
Abstract
The results of different human epidemiological datasets provided the impetus to introduce the now commonly accepted theory coined as ‘developmental programming’, whereby the presence of a stressor during gestation predisposes the growing fetus to develop diseases, such as metabolic dysfunction in later postnatal life. However, in a clinical setting, human lifespan and inaccessibility to tissue for analysis are major limitations to study the molecular mechanisms governing developmental programming. Subsequently, studies using animal models have proved indispensable to the identification of key molecular pathways and epigenetic mechanisms that are dysregulated in metabolic organs of the fetus and adult programmed due to an adverse gestational environment. Rodents such as mice and rats are the most used experimental animals in the study of developmental programming. This review summarises the molecular pathways and epigenetic mechanisms influencing alterations in metabolic tissues of rodent offspring exposed to in utero stress and subsequently programmed for metabolic dysfunction. By comparing molecular mechanisms in a variety of rodent models of in utero stress, we hope to summarise common themes and pathways governing later metabolic dysfunction in the offspring whilst identifying reasons for incongruencies between models so to inform future work. With the continued use and refinement of such models of developmental programming, the scientific community may gain the knowledge required for the targeted treatment of metabolic diseases that have intrauterine origins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Developmental programming of metabolic disease
Metabolic syndrome was characterised by the World Health Organization (WHO) in 1998 as a disease diagnosed by the presence of insulin resistance in addition to two other criteria, namely obesity, hyperlipidemia, hypertension and microalbuminuria [1]. In modern societies, this non-communicable disease has risen in prevalence, with approximately one in four people being afflicted worldwide [2, 3]. Mutations in metabolic genes, as well as life-style factors such as calorie-dense diets and sedentary lifestyle are considered as main risk factors for developing the metabolic syndrome [4,5,6]. However, an often-overlooked risk factor for the development of metabolic syndrome, is the quality of one’s environment during gestation. Exposure to a suboptimal in utero environment due to a pregnancy complication or maternal nutrient deficit or surplus has been associated with the development of cardio-metabolic dysfunction in the offspring in later postnatal life [7,8,9,10]. This paradigm is known as the ‘Developmental origins of health and disease hypothesis’ and was first proposed by Barker and colleagues in 1986. Using birth and death records of men from Hertfordshire, England, Barker and others provided epidemiological evidence associating reduced birthweight, a surrogate for poor intrauterine nutrition and an increased rate of adult mortality from ischaemic heart disease [11]. These findings were also extended to demonstrate similar associations of poor intrauterine nutrition with adult rates of glucose intolerance and type 2 diabetes [12]. Studies of other human cohorts have since then also associated a variety of pregnancy complications such as maternal obesity, gestational diabetes and alcohol consumption to poor offspring metabolic health [13,14,15]. This association may be observed even when there is no birthweight change and may be exposed or exacerbated by unhealthy lifestyle choices, such as the consumption of a high calorie diet postnatally [16, 17].
The method by which early life exposures affect later metabolic health is thought to be mediated via epigenetic processes affecting the structure and function of offspring tissues. However, these processes are difficult to examine in a clinical setting and require the use of animal models. Animal models offer several advantages: gestational variables can be controlled or precisely altered, and the process of developmental programming studied in a comparatively shorter time than in humans. Indeed, numerous animal models of prenatal adversity have been developed in several species. Sheep, pigs, rabbits and guinea pigs are amendable to environmental manipulation and chronic instrumentation and previous reviews have highlighted their usefulness in studying developmental programming [18,19,20,21]. Mice and rats also offer advantages in the study of developmental programming due to the ease of maternal manipulation, availability of molecular tools (including gene manipulation) and short gestation and lifespan. However, rodents and humans vary in the temporal development of metabolic organs. For instance, white adipose tissue is present from birth in humans, but not rodents, even though brown adipose tissue is present in both. The development of the liver and the endocrine pancreas commences and completes earlier in gestation in humans than in mice [22,23,24,25]. Moreover, pancreatic beta cells are functional (i.e. exhibit glucose-stimulated insulin secretion) in humans from early gestation, whereas this only occurs in mice after birth [26,27,28]. However, many genes and signalling pathways governing the development of metabolic organs are shared between humans and mice. Moreover, several key maturational and metabolic events in fetal organs, such as a switch from a glycolytic to a gluconeogenic role in the liver occur around the time of birth in both species [24, 29]. In rodent metabolic organs that continue to develop and differentiate postnatally, environmental conditions during the lactational window can also contribute to the programming. Moreover, in the case of the white adipose tissue, which develops postnatally in rodents, alterations associated with gestational perturbations are likely secondary to programmed changes in the endocrine system occurring prenatally. The aim of this review is to summarise the findings from rodent models that show the involvement of changes in molecular pathways and the epigenetic mechanisms influencing these in offspring programmed for metabolic dysfunction as a result of an adverse in utero environment.
Rodent models of developmental programming
Several models of developmental programming have been developed in mice and rats. The impact of maternal nutrient imbalance (total caloric restriction, high-fat diet and protein restricted diet), inhalation hypoxia, uterine ligation, stress (psychological), glucocorticoid exposure (dexamethasone administration), diabetes (streptozotocin treatment) and alcohol intake on offspring metabolic physiology are shown in Table 1. Of the reviewed studies, the majority were undertaken on male offspring. However, when offspring of both sexes were studied, sexual disparities in metabolism and molecular pathways with maternal manipulations have been identified (Table 1). These models have clinical relevance as undernutrition, specific nutrient deficiencies, obesity and diabetes are seen in women in both developed and developing countries and are associated with an increase in the propensity of their offspring to develop metabolic disease in later life [30,31,32,33]. In addition, hypoxia is a main cause of fetal growth retardation in women living at a high altitude, as well as those at sea level with sleep apnoea or exposed to environmental pollution or tobacco smoke [34,35,36,37]. Reductions in utero-placental blood flow can be seen in human pregnancy complications and despite the warnings of health impacts on the unborn child, alcohol may still be consumed by women during pregnancy [38, 39]. Finally, endogenous glucocorticoids are increased by maternal stress and synthetic glucocorticoids are administered to women with asthma or at risk of preterm birth [40,41,42]. However, regardless of the type of maternal insult during pregnancy, changes in offspring metabolism are linked to alterations in structure and molecular profile of key metabolic organs, namely the pancreas, liver, skeletal muscle and white adipose tissue (Table 1). Moreover, it is important to note that other maternal manipulations, including exposure to endocrine disruptors (bisphenol A), arsenite, nitric oxide synthase inhibitors (NG-nitro-L-arginine methyl ester), lipopolysaccharide, caffeine, vitamin D and iron deficiency, microbiota alterations, disruptions in the circadian rhythm, cigarette smoke and air pollutants (diesel exhaust particles) have also been linked to metabolic aberrations in rodent offspring, but are reviewed elsewhere [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59].
Pancreatic insulin production
Offspring exposed to in utero stress frequently show an altered capacity to produce insulin. Hence, defects in the endocrine pancreas are thought to at least partially contribute to the programming of perturbed glucose handling in these offspring. For instance, rat offspring exposed to maternal undernutrition and protein restriction show decreased pancreatic insulin expression in line with their hypoinsulinaemic state [62, 66, 70, 90]. Moreover, offspring from maternal undernutrition and protein restriction show glucose intolerance at 3 weeks and 4 months, respectively [62, 90]. In pups from undernourished dams, the reduction in insulin expression can be seen as early as on day 0 (at birth) and may be due to enhanced pancreatic expression of ghrelin [66, 70], a known inhibitor insulin synthesis [155]. Conversely, adult offspring from mothers fed a high-fat diet have increased pancreatic insulin expression [103], in agreement with their hyperinsulinaemic profile [62, 156,157,158,159,160,161,162,163]. An upregulation of pancreatic insulin production may although be secondary to programmed changes in peripheral insulin sensitivity or hepatic insulin clearance in the offspring from high-fat diet-fed mothers ([164, 165] and described below).
In models of maternal nutrient perturbation, programmed changes in offspring insulin production may also be attributed to alterations in pancreatic islet mass. For instance, adult offspring exposed to a maternal high-fat diet in utero have an increase in pancreatic β cell mass in accordance with their elevated circulating insulin concentration [103]. Conversely, a reduction in β cell mass and proliferation has been observed in embryos and adult offspring exposed to protein restriction and undernutrition in utero [66, 91, 129, 166]. Decreased β cell mass is also observed in the offspring of mothers treated with dexamethasone or have diabetes (due to streptozotocin treatment) or uterine artery ligation during gestation [116, 129, 144]. In the case of maternal dexamethasone treatment, this is associated with diminished pancreatic insulin secretion [129]. Reductions in the expression of transcription factors important for β cell neogenesis, namely pancreatic and duodenal homeobox 1 (Pdx-1) and Maf bZip transcription factor A (Maf-a) have been observed in the fetus and adult offspring subjected to maternal protein restriction, undernutrition, uterine artery ligation and dexamethasone treatment [62, 66, 90, 91, 117, 119, 129, 166]. Similarly, other regulators of pancreatic islet cell development, namely Nk6 homeobox 1 (Nkx6-1), paired box 6 (Pax6) and neuronal differentiation 1 (Neuro d1) are downregulated in 7-day-old offspring exposed to maternal undernutrition or dexamethasone administration [62, 129]. Offspring of dams fed a high-fat diet also demonstrate changes in Pdx-1 expression, although this is sex-dependent; females showed increased and males showed decreased pancreatic Pdx-1, with a concomitant increase or decrease in pancreatic islet area, respectively [115]. A similar sexual disparity is also observed in the offspring of maternal protein restriction, with only males displaying a decrease in pancreatic islet mass [85]. The expression of factors involved in protein synthesis, cell senescence and cell cycle control have also been implicated in the programming of the offspring pancreas. For instance, a reduction in the mechanistic target of rapamycin (mTOR) pathway, which regulates protein synthesis, as well as markers of cellular senescence, namely cyclin dependent kinase inhibitor 1A (p21) and cyclin dependent kinase inhibitor 2A (p16), are upregulated in the pancreas of adult offspring exposed to maternal protein restriction during pregnancy [87, 91]. Moreover, in offspring from streptozotocin treated dams there is a reduction in the abundance of cell cycle regulators, namely cyclin dependent kinase 4 (Cdk4) and E2f1 in the pancreas [143]. In these offspring from diabetic dams, a reduction in the major subunit of the ATP-sensitive potassium channel, Kir6.2 is also observed alongside reductions in the expression of the ion channels ATP binding cassette subfamily C member 8 (Abcc8), calcium voltage-gated channel subunit alpha1 C (Cav1.2) and calcium voltage-gated channel subunit alpha1 E (Cav2.3) [143, 145].
Increased oxidative stress and reduced mitochondrial function are also implicated in the defective pancreatic formation and function in offspring programmed by an adverse gestational environment. For instance, the mitochondrial uncoupling protein, Ucp2 and cellular energy (ATP) content of islets are decreased in offspring exposed to maternal undernutrition or a high-fat diet during gestation [67]. Interestingly, the reduction in Ucp2 expression in response to maternal undernutrition is sex-dependent, with only males showing a downregulation in this gene. Despite this sexual-disparity in Ucp2 expression, the islets of both male and female offspring failed to increase ATP content in response to glucose [67]. Islet ATP content is also reduced in the pancreas of offspring from rat mothers with uterine artery ligation; however, this is accompanied by increased abundance of the mitochondrial antioxidant enzyme, manganese superoxide dismutase (MnSOD) [116]. An increase in cytokines, such as interleukin 1-1a (Il1-1a) and colony stimulating factor 3 (Csf3) is also seen in the pancreas of offspring from a maternal high-fat diet [105]. There are also programmed changes in the ability of the pancreas to metabolise glucose as a result of a maternal dietary insult during pregnancy however the nature of this effect depends on the insult and offspring sex. For instance, the expression of the glucose transporter, Glut2 is reduced in the islets of female offspring from mothers fed a high-fat diet, increased in female offspring from mothers fed a low-protein diet and elevated in male offspring from mothers undernourished during pregnancy [67, 85]. There are also alterations in the sensing and handling of glucose in the pancreas of offspring exposed to maternal undernutrition or alcohol intake in utero, as indicated by a reduction in glucagon and glucokinase (Gck) expression, respectively [62, 67]. Moreover, in islets of offspring exposed to a maternal high-fat diet, there are programmed changes in the abundance of insulin signalling pathway components, with reduced insulin receptor substrate 1(IRS1), phosphatidylinositol 3-kinase (PI3K) and phosphorylated protein kinase B (p-AKT) [103]. Thus, several molecular pathways involved in pancreatic development, insulin production and glucose handling are programmed by exposure to a suboptimal in utero environment, although the specific nature of effects can be partly dependent on offspring sex.
