/oncotarget.23752
Mukherjee N, Skees J, Todd KJ, West DA, Lambert KA, Robinson WA, Amato CM, Couts KL, Van Gulick R, MacBeth M, Nassar K, Tan AC, Zhai Z, Fujita M, Bagby SM, Dart CR, Lambert JR, Norris DA, Shellman YG (2020) MCL1 inhibitors S63845/MIK665 plus Navitoclax synergistically kill difficult-to-treat melanoma cells. Cell Death Dis 11(6):443. https://doi.org/10.1038/s41419-020-2646-2
Freund A, Laberge RM, Demaria M, Campisi J (2012) Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23(11):2066–2075. https://doi.org/10.1091/mbc.E11-10-0884
Woldhuis RR, de Vries M, Timens W, van den Berge M, Demaria M, Oliver BGG, Heijink IH, Brandsma CA (2020) Link between increased cellular senescence and extracellular matrix changes in COPD. Am J Physiol Lung Cell Mol Physiol 319(1):L48–L60. https://doi.org/10.1152/ajplung.00028.2020
Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, O’Loghlen A (2017) Integrin beta 3 regulates cellular senescence by activating the TGF-beta pathway. Cell Rep 18(10):2480–2493. https://doi.org/10.1016/j.celrep.2017.02.012
Binet R, Ythier D, Robles AI, Collado M, Larrieu D, Fonti C, Brambilla E, Brambilla C, Serrano M, Harris CC, Pedeux R (2009) WNT16B is a new marker of cellular senescence that regulates p53 activity and the phosphoinositide 3-kinase/AKT pathway. Cancer Res 69(24):9183–9191. https://doi.org/10.1158/0008-5472.CAN-09-1016
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M (2019) Cellular senescence: defining a path forward. Cell 179(4):813–827. https://doi.org/10.1016/j.cell.2019.10.005
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479(7372):232–236. https://doi.org/10.1038/nature10600
Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, Kubo Y, Mann DJ, Ohmura M, Hirao A, Saya H, Arase S, Hayashi Y, Nakao K, Matsumoto M, Ohtani N, Hara E (2009) Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J Cell Biol 186(3):393–407. https://doi.org/10.1083/jcb.200904105
Ohtani N, Imamura Y, Yamakoshi K, Hirota F, Nakayama R, Kubo Y, Ishimaru N, Takahashi A, Hirao A, Shimizu T, Mann DJ, Saya H, Hayashi Y, Arase S, Matsumoto M, Kazuki N, Hara E (2007) Visualizing the dynamics of p21(Waf1/Cip1) cyclin-dependent kinase inhibitor expression in living animals. Proc Natl Acad Sci U S A 104(38):15034–15039. https://doi.org/10.1073/pnas.0706949104
Saleh T, Bloukh S, Carpenter VJ, Alwohoush E, Bakeer J, Darwish S, Azab B, Gewirtz DA (2020) Therapy-induced senescence: an “old” friend becomes the enemy. Cancers (Basel). https://doi.org/10.3390/cancers12040822
Wang B, Kohli J, Demaria M (2020) Senescent cells in cancer therapy: friends or foes? Trends Cancer. https://doi.org/10.1016/j.trecan.2020.05.004
Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H, Campisi J (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7(2):165–176. https://doi.org/10.1158/2159-8290.CD-16-0241
Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, de Oliveira RL, Morris B, Gadiot J, Wang W, du Chatinier A, Wang L, Gao D, Evers B, Jin G, Xue Z, Schepers A, Jochems F, Sanchez AM, Mainardi S, Te Riele H, Beijersbergen RL, Qin W, Akkari L, Bernards R (2019) Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 574(7777):268–272. https://doi.org/10.1038/s41586-019-1607-3
Ungerleider NA, Rao SG, Shahbandi A, Yee D, Niu T, Frey WD, Jackson JG (2018) Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res 20(1):115. https://doi.org/10.1186/s13058-018-1044-5
Wang L, Leite de Oliveira R, Wang C, Fernandes Neto JM, Mainardi S, Evers B, Lieftink C, Morris B, Jochems F, Willemsen L, Beijersbergen RL, Bernards R (2017) High-throughput functional genetic and compound screens identify targets for senescence induction in cancer. Cell Rep 21(3):773–783. https://doi.org/10.1016/j.celrep.2017.09.085
Lan L, Holland JD, Qi J, Grosskopf S, Rademann J, Vogel R, Gyorffy B, Wulf-Goldenberg A, Birchmeier W (2015) Shp2 signaling suppresses senescence in PyMT-induced mammary gland cancer in mice. EMBO J 34(11):1493–1508. https://doi.org/10.15252/embj.201489004