Hepatic insulin sensitivity
As a site for glucose uptake, storage and production, the liver plays a vital role in maintaining whole body glucose homeostasis. As such, a reduction in hepatic insulin sensitivity is associated with the development of the metabolic syndrome. Hepatic insulin sensitivity can be indicated by the levels of the insulin receptor, its downstream signalling proteins and genes governing glucose and lipid handling. In rodents, maternal protein restriction or high-fat diet during pregnancy are associated with reduced liver expression of insulin receptor (INSR), IRS1 and insulin like growth factor 1 receptor (IGF1R) in the fetus and adult offspring [96, 104, 109]. There are also reductions in the abundance of AKT proteins (total and activated/phosphorylated) in the liver of offspring exposed to hypoxia during gestation that are exposed by feeding a high-fat diet postnatally [122, 124]. The levels of the INSR and phosphorylated AKT are instead elevated in the liver of offspring exposed to maternal protein restriction [76, 80], even though these offspring are insulin resistant [80]. Together, these findings illustrate that there can be a disconnect between hepatic and body insulin sensitivity. Moreover, changes in the insulin signalling pathway in the liver may serve to compensate for programmed defects in offspring metabolism as a result of an adverse gestational environment.
Hepatic gluconeogenesis
Dysregulations in the pathways controlling hepatic gluconeogenesis are frequently implicated in the development of metabolic disease [167, 168]. For instance, there are alterations in the expression and activity of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), which are two key enzymes involved in gluconeogenesis [169]. Increased expression of PEPCK and G6Pase by the liver is generally observed in rodent offspring of mothers exposed to undernutrition, protein restriction, alcohol, psychological stress, dexamethasone or streptozotocin treatment [62, 77, 78, 88, 93, 126, 127, 131, 132, 137, 140, 149, 151, 152, 154]. In some cases, the nature of the effect is determined by offspring sex (Table 1). For instance, PEPCK expression and concomitant hyperglycaemia are increased in male, but not female offspring exposed to dexamethasone in utero [131]. Furthermore, there are sex-dependent changes in the expression of hepatic gluconeogenic genes including the transcription factor forkhead box o1 (Foxo1) in the offspring exposed to maternal high-fat diet, diabetes or alcohol [103, 140, 154]. In another study, female offspring exposed to alcohol prenatally showed increased hepatic gluconeogenesis in association with an elevated expression of genes including, phosphatase and tensin homolog (Pten) and telomere repeat binding factor 3 (Trb3) [150]. Conversely, male offspring from mothers exposed to inhalation hypoxia have decreased hepatic gluconeogenic capacity (as indicated by G6Pase expression), in line with their hypoglycemic state [123]. Hepatic glucokinase (Gck) expression is, however, decreased in offspring of both sexes exposed to maternal alcohol intake or protein restriction during gestation [77, 78, 151]. Together, these data indicate that the programmed changes in offspring hepatic gluconeogenesis are dependent on the type of maternal manipulation, as well as offspring sex.
In addition to alterations in gluconeogenesis, proteins involved in glucose uptake/transport, namely protein kinase C (PKC) and glucose transporter (GLUT) [170, 171], are frequently altered in the liver of offspring subjected to a suboptimal intrauterine environment. For instance, PKCζ is increased in the liver of rodent offspring exposed to maternal undernutrition or high-fat diet, but is unchanged or decreased in those from hypoxic dams [61, 109, 122]. Moreover, offspring from hypoxic dams and that are fed a high-fat diet from weaning show elevated hepatic PKCθ abundance in accordance with their greater sensitivity to develop metabolic syndrome [124]. Similarly, offspring exposed to maternal alcohol intake during gestation and then fed a high-fat diet after weaning, show upregulated hepatic Glut2 expression, with a corresponding increase in plasma glucose [154]. Overall, adverse in utero environments can program defects in liver glucose handling in the offspring with consequences for metabolic physiology.
Hepatic lipid metabolism
In offspring exposed to suboptimal maternal environments including undernutrition, protein restriction, high-fat diet, hypoxia and dexamethasone treatment, hepatic and circulating lipids, such as cholesterol are increased [61, 68, 69, 73, 74, 76, 86, 89, 99, 108, 109, 111, 122, 124]. Dysregulated hepatic lipid metabolism and an increase in circulating lipids are associated in the development of metabolic disease. As such, key transcription factors involved in hepatic lipid metabolism, namely sterol regulatory element-binding protein 1 (SREBP1) and the peroxisome proliferator-activated receptor (PPAR) ligands (PPARα, PPARγ and PPARδ), are thought to play a critical role. Indeed, mice genetically deficient in PPARγ develop insulin resistance [172] and PPAR agonists such as rosiglitazone and pioglitazone are widely used in the treatment of type 2 diabetes [173]. Neonates and adults from undernourished or dexamethasone-exposed dams show reduced hepatic PPARγ expression [73, 133]. There is also a reduction in hepatic PPARα in offspring exposed to maternal undernutrition [73]. However, offspring exposed to maternal low-protein or high-fat diets or psychological stress display increased expression of both Pparα and Pparγ [79, 92, 108, 138]. The liver X receptor alpha (LXR-α) is a transcription factor that increases lipogenic enzyme expression and reduces gluconeogenesis [174, 175]. In the liver of offspring exposed to maternal protein restriction, Lxr-α is decreased in association with increased G6Pase expression [88]. However, in offspring exposed to maternal undernutrition, hepatic Lxr-α is instead increased and is associated with enhanced expression of the lipogenic gene, Lpl [69]. There is also an increase in the expression of lipolytic genes, namely Srebp1c, fatty acid synthase (Fas), and hepatic lipase, in the liver of fetal and adult offspring exposed to maternal undernutrition or alcohol [73, 74, 154]. The increase in hepatic lipolysis is consistent with the elevated hepatic triglycerides and lipid content observed in these offspring [73, 74]. Conversely, a reduction in Srebp1c is observed in adult offspring exposed to a low-protein diet in utero regardless of sex, despite only male offspring displaying hyperinsulinemia [95]. In response to a maternal low-protein during gestation, there is a decrease in the expression of cytochrome P450 family 7 subfamily A (Cyp7a1), a gene involved in the metabolism of cholesterol, in the fetal liver [79, 86, 89]. Postnatally, hepatic Cyp7a1 expression continued to be altered; however, the nature of the change was dependent on whether the maternal protein restriction was or was not extended into the lactational period; Cyp7a1 expression was instead restored to normal or increased in the offspring of mothers fed a normal protein diet during lactation but remained decreased in offspring of mothers fed the low-protein diet in lactation [86]. Changes in hepatic cholesterol handling were likely responsible for the aberrant levels of cholesterol in the plasma and liver of these offspring, with most pronounced effects observed in those from mothers who were protein restricted both during gestation and lactation [86, 89]. Thus, a poor intrauterine environment can program defects in the lipid handling of the liver of offspring. Moreover, adverse environmental conditions that continue into the lactational period may propagate or amplify changes in offspring metabolic physiology that were programmed in utero.
Hepatic inflammation
Impairments in lipid handling can be associated with the activation of inflammatory pathways in the liver that also contribute to the pathogenesis of metabolic disease. In offspring exposed to maternal protein restriction, activation of c-jun N-terminal kinase (JNK), a protein involved in inflammatory signalling is increased in the liver and linked to a decreased expression of Cyp7a1 and hepatocyte nuclear factor 4α (HNF4α) [89]. An increase in JNK activation is also observed in the liver of offspring exposed to a maternal high-fat diet [106, 107]. Tumour necrosis factor alpha (TNFα) activates the phosphorylation of JNK [176, 177], and increased hepatic Tnfα is also seen in offspring exposed to maternal protein restriction [89]. This effect is observed as early as on embryonic day 20 in the fetus and maintained until adulthood [89]. Exposure to maternal dexamethasone treatment is also associated with activation of inflammatory pathways the offspring liver, with elevated expression of the inflammatory markers, Cd36 and Tnfα observed postnatally [65, 133]. Activation of JNK phosphorylation also occurs through glucocorticoid signalling and as such, changes in glucocorticoid production and activity have been implicated in the development of insulin resistance [178]. Thus, its perhaps not surprising that in studies of maternal protein restriction, stress and dexamethasone treatment, the expression of the glucocorticoid receptor (Gr) and the enzyme involved in converting inactive cortisone to cortisol (11βHsd1), are increased in the liver (as well as adipose and skeletal muscle) of adult offspring in association with both enhanced inflammatory state and impaired glucose tolerance [88, 92, 93, 130, 135, 138]. Moreover, 11βHsd1 is up-regulated in the adipose of male offspring in response to maternal dexamethasone treatment [134] and intrauterine glucocorticoid over-exposure due to the administration of carbenoxolone, an inhibitor of placental 11βHDS2 causes hyperglycaemia in male offspring [179, 180]. Thus, adverse gestational environments can program changes in glucocorticoid signalling and inflammatory pathways in the liver of the offspring and contribute to impaired glucose homeostasis.
Hepatic development
The upregulation of inflammatory markers in the offspring exposed to maternal high-fat diet or dexamethasone treatment is associated with the development of fatty liver (hepatic steatosis) in the offspring [99, 108, 133, 181]. The development of hepatic steatosis in offspring exposed to a maternal high-fat diet is also related to a reduction in the expression of antioxidant defence genes, such as Cu/Zn superoxide dismutase (Sod1) and glutathione peroxidase-1 (Gpx1) and an increase in genes involved in the cellular senescence pathway, such as p16 [99]. Due to a poor environment during gestation, there may also be greater sensitivity of the offspring to develop diet-induced hepatic steatosis postnatally. Indeed, offspring exposed to maternal alcohol intake during pregnancy have a greater propensity to develop liver steatosis with a high-fat diet after weaning [154]. There are also alterations in liver morphology in response to maternal hypoxia, undernutrition and high-fat diet, with a reduction in offspring liver size being observed [182,183,184]. A maternal high-fat diet or diabetes (via streptozotocin treatment) is additionally associated with increased expression of the cell cycle inhibitor, cyclin dependent kinase inhibitor 1A (Cdkn1), and a reduction in the developmental gene, Wnt1 in the liver of exposed offspring [100, 101, 140].
Insulin-like growth factor 1 (IGF1) is produced by the liver and acts in an autocrine/paracrine manner to promote offspring growth [185, 186]. In offspring from mothers with uterine artery ligation, poor fetal and postnatal growth are associated with the decreased hepatic expression and circulating levels of IGF1 [121]. Conversely, in offspring exposed to a protein restriction, high-fat diet or alcohol prenatally, hepatic IGF1 and IGF acid-liable subunit (ALS), as well as circulating IGF1 are increased and likely drive the postnatal catch up growth seen [68, 82, 154]. Exposure to maternal protein restriction, undernutrition, a high-fat diet or diabetes in utero, is also associated with programmed changes in the expression of the IGF binding protein, Igfbp1 in the offspring liver; however, the specific nature of the impact depends on the maternal insult and study age [68, 82, 140]. The IGF1 axis is also implicated in the regulation of insulin sensitivity and fat and carbohydrate metabolism [187, 188]. Thus, programmed changes in the IGF1 system as a result of poor intrauterine environment could contribute to growth and metabolic health outcomes in the offspring.
Adipose lipid metabolism and morphology
Alterations in adipocyte differentiation and lipogenesis are associated with the development of metabolic disease and changes in PPAR expression are implicated in this pathogenic process [62, 189]. Reduced PPARγ expression is observed in the adipose tissue of rodent offspring as a result of maternal undernutrition and a high-fat diet during pregnancy [71, 112, 113]. The observed reduction in PPARγ expression is dependent on sex, with only male offspring from a maternal high-fat diet being affected [112]. However, PPARγ and PPARα expression are upregulated in the adipose tissue of offspring in other studies of maternal undernutrition and high-fat diet, as well as in response to protein restriction or dexamethasone treatment during gestation [62, 114, 134]. Moreover, the expression of PPAR target genes, namely Srebp1, Fas, Lpl, hepatic lipase and stearoyl-CoA desaturase-1 (Scd1) are upregulated in the adipose of offspring exposed to maternal undernutrition, a high-fat diet or stress [84, 112,113,114, 136]. Variations in offspring adipose Ppar expression between studies may relate to the timing and specific nutritional manipulation utilised, the fat depot studied, the age and sex of the offspring, whether birth weight was reduced and/or followed by catch-up growth, as well as whether obesity and insulin resistance also ensued in the model. Nonetheless, changes in offspring adipose Ppar expression were associated with an increase adipocyte size [63, 71] and body adiposity in offspring exposed to maternal undernutrition, protein restriction or high-fat diet during gestation [71, 84, 106, 107, 112].