Walter RFH, Werner R, Wessolly M, Mairinger E, Borchert S, Schmeller J, Kollmeier J, Mairinger T, Hager T, Bankfalvi A, Christoph DC, Eberhardt WEE, Plones T, Aigner C, Schmid KW, Wohlschlaeger J, Mairinger FD (2018) Inhibition of MDM2 via Nutlin-3A: a potential therapeutic approach for pleural mesotheliomas with MDM2-induced inactivation of wild-type P53. J Oncol 2018:1986982. https://doi.org/10.1155/2018/1986982
Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, Gao B (2012) Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 56(3):1150–1159. https://doi.org/10.1002/hep.25744
Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Munoz-Martin M, Blanco-Aparicio C, Pastor J, Gomez-Lopez G, De Martino A, Blasco MA, Abad M, Serrano M (2016) Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. https://doi.org/10.1126/science.aaf4445
Sinclair DA, Guarente L (1997) Extrachromosomal rDNA circles–a cause of aging in yeast. Cell 91(7):1033–1042. https://doi.org/10.1016/s0092-8674(00)80493-6
Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, Guarente L (1997) Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 89(3):381–391. https://doi.org/10.1016/s0092-8674(00)80219-6
Palacios JA, Herranz D, De Bonis ML, Velasco S, Serrano M, Blasco MA (2010) SIRT1 contributes to telomere maintenance and augments global homologous recombination. J Cell Biol 191(7):1299–1313. https://doi.org/10.1083/jcb.201005160
Chen J, Xie JJ, Jin MY, Gu YT, Wu CC, Guo WJ, Yan YZ, Zhang ZJ, Wang JL, Zhang XL, Lin Y, Sun JL, Zhu GH, Wang XY, Wu YS (2018) Sirt6 overexpression suppresses senescence and apoptosis of nucleus pulposus cells by inducing autophagy in a model of intervertebral disc degeneration. Cell Death Dis 9(2):56. https://doi.org/10.1038/s41419-017-0085-5
Fontana L, Nehme J, Demaria M (2018) Caloric restriction and cellular senescence. Mech Ageing Dev 176:19–23. https://doi.org/10.1016/j.mad.2018.10.005
Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430(7000):686–689. https://doi.org/10.1038/nature02789
Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, Grassi D, Wentworth M, Burd CE, Arriaga EA, Ladiges WL, Tchkonia T, Kirkland JL, Robbins PD, Niedernhofer LJ (2018) Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36:18–28. https://doi.org/10.1016/j.ebiom.2018.09.015
Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17(11):679–690. https://doi.org/10.1038/nrm.2016.93
Volonte D, Zou H, Bartholomew JN, Liu Z, Morel PA, Galbiati F (2015) Oxidative stress-induced inhibition of Sirt1 by caveolin-1 promotes p53-dependent premature senescence and stimulates the secretion of interleukin 6 (IL-6). J Biol Chem 290(7):4202–4214. https://doi.org/10.1074/jbc.M114.598268
Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y, Kaneki M (2006) Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25(2):176–185. https://doi.org/10.1038/sj.onc.1209049
Brooks CL, Gu W (2009) How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer 9(2):123–128. https://doi.org/10.1038/nrc2562
Karnewar S, Neeli PK, Panuganti D, Kotagiri S, Mallappa S, Jain N, Jerald MK, Kotamraju S (2018) Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: relevance in age-associated vascular dysfunction. Biochim Biophys Acta Mol Basis Dis 1864(4 pt A):1115–1128. https://doi.org/10.1016/j.bbadis.2018.01.018
Mokhberian N, Bolandi Z, Eftekhary M, Hashemi SM, Jajarmi V, Sharifi K, Ghanbarian H (2020) Inhibition of miR-34a reduces cellular senescence in human adipose tissue-derived mesenchymal stem cells through the activation of SIRT1. Life Sci 257:118055. https://doi.org/10.1016/j.lfs.2020.118055
Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, Kratzat S, Purfurst B, Walenta S, Mueller-Klieser W, Graler M, Hummel M, Keller U, Buck AK, Dorken B, Willmitzer L, Reimann M, Kempa S, Lee S, Schmitt CA (2013) Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501(7467):421–425. https://doi.org/10.1038/nature12437
Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, Wolter K, Pombo J, Irvine EE, Innes AJ, Birch J, Glegola J, Manshaei S, Heide D, Dharmalingam G, Harbig J, Olona A, Behmoaras J, Dauch D, Uren AG, Zender L, Vernia S, Martinez-Barbera JP, Heikenwalder M, Withers DJ, Gil J (2019) Cardiac glycosides are broad-spectrum senolytics. Nat Metab 1(11):1074–1088. https://doi.org/10.1038/s42255-019-0122-z