Through the interaction of PPARγ with CCAAT/enhancer binding proteins (C/EBPβ/ and C/EBPα), adipocyte differentiation occurs [190]. Consistent with an upregulation of PPAR expression and adiposity, C/Ebpα expression is increased in the adipose of offspring born from mothers fed high-fat diet during pregnancy [112, 114]. However, there is also an augmented expression of zinc finger protein 423 (Zfp423), a transcription factor crucial in adipogenic lineage commitment in the adipose of fetuses derived from obese mothers fed a high-fat diet from prior to and during pregnancy [102]. Exposure to maternal undernutrition or dexamethasone treatment in utero is related to upregulated expression of the preadipocyte marker, Dlk1, in the white adipose tissue of offspring postnatally [62]. Collectively, these studies indicate that an insult during in utero development can advance white adipocyte differentiation in the offspring. Maternal undernutrition during pregnancy is also associated with the browning of white adipose tissue in offspring, as indicated by an increase in mitochondrial uncoupling protein 1 (Ucp1) and neuropeptide Y (Npy) expression [75]. Together, these data suggest that molecular pathways governing adipocyte differentiation and overall adipose morphology can be dysregulated in offspring exposed to in utero stressors.
Adipokines such as leptin and adiponectin are implicated in the control of lipid storage, as well as insulin sensitivity. Importantly, adipokines are shown to be altered in the adipose of offspring as result of an adverse environment in utero. For instance, offspring from mothers fed a high-fat diet show increased leptin and reduced adiponectin (Adipoq) expression in their white adipose tissue [110, 112]. Moreover, the expression of another adipokine, apelin, is increased in the white adipose of offspring exposed to undernutrition in utero and exacerbated by a postnatal high-fat diet [64]. Conversely, maternal undernutrition, alcohol exposure or dexamethasone treatment during gestation is associated with decreased leptin and increased Adipoq expression in the white adipose tissue of offspring [62, 75, 146]. Adipokines are also known to induce inflammation and contribute to the pathogenesis of metabolic disease [191]. Indeed, offspring from high-fat diet-fed or diabetic mothers show enhanced expression of inflammatory markers, like JNK and Tnfα in their white adipose tissue [106, 107, 144]. Thus, adipocyte development, adipokine production and inflammatory state are programmed by, and likely contribute to, the development of insulin resistance frequently observed in offspring subjected to a suboptimal intrauterine environment.
Adipose and skeletal muscle insulin resistance
In addition to the liver, the adipose tissue and skeletal muscle respond to insulin and play a key role in maintaining glucose homeostasis in the body. Moreover, there are alterations in the insulin sensitivity of these tissues in the offspring as a result of maternal manipulation during gestation. For instance, maternal protein restriction during gestation is associated with increased InsRβ in the white adipose of adult offspring and increased glucose tolerance [83, 109]. Conversely, exposure to a maternal high-fat diet is associated with increased INSRβ and p85 in the skeletal muscle and unaltered insulin sensitivity in adult offspring [109]. Moreover, INSR levels are decreased in the muscle, liver and adipose tissue in offspring exposed to maternal dexamethasone, ethanol exposure, diabetes or a high-fat diet in utero, although glucose tolerance was not altered in some instances [63, 109, 120, 128, 139, 148]. Generally, a decrease in total and activated AKT are observed in the adipose and muscle of offspring exposed to maternal hypoxia, high-fat diet or undernutrition during pregnancy [71, 98, 122, 125]. Maternal alcohol intake during gestation is also linked to altered levels of both total and activated AKT in the adipose tissue of offspring; however, the specific effect is dependent on the offspring sex and resulting metabolic phenotype [151, 153]. As evident from the aforementioned studies, different imposed maternal manipulations can program alterations in skeletal muscle and white adipose tissue insulin signalling in both male and female offspring. However, how these molecular alterations relate to changes in glucose and insulin dynamics in the offspring in vivo, is less clear.
Adipose and skeletal muscle glucose uptake
The skeletal muscle and adipose tissue are key metabolic organs involved in the uptake and storage of glucose through the action of insulin [171]. In offspring exposed to maternal undernutrition, protein restriction or alcohol during gestation, the abundance of PKCζ is decreased in the muscle and adipose [63, 80, 147]. Moreover, the mRNA expression, abundance and/or translocation of the insulin-sensitive glucose transporter, GLUT4, is reduced in the skeletal muscle and white adipose tissue of offspring exposed to undernutrition, hypoxia or alcohol in utero [60, 71, 122, 146, 147]. In the case of maternal undernutrition, this effect may be influenced by offspring sex [72]. Indeed, Glut4 expression is specifically decreased in the muscle of female rats exposed to a maternal undernutrition or low-protein diet during gestation [60, 94]. However, GLUT4 abundance is decreased in the muscle of male rats from diabetic dams but increased in the adipose tissue [139, 141, 142]. The abundance of the insulin-independent glucose transporter, GLUT1 is increased in the skeletal muscle of offspring in response to maternal undernutrition or uterine ligation [60, 118]. Thus, it is evident that poor maternal environments during gestation program perturbations in the uptake of glucose by skeletal muscle and adipose tissue in the offspring; however, these effects may also be dependent on offspring sex.
In addition to glucose uptake, there are programmed changes in the mitochondrial capacity in the skeletal muscle and white adipose tissue of the offspring in response to a suboptimal intrauterine environment. For example, a maternal low-protein diet during pregnancy is associated with reduced sirtuin protein 3 (Sirt3) expression and mitochondrial respiration in the skeletal muscle of offspring [81]. There is also an increase in Ucp3 expression, a greater density of type II muscle fibres and a reduction in the rate of fatty acid oxidation in the soleus muscle of offspring exposed to a maternal low-protein diet [81, 97]. Expression of mitochondrial electron transfer complexes are reduced in the muscle of offspring from high-fat diet-fed dams [98]. Moreover, Ucp1 is increased and mitochondrial biogenesis reduced (indicated by the lower expression of PPARG coactivator 1 alpha, Pgc1a) in the white adipose tissue of offspring from dams undernourished during gestation [71, 75]. A reduction in Pgc1α is also observed in the liver of female offspring from mothers exposed to stress [135]. Thus, poor maternal nutritional intake or stress during pregnancy can lead to programmed changes in the mitochondrial capacity of metabolic tissues in the offspring. Such changes in mitochondrial capacity would be expected to mediate alterations in metabolic fitness of the offspring.
The role of epigenetic mechanisms in the programming of the fetus
The term epigenetics, meaning “above the gene”, is used to describe phenotypic modification as a result of changes in gene expression without an alteration to the DNA sequence [192]. In the context of the developmental programming field, epigenetic alterations serve as a useful mechanism for the fetus to adapt to a changing environment in utero. However, epigenetic alterations occurring during gestation may be detrimental after birth where the postnatal environment, including nutrient availability, may be different to that prenatally. The most described epigenetic alterations to gene expression include DNA methylation, posttranslational histone modification and small noncoding RNA. The involvement of changes in DNA methylation, histone modifications and small noncoding RNAs in mediating changes in the function of metabolic tissues in offspring programmed as a result of environmental manipulation during gestation are summarised in Tables 2, 3 and 4, respectively. However, it is important to note that the three types of epigenetic alterations are not distinct from one another. Instead, they often occur simultaneously and influence one another in the developmental programming of offspring metabolic phenotype.
Alterations in DNA methylation in response to in utero stressors
DNA methylation occurs when methyl groups are added to DNA, usually on the 5′-position of cytosine residues of a CpG island. The resulting heterochromatin condensation from DNA methylation is thought to be associated with a decrease in gene transcription [205]. However, DNA methylation is more complex than this, with increased methylation also associated with increased gene expression [206]. Alterations in DNA methylation are catalysed by the DNA methyltransferase enzymes (DNMTs), of which there are three types, DNMT1, DNMT3A and DNMT3B [207, 208]. The most studied epigenetic alteration in the developmental programming field is the methylation level of promoters of genes involved in lipid and glucose handling (Table 2).
Alterations in the methylation status of genes involved in hepatic development have often been reported in offspring exposed to nutrient perturbations during gestation. For instance, neonates from high-fat diet-fed mothers who subsequently showed reduced hepatic size and growth, displayed hypomethylation and increased expression of Cdkn1a in their liver [100]. Moreover, offspring from dams fed a low-protein diet during gestation showed increased methylation of the Igf2/H19 imprinting control region (ICR) in line with enhanced Igf2 expression in the liver. This was related to increased H19 and Igf2 expression in these day 0 male offspring [196]. Another study which showed a similar alteration in the Igf2/H19 ICR upon maternal low-protein diet also illustrated insulin resistance and increased adipose expansion in offspring, implicating modifications in Igf2/H19 methylation in adipose growth [193]. Moreover, alterations in the methylation of metabolic genes, including those for the insulin signalling pathway, such as PI3Ks, occur in the liver of offspring exposed to a maternal high-fat diet [202]. Offspring of mothers exposed to a low-protein diet during gestation show decreased methylation, associated with lower Dnmt1 expression and increased hepatic Gr expression [93]. This reduction in Gr promoter methylation can also be observed in the grand-offspring (F2 generation) [209]. A maternal low-protein diet during pregnancy is also associated with the hypermethylation of the promoter for the Lxr-α gene, which encodes a nuclear receptor involved in fatty acid and cholesterol metabolism in the fetal liver [197]. Moreover, hypoxia during pregnancy causes modifications of histone H3, which surrounds the G6Pase gene promoter and is associated with a reduction of G6Pase levels in offspring with metabolic consequences, such as decreased circulating glucose [123]. In addition to alterations in glucose metabolism, fetal nutrient and oxygen deprivation achieved through maternal uterine artery ligation also has an impact on the pancreatic islets of offspring, with altered DNA methylation being linked to changes in the expression of genes involved in insulin secretion, islet proliferation and reduced islet mass [203, 204]. In the pancreas of offspring from diabetic mothers a reduction in the ion channels Abcc8, Cav1.2 and Cav1.3 is associated with the hypomethylation of these genes [145].
Alterations in DNA methylation also dictate the expression of genes involved in lipid metabolism in offspring born to mothers with suboptimal nutrition during pregnancy. A high-fat diet in utero causes epigenetic alterations in offspring adipocytokine genes in the visceral white adipose depot, with the hypermethylation of adiponectin and leptin receptor and hypomethylation of leptin observed. Such alterations in methylation of adiponectin and leptin receptors are associated with increased plasma triglycerides and cholesterol [110]. Conversely, exposure to a maternal low-protein diet in utero is associated with a decrease in methylation of the leptin gene promoter in adipose tissue [198]. Gestational hypoxia also causes hypermethylation of the adiponectin gene promoter with concomitant decrease in adiponectin levels in male offspring, who are more susceptible to metabolic syndrome in adulthood, as indicated by hyperleptinaemia and decreased insulin sensitivity. It additionally alters the methylation patterns of genes involved in metabolic regulation, including methionine degradation and cysteine biosynthesis in white adipose tissue of offspring exposed to maternal hypoxia in utero [125, 210]. An increase in the expression of the inflammatory marker, Tnfα is associated with a reduction in the methylation of its promoter in offspring from dams with diabetes [144]. Altered Pgc1α expression in the skeletal muscle of offspring exposed to protein restriction in utero is associated with the hypermethylation of its promoter [194]. Moreover, in offspring exposed to a maternal high-fat diet, hypermethylation of the Pparγ promoter or Pparγ gene is observed in the liver and adipose, respectively [113, 199]. In contrast, a reduction in Pparα promoter methylation is seen in the liver of offspring exposed to a maternal low-protein diet during gestation [195]. In offspring exposed to a maternal protein restriction, the alterations in PGC1α and PPARγ methylation are associated with reduced glucose tolerance and increased circulating triacylglycerols, respectively [194].
The expression of key genes involved in adipocyte differentiation are also impacted by changes in methylation caused by poor maternal environments. For instance, methylation is reduced in association with increased expression of Zfp423 in the fetus by a maternal obesogenic diet [102]. As mentioned previously, Zfp423 is involved in adipogenic lineage commitment and changes to its methylation and expression by a maternal obesogenic diet were coupled to advanced adipogenesis in the offspring at weaning and impaired expansion capacity with the introduction of a high-fat diet postnatally [200]. A prenatal low-protein diet is also associated with altered adipocyte differentiation; however, this was associated with increased methylation and expression of the Igf2 gene in the offspring adipose tissue, as well as elevated adiposity [193]. There was no interactive effect on adiposity, and Igf2 gene methylation and expression in the offspring exposed to a maternal low-protein diet, by superimposing a high-fat diet postnatally [193].
Identifying the role of changes in genome methylation in animal models of offspring metabolic programming is critical in understanding how to reverse or prevent such programming from occurring. The provision of a methyl donor supplement to the mothers fed a high-fat diet prevents the changes in offspring adipose tissue DNA methylation and expression of genes, including Fas, leptin and Pparγ [201]. The supplement also mitigates the negative metabolic outcomes such as insulin resistance that are induced in the offspring by maternal high-fat diet [201]. Moreover, methyl donors also normalise the methylation level of the leptin gene promoter and reduce offspring leptin concentrations as a result of maternal protein restriction [211]. Changes in the expression and methylation patterns of genes involved in fatty acid metabolism upon methyl-donor group supplementation further demonstrate the importance of methylation as an epigenetic regulator of offspring programming [201, 211]. Maternal intake of folate, a vitamin which is a methyl group donor, has also been studied for its ability to restore normal levels of DNA methylation in offspring tissues that arise as a result of developmental programming. For instance, in a model of maternal protein restriction, folate supplementation largely ameliorates the alterations in genome-wide methylation [212], methylation of the H19/Igf2 ICR [196] and Pparα and Gr gene methylation and expression in the offspring [92]. These findings suggest that maternal folate supplementation during pregnancy may have benefit in mitigating negative epigenetic modifications mediating programming effects in offspring tissues as a result of in utero insults.
Alterations in histone modifications in response to in utero stressors
In the nucleus of eukaryotic cells, DNA is packaged into nucleosomes by wrapping around an octameric histone complex comprised of two copies of H2A, H2B, H3 and H4 histones [222]. Acetylation, methylation, phosphorylation, ubiquitination, sumoylation, glycosylation and ADP-ribosylation occur at the amino termini of histones. Such post-translational modifications alter chromatin structure and the accessibility of transcription factors and DNA binding elements, thus regulating gene expression [223, 224]. The two most common post-translational modifications are acetylation and methylation, whereby acetylation of lysine within the H3 histone activates transcription through inhibition of the secondary and tertiary nucleosome structure and thus, results in chromatin decondensation [225]. Conversely, the methylation of arginine and lysine residues cause chromatin condensation and transcriptional repression [226]. Moreover, post-translational modifications include the addition of marks which activate transcription (H3K9ac, H3K4me3) or marks which repress transcription (H3K27me3, H3K9me2) [227].
Histone alterations appear to mediate the effects of suboptimal gestational environments on offspring pancreatic development and function (Table 3). In offspring exposed to reduced uteroplacental blood flow during gestation, the reduced islet Pdx-1 expression is associated with a decrease in acetylation of H3 and H4 histones in the fetus and altered methylation of the Pdx-1proximal promoter in adult diabetic offspring [117]. These epigenetic modifications in the exposed offspring could be reversed by using a glucagon-like peptide-1 analogue, exendin-4 (Ex-4), which restored histone acetylation and normalised DNA methylation in the islets at the corresponding ages [221]. A maternal low-protein diet also impacts β cell function in adult offspring through changes in histone modifications. In particular, the decreased expression of the transcription factor, hepatocyte nuclear factor 4-α (Hnf4α) in offspring pancreatic islets, is associated with decreased active and enriched repressive histone marks at regulatory regions of the gene [213].
Histone alterations also play a critical role in the programming of offspring insulin-regulated glucose uptake and metabolism in skeletal muscle. For instance, the alterations in Glut4 expression in the muscle seen specifically in female rats exposed to a maternal low-protein diet or undernutrition during gestation is related to changes in histone acetylation and methylation at the Glut4 promoter [72, 94]. Moreover, in the muscle of female and not male offspring, exposure to a maternal low-protein diet during gestation is associated with increased H3 and H4 histone acetylation, as well as expression of the C/Ebpβ, a transcription factor involved in the control of skeletal muscle carbohydrate and amino acid metabolism [215]. In female offspring exposed to maternal protein restriction, the alterations in histone modifications at the Glut4 and C/Ebpβ promoters is coupled to increased glycogen content of the skeletal muscle [94]. Together, these studies indicate that changes in histone modifications underlie sex-related differences in the programming of offspring skeletal muscle in response to maternal malnutrition.
Alterations in the expression of genes involved in hepatic development and gluconeogenesis in offspring programmed by a poor intrauterine environment are also mediated by histone modifications. For instance, in offspring of dams with maternal uterine artery ligation, reduced hepatic Igf1 expression is related to alterations in the H3 histone methylation in both sexes and disrupted H3 acetylation in females specifically [121]. Moreover, the decreased hepatic Wnt1 expression in pups from dams fed a high-fat diet is linked to altered acetylation of the H4 histone at the Wnt1 promoter [101]. The liver of pups born from high-fat diet-fed dams also have alterations in histone modifications and these are associated with increased expression of the gluconeogenic genes, Pck1 [216] and Pepck [217]; however, the latter effect is only observed in female offspring [217]. In female offspring exposed to alcohol prenatally, enhanced expression of the Pten and Trb3 by the liver is associated with a reduced acetylation [150]. Furthermore, in offspring from dams fed a low-protein diet, reduced hepatic Cyp7a1 expression is associated with repressive histone modifications of its promoter and an increase in circulating cholesterol in these offspring [86].
Alterations in histone modifications influence fatty acid metabolism gene expression in the adipose tissue of offspring born to dams with nutritional alterations during gestation. As previously mentioned, an in utero high-fat diet programs alterations in adiponectin, leptin and Pparγ expression in the adipose tissue of offspring. Decreased adiponectin and increased leptin expression in the offspring adipose tissue is associated with altered levels of acetyl H3K9 and dimethyl H3K9 at their gene promoters [220]. A reduction in Pparγ2 gene expression in the adipose of offspring born to obese dams is also associated with histone modifications; with adult offspring displaying a reduction in the active mark, H3AC, at the Pparγ2 promoter [113]. Histone modifications also occur in the adipose tissue of offspring born to dams fed a low-protein diet and given a high-fat diet postnatally. In particular, the reduction in expression of the beige adipocyte marker fibroblast growth factor 21 (Fgf21) in these offspring is associated with an increase in histone methyltransferase G9a [214, 219, 220]. Thus, histone modifications are implicated in the programming of offspring adipose tissue function, lipid metabolism and phenotype.
The reversal or prevention of changes in histone modifications in offspring tissues as a consequence of developmental programming has been achieved through maternally administered interventions. For instance, preconception weight loss in obese mice prevents the changes in lysine acetyltransferase expression and associated reduction in glucokinase expression in the fetal liver [218]. In addition, administration of constitutive androstane receptor (CAR) ligand (normally used to improve insulin sensitivity in the context of obesity) to dams fed a high-fat diet during gestation ameliorates the changes in acetyl H3K9 and dimethyl H3K9 that are associated with the adiponectin and leptin gene promoters and normalises circulating concentrations in the offspring [219]. These data suggest that in a clinical setting, maternal weight loss and ligands like CAR may offer additional benefit, by mitigating the epigenetic modifications and consequent negative metabolic outcomes in the offspring.
Alterations in small noncoding RNA in response to in utero stressors
Non-coding RNAs are major regulators of gene expression [240,241,242]. In general, non-coding RNAs have been categorised based on their size and include miRNAs (~ 22 nt), piRNAs (24–30 nt), snoRNAs (60–300 nt) and long non-coding RNA (lncRNAs) (> 200 nt) [243]. miRNAs play a critical role in the epigenetic control of gene expression through regulating DNA methylation and histone modification [244]. Moreover, lncRNAs are involved in genomic imprinting and DNA methylation. For instance, methylation of the paternally inherited allele of the H19 lncRNA supresses its transcription [245]. Furthermore, lncRNAs affect the function of miRNAs and vice versa. For example, H19 contains binding sites for the let-7 family of miRNAs and influences their availability [246]. In addition to regulating the expression of genes governing biological processes, non-coding RNAs have been implicated in the development of metabolic disease [247, 248]. However, the role of non-coding RNAs in developmental programming of metabolic disease has been comparatively less studied, with most studies to date looking at the role of miRNAs.
miRNAs are critical in the programming of insulin production in offspring exposed to in utero stress (Table 4). Alterations in the levels of the pancreatic islet-specific miRNA-375, which regulates genes involved in pancreatic islet expansion, occur in response to insulin resistance and contribute to the development of type 2 diabetes in vivo [248,249,250,251]. The abundance of miRNA-375 is higher in the islets of rat foetuses and adult offspring exposed to a maternal low-protein diet in utero [229]. Elevated miRNA-375 levels are associated with impaired islet Pdx-1 expression, proliferation and insulin secretion and the development of diabetes in these offspring [229]. Moreover, normalizing miR-375 levels in the islets derived from adult offspring of protein-restricted dams restores their capacity to secrete insulin and proliferate in vitro [229]. Changes in miRNA expression are also implicated in the development of peripheral insulin resistance in offspring exposed to an obesogenic diet in utero. In particular, the decrease in the insulin signalling protein, IRS1, in the adipose tissue of offspring exposed to a maternal high-fat diet is linked to an upregulation of miR-126 [233].
Pathological processes relating to metabolic dysfunction such as altered lipid storage and subsequent lipotoxicity and insulin resistance are also regulated by miRNAs in the adipose tissue. For instance, an upregulation of miR-483-3p is observed in the white adipose of adult rats exposed to a maternal low-protein diet [230]. In vitro, miR-483-3p inhibits adipocyte differentiation and lipid accumulation through reducing growth differentiation factor-3 (Gdf3) expression, and in turn, Fabp4 and Pparγ [230]. Defects in the formation of brown adipose tissue in the fetus may also contribute to the later development of poor lipid handling and genesis of metabolic syndrome as a result of poor conditions during in utero development. For instance, defects in brown tissue formation, mitochondriogenesis and mitochondrial capacity in fetuses from obese mouse mothers is associated with an upregulation of miR-204-5p, which negatively regulates Pgc1α and Sirt1 expression [239]. Thus, miRNAs are implicated in key metabolic alterations to offspring adipose tissue, namely reduced browning and increased lipid accumulation.
Alterations in lipid metabolism in the liver of offspring exposed to poor gestational conditions may also be partly induced by alterations in miRNA levels. In the liver of adult offspring of mothers given a high-fat diet or exposed to caloric restriction during gestation, miR-122 levels are decreased in association with increased expression of genes involved in lipid synthesis (e.g, Pparα, Pparβ, Scd1) and fatty acid oxidation (e.g, Pgc1α and Cpt-1α) and an associated reduction in hepatic triglycerides [228, 234,235,236]. In offspring from mothers fed a high-fat diet during gestation, the reduction in hepatic miR-122 expression is not prevented by cross-fostering to mothers fed a control diet during lactation [237]. In offspring of mothers given a low-protein or high-calorie diet during gestation, the hepatic inflammation observed appears to be mediated by dysregulated expression of several miRNAs that contribute to the increased expression of Tnfα and Il-6 genes [232, 238]. Together, these studies highlight that miRNAs may serve as valuable therapeutic targets to prevent the developmental programming of metabolic syndrome in offspring exposed to adverse gestational environments.
Whilst the role of other of non-coding RNAs species in the programming of offspring metabolic disease has been less studied, lncRNAs have been shown to regulate several metabolic pathways including gluconeogenesis and fatty acid metabolism [252, 253]. Moreover, in offspring exposed to a low-protein diet in utero, defective β cell expansion is associated with the decreased expression of the lncRNA, H19 [231]. In part, the association between H19 expression and β cell expansion is thought to be related to loss of H19-mediated inhibition of Let-7, a negative regulator of PI3K-AKT signalling [231]. Together, these results indicate the importance of lncRNA in influencing metabolic processes in the programming of offspring phenotype.