Soto-Gamez A, Demaria M (2017) Therapeutic interventions for aging: the case of cellular senescence. Drug Discov Today 22(5):786–795. https://doi.org/10.1016/j.drudis.2017.01.004
Zhang X, Zhang S, Liu X, Wang Y, Chang J, Zhang X, Mackintosh SG, Tackett AJ, He Y, Lv D, Laberge RM, Campisi J, Wang J, Zheng G, Zhou D (2018) Oxidation resistance 1 is a novel senolytic target. Aging Cell 17(4):e12780. https://doi.org/10.1111/acel.12780
He Y, Li W, Lv D, Zhang X, Zhang X, Ortiz YT, Budamagunta V, Campisi J, Zheng G, Zhou D (2020) Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell 19(3):e13117. https://doi.org/10.1111/acel.13117
Wakita M, Takahashi A, Sano O, Loo TM, Imai Y, Narukawa M, Iwata H, Matsudaira T, Kawamoto S, Ohtani N, Yoshimori T, Hara E (2020) A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun 11(1):1935. https://doi.org/10.1038/s41467-020-15719-6
Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, Kulick A, Houlihan S, Peerschke E, Friedman SL, Ponomarev V, Piersigilli A, Sadelain M, Lowe SW (2020) Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583(7814):127–132. https://doi.org/10.1038/s41586-020-2403-9
Guerrero A, Guiho R, Herranz N, Uren A, Withers DJ, Martinez-Barbera JP, Tietze LF, Gil J (2020) Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 19(4):e13133. https://doi.org/10.1111/acel.13133
Ozsvari B, Nuttall JR, Sotgia F, Lisanti MP (2018) Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging (Albany NY) 10(11):3294–3307. https://doi.org/10.18632/aging.101633
Laberge RM, Zhou L, Sarantos MR, Rodier F, Freund A, de Keizer PL, Liu S, Demaria M, Cong YS, Kapahi P, Desprez PY, Hughes RE, Campisi J (2012) Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 11(4):569–578. https://doi.org/10.1111/j.1474-9726.2012.00818.x
Perrott KM, Wiley CD, Desprez PY, Campisi J (2017) Apigenin suppresses the senescence-associated secretory phenotype and paracrine effects on breast cancer cells. Geroscience 39(2):161–173. https://doi.org/10.1007/s11357-017-9970-1
Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, Giorgadze N, Jensen MD, LeBrasseur NK, Kirkland JL (2015) JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A 112(46):E6301-6310. https://doi.org/10.1073/pnas.1515386112
Freund A, Patil CK, Campisi J (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30(8):1536–1548. https://doi.org/10.1038/emboj.2011.69
Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, Dharmalingam G, Faull P, Carroll T, Martinez-Barbera JP, Cutillas P, Reisinger F, Heikenwalder M, Miller RA, Withers D, Zender L, Thomas GJ, Gil J (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17(9):1205–1217. https://doi.org/10.1038/ncb3225
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460(7253):392–395. https://doi.org/10.1038/nature08221
Biermann M, Maueroder C, Brauner JM, Chaurio R, Janko C, Herrmann M, Munoz LE (2013) Surface code–biophysical signals for apoptotic cell clearance. Phys Biol 10(6):065007. https://doi.org/10.1088/1478-3975/10/6/065007
Jun JI, Kim KH, Lau LF (2015) The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat Commun 6:7386. https://doi.org/10.1038/ncomms8386
Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, Schadt EE, Quertermous T, Betancur P, Maegdefessel L, Matic LP, Hedin U, Weissman IL, Leeper NJ (2016) CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536(7614):86–90. https://doi.org/10.1038/nature18935
Overhoff MG, Garbe JC, Koh J, Stampfer MR, Beach DH, Bishop CL (2014) Cellular senescence mediated by p16INK4A-coupled miRNA pathways. Nucleic Acids Res 42(3):1606–1618. https://doi.org/10.1093/nar/gkt1096
Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J (2003) Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22(16):4212–4222. https://doi.org/10.1093/emboj/cdg417
Di Mitri D, Azevedo RI, Henson SM, Libri V, Riddell NE, Macaulay R, Kipling D, Soares MV, Battistini L, Akbar AN (2011) Reversible senescence in human CD4+CD45RA+CD27- memory T cells. J Immunol 187(5):2093–2100. https://doi.org/10.4049/jimmunol.1100978
Artero-Castro A, Kondoh H, Fernandez-Marcos PJ, Serrano M, Ramon y Cajal S, Lleonart ME, (2009) Rplp1 bypasses replicative senescence and contributes to transformation. Exp Cell Res 315(8):1372–1383. https://doi.org/10.1016/j.yexcr.2009.02.007