Conclusions and future directions
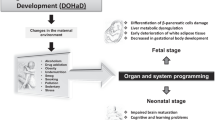
This review focused on the contribution of suboptimal maternal environments (in utero stress) to the developmental programming of offspring metabolic health via changes in key metabolic tissues in the first generation. Data from rodent models show that in utero stress can lead to epigenetic changes in key metabolic organs, which impact growth, morphology, insulin sensitivity, nutrient handing, metabolism and inflammation with consequences for the metabolic physiology of the offspring (Fig. 1). However, the placenta is the interface between the mother and fetus and mediates maternal insults imposed through the regulation of fetal nutrient provision. Thus, although not addressed in the current review, the placenta will have contributed to the programming of offspring metabolic disease as a result of in utero stress (reviewed in [254,255,256]). Moreover, half of the placental and fetal genome derives from the father. A significant paternal contribution to the developmental programming of offspring metabolism has recently come to light, with paternal exposure to alcohol or a high-fat or low-protein diet also programming adult offspring metabolism [257,258,259]. The mechanisms by which this occurs have been reviewed elsewhere [260,261,262,263,264] and above all highlight the need to also consider paternal environmental exposures when elucidating the mechanisms underlying the programming of metabolic disturbances in the offspring. Furthermore, the transmission of metabolic dysfunctions has been observed across subsequent generations even in the absence of further insults and normal environmental conditions via epigenetic processes (discussed previously; [265,266,267]). Moreover, in utero stressors can also program defects in the function in other systems that may interact with the metabolic system of the offspring, including the reproductive, neuroendocrinological, microbiome and vasculature systems [268,269,270,271,272,273,274]. To date, the contribution of alterations in the methylome of fetal and adult tissues to the programming of offspring metabolic phenotype has been most widely studied although other epigenetic factors, such as histone modifications and non-coding RNA are also emerging as key players in this process and require further investigation. However, other mediators such as modification to mitochondrial DNA (which are normally inherited from the mother), de novo point mutations in the germline or somatic cells and vertical transmission of microorganisms to offspring can contribute to the developmental programming of metabolic syndrome postnatally [116, 275,276,277].

The effect of in utero stressors on epigenetic, gene expression and structural alterations in metabolic organs of offspring. Δ, change in; HFD, high-fat diet
Undeniably, studies in rodent models have been useful in showing causation between specific gestational insults and offspring outcomes by selectively manipulating a variable in a controlled way. Indeed, the studies in this review illustrate that in many instances a variety of maternal manipulations imposed on rodents act to perturb the same metabolic pathways, as postulated by the gatekeeping hypothesis [278]. However, it is important to note that in some instances, there are inconsistencies in the changes observed for molecular pathways in the same, let alone different rodent model of in utero stress, making it challenging to draw firm conclusions on the commonality of pathways involved. Moreover, studies have typically been limited by not considering offspring sex or only studying the outcomes of in utero stress on the male offspring (due to challenges faced by the need to control for the influence of the oestrus cycle on metabolic function in females). Indeed, in the few studies that have studied both female and male offspring, sexually-dimorphic metabolic outcomes of developmental programming have frequently been observed and may be attributed to a variety of factors, including the effect of sex chromosome-linked genes in the placenta, as well as sex differences in the temporal development of metabolic organs and fetal and adult levels of hormones like corticosterone/cortisol and sex steroids [279,280,281,282,283]. Thus, to further address the molecular mechanisms underlying the developmental programming of offspring metabolic health, we recommend that both male and female offspring are studied in future work. Differences in strain, husbandry, control diet composition, severity and duration of the insult, cross fostering techniques and standardisation of litter size postnatally, are also parameters that have been shown to influence metabolic health outcomes [284,285,286,287,288]. For instance, methods to impose “maternal stress” in rodent models of programming range from saline injection, to swimming and physical restraint, which are likely to have differing impacts on raising endogenous corticosterone/cortisol in the mother. Whilst in most studies, the maternal insult is confined to the gestational period, in some instances, insults are continued into the lactational period. As such it is difficult to discriminate whether the programming of offspring metabolic physiology is occurring during gestation or lactation and studies should be designed to more directly address this in future work. Furthermore, future studies should be aimed at more accurately depicting clinical settings, where changes in prenatal environment can be due to a variety of causes, operating at different times in gestation, which may together impact and drive resulting offspring metabolic outcomes. Moreover, future research may also benefit from developing diagnostic criteria to grade metabolic health at certain ages and stages of the disease process in rodent species, as is done in a clinical setting. However, taken together, rodent models have undoubtedly provided significant insight into understanding the ontogeny and molecular mechanisms by which offspring metabolic health may be programmed by exposure to in utero stress. Further, with their continued use, rodent models may provide the knowledge needed to improve or prevent the propagation of metabolic diseases that are reaching epidemic proportions in many countries across the world.
References
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15(7):539–553. 10.1002/(sici)1096-9136(199807)15:7<539::Aid-dia668>3.0.Co;2-s
Saklayen MG (2018) The global epidemic of the metabolic syndrome. Curr Hypertens Rep 20(2):12. https://doi.org/10.1007/s11906-018-0812-z
O'Neill S, O'Driscoll L (2015) Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev 16(1):1–12. https://doi.org/10.1111/obr.12229
Kolb H, Martin S (2017) Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med 15(1):131. https://doi.org/10.1186/s12916-017-0901-x
Povel CM, Boer JM, Reiling E, Feskens EJ (2011) Genetic variants and the metabolic syndrome: a systematic review. Obes Rev 12(11):952–967. https://doi.org/10.1111/j.1467-789X.2011.00907.x
Pollex RL, Hegele RA (2006) Genetic determinants of the metabolic syndrome. Nat Clin Pract Cardiovasc Med 3(9):482–489. https://doi.org/10.1038/ncpcardio0638
Barouki R, Gluckman PD, Grandjean P, Hanson M, Heindel JJ (2012) Developmental origins of non-communicable disease: implications for research and public health. Environ Health 11:42. https://doi.org/10.1186/1476-069x-11-42
Hoffman DJ, Reynolds RM, Hardy DB (2017) Developmental origins of health and disease: current knowledge and potential mechanisms. Nutr Rev 75(12):951–970. https://doi.org/10.1093/nutrit/nux053
Lee HS (2015) Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients 7(11):9492–9507. https://doi.org/10.3390/nu7115467
Wadhwa PD, Buss C, Entringer S, Swanson JM (2009) Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med 27(5):358–368. https://doi.org/10.1055/s-0029-1237424
Barker DJ, Osmond C (1986) Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1(8489):1077–1081
Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD (1991) Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303(6809):1019–1022. https://doi.org/10.1136/bmj.303.6809.1019
Tan HC, Roberts J, Catov J, Krishnamurthy R, Shypailo R, Bacha F (2015) Mother's pre-pregnancy BMI is an important determinant of adverse cardiometabolic risk in childhood. Pediatr Diabetes 16(6):419–426. https://doi.org/10.1111/pedi.12273
Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G (1997) Glucose tolerance and insulin secretion in children of mothers with pregestational IDDM or gestational diabetes. Diabetologia 40(9):1094–1100. https://doi.org/10.1007/s001250050792
Fuglestad AJ, Boys CJ, Chang PN, Miller BS, Eckerle JK, Deling L, Fink BA, Hoecker HL, Hickey MK, Jimenez-Vega JM, Wozniak JR (2014) Overweight and obesity among children and adolescents with fetal alcohol spectrum disorders. Alcohol Clin Exp Res 38(9):2502–2508. https://doi.org/10.1111/acer.12516
Li Y, Ley SH, VanderWeele TJ, Curhan GC, Rich-Edwards JW, Willett WC, Forman JP, Hu FB, Qi L (2015) Joint association between birth weight at term and later life adherence to a healthy lifestyle with risk of hypertension: a prospective cohort study. BMC Med 13:175. https://doi.org/10.1186/s12916-015-0409-1
Li Y, Ley SH, Tobias DK, Chiuve SE, VanderWeele TJ, Rich-Edwards JW, Curhan GC, Willett WC, Manson JE, Hu FB, Qi L (2015) Birth weight and later life adherence to unhealthy lifestyles in predicting type 2 diabetes: prospective cohort study. BMJ 351:h3672. https://doi.org/10.1136/bmj.h3672
Lopez-Tello J, Arias-Alvarez M, Gonzalez-Bulnes A, Sferuzzi-Perri AN (2019) Models of Intrauterine growth restriction and fetal programming in rabbits. Mol Reprod Dev. https://doi.org/10.1002/mrd.23271
Reynolds LP, Borowicz PP, Caton JS, Vonnahme KA, Luther JS, Hammer CJ, Maddock Carlin KR, Grazul-Bilska AT, Redmer DA (2010) Developmental programming: the concept, large animal models, and the key role of uteroplacental vascular development. J Anim Sci 88(13 Suppl):E61–72. https://doi.org/10.2527/jas.2009-2359
Gonzalez-Bulnes A, Chavatte-Palmer P (2017) Contribution of large animals to translational research on prenatal programming of obesity and associated diseases. Curr Pharm Biotechnol 18(7):541–551. https://doi.org/10.2174/1389201018666170811150920
Morrison JL, Botting KJ, Darby JRT, David AL, Dyson RM, Gatford KL, Gray C, Herrera EA, Hirst JJ, Kim B, Kind KL, Krause BJ, Matthews SG, Palliser HK, Regnault TRH, Richardson BS, Sasaki A, Thompson LP, Berry MJ (2018) Guinea pig models for translation of the developmental origins of health and disease hypothesis into the clinic. J Physiol 596(23):5535–5569. https://doi.org/10.1113/jp274948
Ailhaud G, Grimaldi P, Négrel R (1992) Cellular and molecular aspects of adipose tissue development. Annu Rev Nutr 12:207–233. https://doi.org/10.1146/annurev.nu.12.070192.001231
Burchell A, Gibb L, Waddell ID, Giles M, Hume R (1990) The ontogeny of human hepatic microsomal glucose-6-phosphatase proteins. Clin Chem 36(9):1633–1637
Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 6(3):208–216. https://doi.org/10.1016/j.cmet.2007.08.006
Gordillo M, Evans T, Gouon-Evans V (2015) Orchestrating liver development. Development 142(12):2094–2108. https://doi.org/10.1242/dev.114215
Romer AI, Sussel L (2015) Pancreatic islet cell development and regeneration. Curr Opin Endocrinol Diabetes Obes 22(4):255–264. https://doi.org/10.1097/med.0000000000000174
Fowden AL, Hill DJ (2001) Intra-uterine programming of the endocrine pancreas. Br Med Bull 60:123–142. https://doi.org/10.1093/bmb/60.1.123
Pan FC, Wright C (2011) Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 240(3):530–565. https://doi.org/10.1002/dvdy.22584
Sadava D, Frykman P, Harris E, Majerus D, Mustard J, Bernard B (1992) Development of enzymes of glycolysis and gluconeogenesis in human fetal liver. Biol Neonate 62(2–3):89–95. https://doi.org/10.1159/000243859
Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L, Damm P (2009) Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J Clin Endocrinol Metab 94(7):2464–2470. https://doi.org/10.1210/jc.2009-0305
Roseboom TJ, Painter RC, van Abeelen AF, Veenendaal MV, de Rooij SR (2011) Hungry in the womb: what are the consequences? Lessons from the Dutch famine. Maturitas 70(2):141–145. https://doi.org/10.1016/j.maturitas.2011.06.017
Roseboom T, de Rooij S, Painter R (2006) The Dutch famine and its long-term consequences for adult health. Early Hum Dev 82(8):485–491. https://doi.org/10.1016/j.earlhumdev.2006.07.001
Chandler-Laney PC, Bush NC, Granger WM, Rouse DJ, Mancuso MS, Gower BA (2012) Overweight status and intrauterine exposure to gestational diabetes are associated with children's metabolic health. Pediatr Obes 7(1):44–52. https://doi.org/10.1111/j.2047-6310.2011.00009.x
Moore LG, Charles SM, Julian CG (2011) Humans at high altitude: hypoxia and fetal growth. Respir Physiol Neurobiol 178(1):181–190. https://doi.org/10.1016/j.resp.2011.04.017
Micheli K, Komninos I, Bagkeris E, Roumeliotaki T, Koutis A, Kogevinas M, Chatzi L (2011) Sleep patterns in late pregnancy and risk of preterm birth and fetal growth restriction. Epidemiology 22(5):738–744. https://doi.org/10.1097/EDE.0b013e31822546fd
Rich DQ, Demissie K, Lu SE, Kamat L, Wartenberg D, Rhoads GG (2009) Ambient air pollutant concentrations during pregnancy and the risk of fetal growth restriction. J Epidemiol Community Health 63(6):488–496. https://doi.org/10.1136/jech.2008.082792
Jauniaux E, Burton GJ (2007) Morphological and biological effects of maternal exposure to tobacco smoke on the feto-placental unit. Early Hum Dev 83(11):699–706. https://doi.org/10.1016/j.earlhumdev.2007.07.016
Popova S, Lange S, Probst C, Gmel G, Rehm J (2017) Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: a systematic review and meta-analysis. Lancet Glob Health 5(3):e290–e299. https://doi.org/10.1016/s2214-109x(17)30021-9
Arduini D, Rizzo G, Romanini C, Mancuso S (1987) Utero-placental blood flow velocity waveforms as predictors of pregnancy-induced hypertension. Eur J Obstet Gynecol Reprod Biol 26(4):335–341. https://doi.org/10.1016/0028-2243(87)90131-6
Roberts D, Brown J, Medley N, Dalziel SR (2017) Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev 3:Cd004454. https://doi.org/10.1002/14651858.CD004454.pub3
Blais L, Beauchesne MF, Rey E, Malo JL, Forget A (2007) Use of inhaled corticosteroids during the first trimester of pregnancy and the risk of congenital malformations among women with asthma. Thorax 62(4):320–328. https://doi.org/10.1136/thx.2006.062950
Takahashi LK, Turner JG, Kalin NH (1998) Prolonged stress-induced elevation in plasma corticosterone during pregnancy in the rat: implications for prenatal stress studies. Psychoneuroendocrinology 23(6):571–581. https://doi.org/10.1016/s0306-4530(98)00024-9
Chen HE, Lin YJ, Lin IC, Yu HR, Sheen JM, Tsai CC, Huang LT, Tain YL (2019) Resveratrol prevents combined prenatal N(G)-nitro-l-arginine-methyl ester (L-NAME) treatment plus postnatal high-fat diet induced programmed hypertension in adult rat offspring: interplay between nutrient-sensing signals, oxidative stress and gut microbiota. J Nutr Biochem 70:28–37. https://doi.org/10.1016/j.jnutbio.2019.04.002
Tain YL, Huang LT, Lee CT, Chan JY, Hsu CN (2015) Maternal citrulline supplementation prevents prenatal N(G)-nitro-l-arginine-methyl ester (L-NAME)-induced programmed hypertension in rats. Biol Reprod 92(1):7. https://doi.org/10.1095/biolreprod.114.121384
Hao XQ, Du JX, Li Y, Li M, Zhang SY (2014) Prenatal exposure to lipopolysaccharide combined with pre- and postnatal high-fat diet result in lowered blood pressure and insulin resistance in offspring rats. PLoS ONE 9(2):e88127. https://doi.org/10.1371/journal.pone.0088127
Chen H, Iglesias MA, Caruso V, Morris MJ (2011) Maternal cigarette smoke exposure contributes to glucose intolerance and decreased brain insulin action in mice offspring independent of maternal diet. PLoS ONE 6(11):e27260. https://doi.org/10.1371/journal.pone.0027260
Holloway AC, Lim GE, Petrik JJ, Foster WG, Morrison KM, Gerstein HC (2005) Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia 48(12):2661–2666. https://doi.org/10.1007/s00125-005-0022-5
Bolton JL, Auten RL, Bilbo SD (2014) Prenatal air pollution exposure induces sexually dimorphic fetal programming of metabolic and neuroinflammatory outcomes in adult offspring. Brain Behav Immun 37:30–44. https://doi.org/10.1016/j.bbi.2013.10.029
Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, Bilbo SD (2012) Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. Faseb J 26(11):4743–4754. https://doi.org/10.1096/fj.12-210989
Wei J, Lin Y, Li Y, Ying C, Chen J, Song L, Zhou Z, Lv Z, Xia W, Chen X, Xu S (2011) Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology 152(8):3049–3061. https://doi.org/10.1210/en.2011-0045
Angle BM, Do RP, Ponzi D, Stahlhut RW, Drury BE, Nagel SC, Welshons WV, Besch-Williford CL, Palanza P, Parmigiani S, vom Saal FS, Taylor JA (2013) Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod Toxicol 42:256–268. https://doi.org/10.1016/j.reprotox.2013.07.017
Lejonklou MH, Dunder L, Bladin E, Pettersson V, Rönn M, Lind L, Waldén TB, Lind PM (2017) Effects of low-dose developmental bisphenol a exposure on metabolic parameters and gene expression in male and female fischer 344 rat offspring. Environ Health Perspect 125(6):067018. https://doi.org/10.1289/ehp505
Pei LG, Yuan C, Guo YT, Kou H, Xia LP, Zhang L, Yan YE, Xu D, Wang H (2017) Prenatal caffeine exposure induced high susceptibility to metabolic syndrome in adult female offspring rats and its underlying mechanisms. Reprod Toxicol 71:150–158. https://doi.org/10.1016/j.reprotox.2017.06.045
Zhang H, Chu X, Huang Y, Li G, Wang Y, Li Y, Sun C (2014) Maternal vitamin D deficiency during pregnancy results in insulin resistance in rat offspring, which is associated with inflammation and Iκbα methylation. Diabetologia 57(10):2165–2172. https://doi.org/10.1007/s00125-014-3316-7
Lewis RM, Forhead AJ, Petry CJ, Ozanne SE, Hales CN (2002) Long-term programming of blood pressure by maternal dietary iron restriction in the rat. Br J Nutr 88(3):283–290. https://doi.org/10.1079/bjn2002656
Crowe C, Dandekar P, Fox M, Dhingra K, Bennet L, Hanson M (1995) The effects of anaemia on heart, placenta and body weight, and blood pressure in fetal and neonatal rats. J Physiol 488(2):515–519
Bonaventura MM, Bourguignon NS, Bizzozzero M, Rodriguez D, Ventura C, Cocca C, Libertun C, Lux-Lantos VA (2017) Arsenite in drinking water produces glucose intolerance in pregnant rats and their female offspring. Food Chem Toxicol 100:207–216. https://doi.org/10.1016/j.fct.2016.12.025
Kimura I, Miyamoto J, Ohue-Kitano R, Watanabe K, Yamada T, Onuki M, Aoki R, Isobe Y, Kashihara D, Inoue D, Inaba A, Takamura Y, Taira S, Kumaki S, Watanabe M, Ito M, Nakagawa F, Irie J, Kakuta H, Shinohara M, Iwatsuki K, Tsujimoto G, Ohno H, Arita M, Itoh H, Hase K (2020) Maternal gut microbiota in pregnancy influences offspring metabolic phenotype in mice. Science. https://doi.org/10.1126/science.aaw8429
Varcoe TJ, Wight N, Voultsios A, Salkeld MD, Kennaway DJ (2011) Chronic phase shifts of the photoperiod throughout pregnancy programs glucose intolerance and insulin resistance in the rat. PLoS ONE 6(4):e18504. https://doi.org/10.1371/journal.pone.0018504
Agote M, Goya L, Ramos S, Alvarez C, Gavete ML, Pascual-Leone AM, Escriva F (2001) Glucose uptake and glucose transporter proteins in skeletal muscle from undernourished rats. Am J Physiol Endocrinol Metab 281(5):E1101–1109. https://doi.org/10.1152/ajpendo.2001.281.5.E1101
Norman AM, Miles-Chan JL, Thompson NM, Breier BH, Huber K (2012) Postnatal development of metabolic flexibility and enhanced oxidative capacity after prenatal undernutrition. Reprod Sci 19(6):607–614. https://doi.org/10.1177/1933719111428519
Somm E, Vauthay DM, Guerardel A, Toulotte A, Cettour-Rose P, Klee P, Meda P, Aubert ML, Huppi PS, Schwitzgebel VM (2012) Early metabolic defects in dexamethasone-exposed and undernourished intrauterine growth restricted rats. PLoS ONE 7(11):e50131. https://doi.org/10.1371/journal.pone.0050131
Thompson N, Huber K, Bedurftig M, Hansen K, Miles-Chan J, Breier BH (2014) Metabolic programming of adipose tissue structure and function in male rat offspring by prenatal undernutrition. Nutr Metab (Lond) 11(1):50. https://doi.org/10.1186/1743-7075-11-50
Lecoutre S, Marousez L, Drougard A, Knauf C, Guinez C, Eberle D, Laborie C, Vieau D, Lesage J, Breton C (2017) Maternal undernutrition programs the apelinergic system of adipose tissue in adult male rat offspring. J Dev Orig Health Dis 8(1):3–7. https://doi.org/10.1017/s2040174416000702
Lecoutre S, Montel V, Vallez E, Pourpe C, Delmont A, Eury E, Verbanck M, Dickes-Coopman A, Daubersies P, Lesage J, Laborie C, Tailleux A, Staels B, Froguel P, Breton C, Vieau D (2019) Transcription profiling in the liver of undernourished male rat offspring reveals altered lipid metabolism pathways and predisposition to hepatic steatosis. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.00291.2019
Xu YP, Liang L, Wang XM (2011) The levels of Pdx1/insulin, Cacna1c and Cacna1d, and beta-cell mass in a rat model of intrauterine undernutrition. J Matern Fetal Neonatal Med 24(3):437–443. https://doi.org/10.3109/14767058.2010.497571
Theys N, Ahn MT, Bouckenooghe T, Reusens B, Remacle C (2011) Maternal malnutrition programs pancreatic islet mitochondrial dysfunction in the adult offspring. J Nutr Biochem 22(10):985–994. https://doi.org/10.1016/j.jnutbio.2010.08.015
Smith T, Sloboda DM, Saffery R, Joo E, Vickers MH (2014) Maternal nutritional history modulates the hepatic IGF-IGFBP axis in adult male rat offspring. Endocrine 46(1):70–82. https://doi.org/10.1007/s12020-013-0034-8
Zhu WF, Zhu JF, Liang L, Shen Z, Wang YM (2016) Maternal undernutrition leads to elevated hepatic triglycerides in male rat offspring due to increased expression of lipoprotein lipase. Mol Med Rep 13(5):4487–4493. https://doi.org/10.3892/mmr.2016.5040
Wang X, Liang L, Du L (2007) The effects of intrauterine undernutrition on pancreas ghrelin and insulin expression in neonate rats. J Endocrinol 194(1):121–129. https://doi.org/10.1677/joe-07-0057
Reynolds CM, Li M, Gray C, Vickers MH (2013) Preweaning growth hormone treatment ameliorates adipose tissue insulin resistance and inflammation in adult male offspring following maternal undernutrition. Endocrinology 154(8):2676–2686. https://doi.org/10.1210/en.2013-1146
Raychaudhuri N, Raychaudhuri S, Thamotharan M, Devaskar SU (2008) Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J Biol Chem 283(20):13611–13626. https://doi.org/10.1074/jbc.M800128200
Magee TR, Han G, Cherian B, Khorram O, Ross MG, Desai M (2008) Down-regulation of transcription factor peroxisome proliferator-activated receptor in programmed hepatic lipid dysregulation and inflammation in intrauterine growth-restricted offspring. Am J Obstet Gynecol 199(3):271.e271–275. https://doi.org/10.1016/j.ajog.2008.05.022