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436(7051):720–724. https://doi.org/10.1038/nature03890
Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, Kellogg TA, Khosla S, Koerber DM, Lagnado AB, Lawson DK, LeBrasseur NK, Lerman LO, McDonald KM, McKenzie TJ, Passos JF, Pignolo RJ, Pirtskhalava T, Saadiq IM, Schaefer KK, Textor SC, Victorelli SG, Volkman TL, Xue A, Wentworth MA, Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL (2019) Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47:446–456. https://doi.org/10.1016/j.ebiom.2019.08.069
Robles SJ, Adami GR (1998) Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 16(9):1113–1123. https://doi.org/10.1038/sj.onc.1201862
Galvan MD, Greenlee-Wacker MC, Bohlson SS (2012) C1q and phagocytosis: the perfect complement to a good meal. J Leukoc Biol 92(3):489–497. https://doi.org/10.1189/jlb.0212099
Brown GC, Vilalta A, Fricker M (2015) Phagoptosis—cell death by phagocytosis—plays central roles in physiology, host defense and pathology. Curr Mol Med 15(9):842–851. https://doi.org/10.2174/156652401509151105130628
Herranz N, Gil J (2018) Mechanisms and functions of cellular senescence. J Clin Invest 128(4):1238–1246. https://doi.org/10.1172/JCI95148
Calimport SRG, Bentley BL, Stewart CE, Pawelec G, Scuteri A, Vinciguerra M, Slack C, Chen D, Harries LW, Marchant G, Fleming GA, Conboy M, Antebi A, Small GW, Gil J, Lakatta EG, Richardson A, Rosen C, Nikolich K, Wyss-Coray T, Steinman L, Montine T, de Magalhaes JP, Campisi J, Church G (2019) To help aging populations, classify organismal senescence. Science 366(6465):576–578. https://doi.org/10.1126/science.aay7319
Nishio K, Inoue A, Qiao S, Kondo H, Mimura A (2001) Senescence and cytoskeleton: overproduction of vimentin induces senescent-like morphology in human fibroblasts. Histochem Cell Biol 116(4):321–327. https://doi.org/10.1007/s004180100325
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92(20):9363–9367. https://doi.org/10.1073/pnas.92.20.9363
Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M, Gorgoulis VG (2013) Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 5(1):37–50. https://doi.org/10.18632/aging.100527
Singh M, Piekorz RP (2013) Senescence-associated lysosomal alpha-L-fucosidase (SA-alpha-Fuc): a sensitive and more robust biomarker for cellular senescence beyond SA-beta-Gal. Cell Cycle 12(13):1996. https://doi.org/10.4161/cc.25318
Martens UM, Brass V, Engelhardt M, Glaser S, Waller CF, Lange W, Schmoor C, Poon SS, Landsdorp PM (2000) Measurement of telomere length in haematopoietic cells using in situ hybridization techniques. Biochem Soc Trans 28(2):245–250. https://doi.org/10.1042/bst0280245
Paluvai H, Di Giorgio E, Brancolini C (2020) The histone code of senescence. Cells. https://doi.org/10.3390/cells9020466
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113(6):703–716. https://doi.org/10.1016/s0092-8674(03)00401-x
Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppe JP, Campeau E, Beausejour CM, Kim SH, Davalos AR, Campisi J (2011) DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci 124(Pt 1):68–81. https://doi.org/10.1242/jcs.071340
Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD (2017) The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc 65(10):2297–2301. https://doi.org/10.1111/jgs.14969
Acknowledgements
We thank Prof Federica Marelli-Berg for the critical review of the manuscript. Biorender.com was used in the preparation of figures.
Funding
This work was funded by Medical Research Council (grant MR/K013068/1). CSADW is funded by a BBSRC LIDo studentship (BB/M009513/1).
Author information
Authors and Affiliations
Contributions
CSADW, EP and TMM collected and revised information; TMM wrote manuscript; CSADW, EP and MP revised manuscript.
Corresponding author
Ethics declarations
Conflict of interests
All authors declare no conflict of interests.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davan-Wetton, C.S.A., Pessolano, E., Perretti, M. et al. Senescence under appraisal: hopes and challenges revisited. Cell. Mol. Life Sci. 78, 3333–3354 (2021). https://doi.org/10.1007/s00018-020-03746-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03746-x