Yamada M, Wolfe D, Han G, French SW, Ross MG, Desai M (2011) Early onset of fatty liver in growth-restricted rat fetuses and newborns. Congenit Anom (Kyoto) 51(4):167–173. https://doi.org/10.1111/j.1741-4520.2011.00336.x
Delahaye F, Lukaszewski MA, Wattez JS, Cisse O, Dutriez-Casteloot I, Fajardy I, Montel V, Dickes-Coopman A, Laborie C, Lesage J, Breton C, Vieau D (2010) Maternal perinatal undernutrition programs a "brown-like" phenotype of gonadal white fat in male rat at weaning. Am J Physiol Regul Integr Comp Physiol 299(1):R101–110. https://doi.org/10.1152/ajpregu.00604.2009
Campisano SE, Echarte SM, Podaza E, Chisari AN (2017) Protein malnutrition during fetal programming induces fatty liver in adult male offspring rats. J Physiol Biochem 73(2):275–285. https://doi.org/10.1007/s13105-017-0549-1
Desai M, Byrne CD, Zhang J, Petry CJ, Lucas A, Hales CN (1997) Programming of hepatic insulin-sensitive enzymes in offspring of rat dams fed a protein-restricted diet. Am J Physiol 272(5 Pt 1):G1083–1090. https://doi.org/10.1152/ajpgi.1997.272.5.G1083
Desai M, Byrne CD, Meeran K, Martenz ND, Bloom SR, Hales CN (1997) Regulation of hepatic enzymes and insulin levels in offspring of rat dams fed a reduced-protein diet. Am J Physiol 273(4):G899–904. https://doi.org/10.1152/ajpgi.1997.273.4.G899
Zheng J, Xiao X, Zhang Q, Yu M, Xu J, Wang Z (2015) Maternal protein restriction induces early-onset glucose intolerance and alters hepatic genes expression in the peroxisome proliferator-activated receptor pathway in offspring. J Diabetes Investig 6(3):269–279. https://doi.org/10.1111/jdi.12303
Fernandez-Twinn DS, Wayman A, Ekizoglou S, Martin MS, Hales CN, Ozanne SE (2005) Maternal protein restriction leads to hyperinsulinemia and reduced insulin-signaling protein expression in 21-mo-old female rat offspring. Am J Physiol Regul Integr Comp Physiol 288(2):R368–373. https://doi.org/10.1152/ajpregu.00206.2004
Claycombe KJ, Roemmich JN, Johnson L, Vomhof-DeKrey EE, Johnson WT (2015) Skeletal muscle Sirt3 expression and mitochondrial respiration are regulated by a prenatal low-protein diet. J Nutr Biochem 26(2):184–189. https://doi.org/10.1016/j.jnutbio.2014.10.003
Gosby AK, Stanton LM, Maloney CA, Thompson M, Briody J, Baxter RC, Bryson JM, Denyer GS, Caterson ID (2009) Postnatal nutrition alters body composition in adult offspring exposed to maternal protein restriction. Br J Nutr 101(12):1878–1884. https://doi.org/10.1017/S0007114508135851
Shepherd PR, Crowther NJ, Desai M, Hales CN, Ozanne SE (1997) Altered adipocyte properties in the offspring of protein malnourished rats. Br J Nutr 78(1):121–129. https://doi.org/10.1079/bjn19970124
Guan H, Arany E, van Beek JP, Chamson-Reig A, Thyssen S, Hill DJ, Yang K (2005) Adipose tissue gene expression profiling reveals distinct molecular pathways that define visceral adiposity in offspring of maternal protein-restricted rats. Am J Physiol Endocrinol Metab 288(4):E663–673. https://doi.org/10.1152/ajpendo.00461.2004
Theys N, Bouckenooghe T, Ahn MT, Remacle C, Reusens B (2009) Maternal low-protein diet alters pancreatic islet mitochondrial function in a sex-specific manner in the adult rat. Am J Physiol Regul Integr Comp Physiol 297(5):R1516–1525. https://doi.org/10.1152/ajpregu.00280.2009
Sohi G, Marchand K, Revesz A, Arany E, Hardy DB (2011) Maternal protein restriction elevates cholesterol in adult rat offspring due to repressive changes in histone modifications at the cholesterol 7alpha-hydroxylase promoter. Mol Endocrinol 25(5):785–798. https://doi.org/10.1210/me.2010-0395
Tarry-Adkins JL, Chen JH, Smith NS, Jones RH, Cherif H, Ozanne SE (2009) Poor maternal nutrition followed by accelerated postnatal growth leads to telomere shortening and increased markers of cell senescence in rat islets. Faseb j 23(5):1521–1528. https://doi.org/10.1096/fj.08-122796
Vo TX, Revesz A, Sohi G, Ma N, Hardy DB (2013) Maternal protein restriction leads to enhanced hepatic gluconeogenic gene expression in adult male rat offspring due to impaired expression of the liver X receptor. J Endocrinol 218(1):85–97. https://doi.org/10.1530/JOE-13-0055
Liu X, Qi Y, Tian B, Chen D, Gao H, Xi C, Xing Y, Yuan Z (2014) Maternal protein restriction induces alterations in hepatic tumor necrosis factor-alpha/CYP7A1 signaling and disorders regulation of cholesterol metabolism in the adult rat offspring. J Clin Biochem Nutr 55(1):40–47. https://doi.org/10.3164/jcbn.13-100
Abuzgaia AM, Hardy DB, Arany E (2015) Regulation of postnatal pancreatic Pdx1 and downstream target genes after gestational exposure to protein restriction in rats. Reproduction 149(3):293–303. https://doi.org/10.1530/REP-14-0245
Alejandro EU, Gregg B, Wallen T, Kumusoglu D, Meister D, Chen A, Merrins MJ, Satin LS, Liu M, Arvan P, Bernal-Mizrachi E (2014) Maternal diet-induced microRNAs and mTOR underlie beta cell dysfunction in offspring. J Clin Invest 124(10):4395–4410. https://doi.org/10.1172/JCI74237
Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC (2005) Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr 135(6):1382–1386. https://doi.org/10.1093/jn/135.6.1382
Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC (2007) Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr 97(6):1064–1073. https://doi.org/10.1017/S000711450769196X
Zheng S, Rollet M, Pan YX (2012) Protein restriction during gestation alters histone modifications at the glucose transporter 4 (GLUT4) promoter region and induces GLUT4 expression in skeletal muscle of female rat offspring. J Nutr Biochem 23(9):1064–1071. https://doi.org/10.1016/j.jnutbio.2011.05.013
Erhuma A, Bellinger L, Langley-Evans SC, Bennett AJ (2007) Prenatal exposure to undernutrition and programming of responses to high-fat feeding in the rat. Br J Nutr 98(3):517–524. https://doi.org/10.1017/s0007114507721505
Nusken KD, Schneider H, Plank C, Trollmann R, Nusken E, Rascher W, Dotsch J (2011) Fetal programming of gene expression in growth-restricted rats depends on the cause of low birth weight. Endocrinology 152(4):1327–1335. https://doi.org/10.1210/en.2010-1116
da Silva AR, Guzman-Quevedo O, Perez-Garcia G, Toscano AE, Gois Leandro C, Manhaes-de-Castro R, Bolanos-Jimenez F (2014) Differential developmental programming by early protein restriction of rat skeletal muscle according to its fibre-type composition. Acta Physiol (Oxf) 210(1):70–83. https://doi.org/10.1111/apha.12073
Latouche C, Heywood SE, Henry SL, Ziemann M, Lazarus R, El-Osta A, Armitage JA, Kingwell BA (2014) Maternal overnutrition programs changes in the expression of skeletal muscle genes that are associated with insulin resistance and defects of oxidative phosphorylation in adult male rat offspring. J Nutr 144(3):237–244. https://doi.org/10.3945/jn.113.186775
Zhang X, Strakovsky R, Zhou D, Zhang Y, Pan YX (2011) A maternal high-fat diet represses the expression of antioxidant defense genes and induces the cellular senescence pathway in the liver of male offspring rats. J Nutr 141(7):1254–1259. https://doi.org/10.3945/jn.111.139576
Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH (2011) Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 6(7):e21662. https://doi.org/10.1371/journal.pone.0021662
Yang KF, Cai W, Xu JL, Shi W (2012) Maternal high-fat diet programs Wnt genes through histone modification in the liver of neonatal rats. J Mol Endocrinol 49(2):107–114. https://doi.org/10.1530/jme-12-0046
Yang QY, Liang JF, Rogers CJ, Zhao JX, Zhu MJ, Du M (2013) Maternal obesity induces epigenetic modifications to facilitate Zfp423 expression and enhance adipogenic differentiation in fetal mice. Diabetes 62(11):3727–3735. https://doi.org/10.2337/db13-0433
Bringhenti I, Ornellas F, Mandarim-de-Lacerda CA, Aguila MB (2016) The insulin-signaling pathway of the pancreatic islet is impaired in adult mice offspring of mothers fed a high-fat diet. Nutrition 32(10):1138–1143. https://doi.org/10.1016/j.nut.2016.03.001
Pereira TJ, Fonseca MA, Campbell KE, Moyce BL, Cole LK, Hatch GM, Doucette CA, Klein J, Aliani M, Dolinsky VW (2015) Maternal obesity characterized by gestational diabetes increases the susceptibility of rat offspring to hepatic steatosis via a disrupted liver metabolome. J Physiol 593(14):3181–3197. https://doi.org/10.1113/jp270429
Agarwal P, Brar N, Morriseau TS, Kereliuk SM, Fonseca MA, Cole LK, Jha A, Xiang B, Hunt KL, Seshadri N, Hatch GM, Doucette CA, Dolinsky VW (2019) Gestational diabetes adversely affects pancreatic islet architecture and function in the male rat offspring. Endocrinology 160(8):1907–1925. https://doi.org/10.1210/en.2019-00232
Fante T, Simino LA, Reginato A, Payolla TB, Vitoreli DC, Souza M, Torsoni MA, Milanski M, Torsoni AS (2016) Diet-induced maternal obesity alters insulin signalling in male mice offspring rechallenged with a high-fat diet in adulthood. PLoS ONE 11(8):e0160184. https://doi.org/10.1371/journal.pone.0160184
Payolla TB, Lemes SF, de Fante T, Reginato A, Mendes da Silva C, de Oliveira MT, Rodrigues HG, Torsoni AS, Milanski M, Torsoni MA (2016) High-fat diet during pregnancy and lactation impairs the cholinergic anti-inflammatory pathway in the liver and white adipose tissue of mouse offspring. Mol Cell Endocrinol 422:192–202. https://doi.org/10.1016/j.mce.2015.12.004
Zheng J, Xiao X, Zhang Q, Yu M, Xu J, Wang Z (2014) Maternal high-fat diet modulates hepatic glucose, lipid homeostasis and gene expression in the PPAR pathway in the early life of offspring. Int J Mol Sci 15(9):14967–14983. https://doi.org/10.3390/ijms150914967
Buckley AJ, Keseru B, Briody J, Thompson M, Ozanne SE, Thompson CH (2005) Altered body composition and metabolism in the male offspring of high fat-fed rats. Metabolism 54(4):500–507. https://doi.org/10.1016/j.metabol.2004.11.003
Khalyfa A, Carreras A, Hakim F, Cunningham JM, Wang Y, Gozal D (2013) Effects of late gestational high-fat diet on body weight, metabolic regulation and adipokine expression in offspring. Int J Obes (Lond) 37(11):1481–1489. https://doi.org/10.1038/ijo.2013.12
Seet EL, Yee JK, Jellyman JK, Han G, Ross MG, Desai M (2015) Maternal high-fat-diet programs rat offspring liver fatty acid metabolism. Lipids 50(6):565–573. https://doi.org/10.1007/s11745-015-4018-8
Lecoutre S, Deracinois B, Laborie C, Eberle D, Guinez C, Panchenko PE, Lesage J, Vieau D, Junien C, Gabory A, Breton C (2016) Depot- and sex-specific effects of maternal obesity in offspring's adipose tissue. J Endocrinol 230(1):39–53. https://doi.org/10.1530/JOE-16-0037
Lecoutre S, Pourpe C, Butruille L, Marousez L, Laborie C, Guinez C, Lesage J, Vieau D, Eeckhoute J, Gabory A, Oger F, Eberle D, Breton C (2018) Reduced PPARgamma2 expression in adipose tissue of male rat offspring from obese dams is associated with epigenetic modifications. Faseb J 32(5):2768–2778. https://doi.org/10.1096/fj.201700997R
Desai M, Jellyman JK, Han G, Lane RH, Ross MG (2015) Programmed regulation of rat offspring adipogenic transcription factor (PPARgamma) by maternal nutrition. J Dev Orig Health Dis 6(6):530–538. https://doi.org/10.1017/s2040174415001440
Yokomizo H, Inoguchi T, Sonoda N, Sakaki Y, Maeda Y, Inoue T, Hirata E, Takei R, Ikeda N, Fujii M, Fukuda K, Sasaki H, Takayanagi R (2014) Maternal high-fat diet induces insulin resistance and deterioration of pancreatic beta-cell function in adult offspring with sex differences in mice. Am J Physiol Endocrinol Metab 306(10):E1163–1175. https://doi.org/10.1152/ajpendo.00688.2013
Simmons RA, Suponitsky-Kroyter I, Selak MA (2005) Progressive accumulation of mitochondrial DNA mutations and decline in mitochondrial function lead to beta-cell failure. J Biol Chem 280(31):28785–28791. https://doi.org/10.1074/jbc.M505695200
Park JH, Stoffers DA, Nicholls RD, Simmons RA (2008) Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 118(6):2316–2324. https://doi.org/10.1172/jci33655
Sadiq HF, Das UG, Tracy TF, Devaskar SU (1999) Intra-uterine growth restriction differentially regulates perinatal brain and skeletal muscle glucose transporters. Brain Res 823(1–2):96–103. https://doi.org/10.1016/s0006-8993(99)01145-2
Stoffers DA, Desai BM, DeLeon DD, Simmons RA (2003) Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes 52(3):734–740. https://doi.org/10.2337/diabetes.52.3.734
Germani D, Puglianiello A, Cianfarani S (2008) Uteroplacental insufficiency down regulates insulin receptor and affects expression of key enzymes of long-chain fatty acid (LCFA) metabolism in skeletal muscle at birth. Cardiovasc Diabetol 7:14. https://doi.org/10.1186/1475-2840-7-14
Fu Q, Yu X, Callaway CW, Lane RH, McKnight RA (2009) Epigenetics: intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. Faseb J 23(8):2438–2449. https://doi.org/10.1096/fj.08-124768
Camm EJ, Martin-Gronert MS, Wright NL, Hansell JA, Ozanne SE, Giussani DA (2011) Prenatal hypoxia independent of undernutrition promotes molecular markers of insulin resistance in adult offspring. FASEB J 25(1):420–427. https://doi.org/10.1096/fj.10-158188
Osumek JE, Revesz A, Morton JS, Davidge ST, Hardy DB (2014) Enhanced trimethylation of histone h3 mediates impaired expression of hepatic glucose 6-phosphatase expression in offspring from rat dams exposed to hypoxia during pregnancy. Reprod Sci 21(1):112–121. https://doi.org/10.1177/1933719113492212
Rueda-Clausen CF, Dolinsky VW, Morton JS, Proctor SD, Dyck JR, Davidge ST (2011) Hypoxia-induced intrauterine growth restriction increases the susceptibility of rats to high-fat diet-induced metabolic syndrome. Diabetes 60(2):507–516. https://doi.org/10.2337/db10-1239
Badran M, Yassin BA, Lin DTS, Kobor MS, Ayas N, Laher I (2019) Gestational intermittent hypoxia induces endothelial dysfunction, reduces perivascular adiponectin and causes epigenetic changes in adult male offspring. J Physiol 597(22):5349–5364. https://doi.org/10.1113/jp277936
Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR (1998) Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest 101(10):2174–2181. https://doi.org/10.1172/jci1567
Drake AJ, Walker BR, Seckl JR (2005) Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am J Physiol Regul Integr Comp Physiol 288(1):R34–38. https://doi.org/10.1152/ajpregu.00106.2004
Sheen JM, Hsieh CS, Tain YL, Li SW, Yu HR, Chen CC, Tiao MM, Chen YC, Huang LT (2016) Programming effects of prenatal glucocorticoid exposure with a postnatal high-fat diet in diabetes mellitus. Int J Mol Sci 17(4):533. https://doi.org/10.3390/ijms17040533
Chen YC, Huang YH, Sheen JM, Tain YL, Yu HR, Chen CC, Tiao MM, Kuo HC, Huang LT (2017) Prenatal dexamethasone exposure programs the development of the pancreas and the secretion of insulin in rats. Pediatr Neonatol 58(2):135–144. https://doi.org/10.1016/j.pedneo.2016.02.008
Cleasby ME, Kelly PA, Walker BR, Seckl JR (2003) Programming of rat muscle and fat metabolism by in utero overexposure to glucocorticoids. Endocrinology 144(3):999–1007. https://doi.org/10.1210/en.2002-220559
O'Regan D, Kenyon CJ, Seckl JR, Holmes MC (2004) Glucocorticoid exposure in late gestation in the rat permanently programs gender-specific differences in adult cardiovascular and metabolic physiology. Am J Physiol Endocrinol Metab 287(5):E863–870. https://doi.org/10.1152/ajpendo.00137.2004
Franko KL, Forhead AJ, Fowden AL (2010) Differential effects of prenatal stress and glucocorticoid administration on postnatal growth and glucose metabolism in rats. J Endocrinol 204(3):319–329. https://doi.org/10.1677/JOE-09-0390
Carbone DL, Zuloaga DG, Hiroi R, Foradori CD, Legare ME, Handa RJ (2012) Prenatal dexamethasone exposure potentiates diet-induced hepatosteatosis and decreases plasma IGF-I in a sex-specific fashion. Endocrinology 153(1):295–306. https://doi.org/10.1210/en.2011-1601
Mark PJ, Wyrwoll CS, Zulkafli IS, Mori TA, Waddell BJ (2014) Rescue of glucocorticoid-programmed adipocyte inflammation by omega-3 fatty acid supplementation in the rat. Reprod Biol Endocrinol 12:39. https://doi.org/10.1186/1477-7827-12-39
Brunton PJ, Sullivan KM, Kerrigan D, Russell JA, Seckl JR, Drake AJ (2013) Sex-specific effects of prenatal stress on glucose homoeostasis and peripheral metabolism in rats. J Endocrinol 217(2):161–173. https://doi.org/10.1530/joe-12-0540
Paternain L, de la Garza AL, Batlle MA, Milagro FI, Martínez JA, Campión J (2013) Prenatal stress increases the obesogenic effects of a high-fat-sucrose diet in adult rats in a sex-specific manner. Stress 16(2):220–232. https://doi.org/10.3109/10253890.2012.707708
Franko KL, Forhead AJ, Fowden AL (2017) Effects of stress during pregnancy on hepatic glucogenic capacity in rat dams and their fetuses. Physiol Rep. https://doi.org/10.14814/phy2.13293
Maeyama H, Hirasawa T, Tahara Y, Obata C, Kasai H, Moriishi K, Mochizuki K, Kubota T (2015) Maternal restraint stress during pregnancy in mice induces 11β-HSD1-associated metabolic changes in the livers of the offspring. J Dev Orig Health Dis 6(2):105–114. https://doi.org/10.1017/s2040174415000100
Kamel MA, Helmy MH, Hanafi MY, Mahmoud SA, Abo Elfetooh H (2014) Impaired peripheral glucose sensing in F1 offspring of diabetic pregnancy. J Physiol Biochem 70(3):685–699. https://doi.org/10.1007/s13105-014-0338-z
Inoguchi Y, Ichiyanagi K, Ohishi H, Maeda Y, Sonoda N, Ogawa Y, Inoguchi T, Sasaki H (2019) Poorly controlled diabetes during pregnancy and lactation activates the Foxo1 pathway and causes glucose intolerance in adult offspring. Sci Rep 9(1):10181. https://doi.org/10.1038/s41598-019-46638-2
Oliveira AC, Andreotti S, Chimin P, Sertie RA, Farias Tda S, Torres-Leal FL, de Proenca AR, Campana AB, D'Avila LS, Oliveira KA, Lima FB (2015) Neonatal streptozotocin-induced diabetes in mothers promotes metabolic programming of adipose tissue in male rat offspring. Life Sci 136:151–156. https://doi.org/10.1016/j.lfs.2015.06.024
Dong HP, Tan MZ, Liu QJ, Wang J, Zhong SB (2017) The study on the effect of hyperglycemia on offspring fatty tissue metabolism during pregnancy. Eur Rev Med Pharmacol Sci 21(16):3658–3664
Nazari Z, Nabiuni M, Saeidi M, Golalipour MJ (2017) Gestational diabetes leads to down-regulation of CDK4-pRB-E2F1 pathway genes in pancreatic islets of rat offspring. Iran J Basic Med Sci 20(2):150–154. https://doi.org/10.22038/ijbms.2017.8240
Su R, Yan J, Yang H (2016) Transgenerational glucose intolerance of tumor necrosis factor with epigenetic alteration in rat perirenal adipose tissue induced by intrauterine hyperglycemia. J Diabetes Res 2016:4952801. https://doi.org/10.1155/2016/4952801
Zhu H, Chen B, Cheng Y, Zhou Y, Yan YS, Luo Q, Jiang Y, Sheng JZ, Ding GL, Huang HF (2019) Insulin therapy for gestational diabetes mellitus does not fully protect offspring from diet-induced metabolic disorders. Diabetes 68(4):696–708. https://doi.org/10.2337/db18-1151
Chen L, Nyomba BL (2003) Effects of prenatal alcohol exposure on glucose tolerance in the rat offspring. Metabolism 52(4):454–462. https://doi.org/10.1053/meta.2003.50073
Yao XH, Gregoire Nyomba BL (2007) Abnormal glucose homeostasis in adult female rat offspring after intrauterine ethanol exposure. Am J Physiol Regul Integr Comp Physiol 292(5):R1926–1933. https://doi.org/10.1152/ajpregu.00822.2006
Chen L, Yao XH, Nyomba BL (2005) In vivo insulin signaling through PI3-kinase is impaired in skeletal muscle of adult rat offspring exposed to ethanol in utero. J Appl Physiol (1985) 99(2):528–534. https://doi.org/10.1152/japplphysiol.01098.2004
Yao XH, Chen L, Nyomba BL (2006) Adult rats prenatally exposed to ethanol have increased gluconeogenesis and impaired insulin response of hepatic gluconeogenic genes. J Appl Physiol (1985) 100(2):642–648. https://doi.org/10.1152/japplphysiol.01115.2005
Yao XH, Nyomba BL (2008) Hepatic insulin resistance induced by prenatal alcohol exposure is associated with reduced PTEN and TRB3 acetylation in adult rat offspring. Am J Physiol Regul Integr Comp Physiol 294(6):R1797–1806. https://doi.org/10.1152/ajpregu.00804.2007
Gardebjer EM, Anderson ST, Pantaleon M, Wlodek ME, Moritz KM (2015) Maternal alcohol intake around the time of conception causes glucose intolerance and insulin insensitivity in rat offspring, which is exacerbated by a postnatal high-fat diet. FASEB J 29(7):2690–2701. https://doi.org/10.1096/fj.14-268979
Yao XH, Nguyen HK, Nyomba BL (2013) Prenatal ethanol exposure causes glucose intolerance with increased hepatic gluconeogenesis and histone deacetylases in adult rat offspring: reversal by tauroursodeoxycholic acid. PLoS ONE 8(3):e59680. https://doi.org/10.1371/journal.pone.0059680
Nguyen TMT, Steane SE, Moritz KM, Akison LK (2019) Prenatal alcohol exposure programmes offspring disease: insulin resistance in adult males in a rat model of acute exposure. J Physiol. https://doi.org/10.1113/jp278531
Shen L, Liu Z, Gong J, Zhang L, Wang L, Magdalou J, Chen L, Wang H (2014) Prenatal ethanol exposure programs an increased susceptibility of non-alcoholic fatty liver disease in female adult offspring rats. Toxicol Appl Pharmacol 274(2):263–273. https://doi.org/10.1016/j.taap.2013.11.009
Wierup N, Yang S, McEvilly RJ, Mulder H, Sundler F (2004) Ghrelin is expressed in a novel endocrine cell type in developing rat islets and inhibits insulin secretion from INS-1 (832/13) cells. J Histochem Cytochem 52(3):301–310. https://doi.org/10.1177/002215540405200301
Ellis-Hutchings RG, Zucker RM, Grey BE, Norwood J Jr, Richards JH, Lau C, Rogers JM (2010) Altered health outcomes in adult offspring of Sprague Dawley and Wistar rats undernourished during early or late pregnancy. Birth Defects Res B Dev Reprod Toxicol 89(5):396–407. https://doi.org/10.1002/bdrb.20265
Sampaio de Freitas M, Garcia De Souza EP, Vargas da Silva S, da Rocha KA, da Silva VR, Sanchez Moura A, Barja-Fidalgo C (2003) Up-regulation of phosphatidylinositol 3-kinase and glucose transporter 4 in muscle of rats subjected to maternal undernutrition. BBA Mol Basis Dis 1639(1):8–16. https://doi.org/10.1016/s0925-4439(03)00096-6
Holemans K, Van Bree R, Verhaeghe J, Meurrens K, Van Assche FA (1997) Maternal semistarvation and streptozotocin-diabetes in rats have different effects on the in vivo glucose uptake by peripheral tissues in their female adult offspring. J Nutr 127(7):1371–1376. https://doi.org/10.1093/jn/127.7.1371
Taylor PD, McConnell J, Khan IY, Holemans K, Lawrence KM, Asare-Anane H, Persaud SJ, Jones PM, Petrie L, Hanson MA, Poston L (2005) Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am J Physiol Regul Integr Comp Physiol 288(1):R134–139. https://doi.org/10.1152/ajpregu.00355.2004
Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS (2006) Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab 291(4):E792–799. https://doi.org/10.1152/ajpendo.00078.2006
Zambrano E, Martinez-Samayoa PM, Rodriguez-Gonzalez GL, Nathanielsz PW (2010) Dietary intervention prior to pregnancy reverses metabolic programming in male offspring of obese rats. J Physiol 588(Pt 10):1791–1799. https://doi.org/10.1113/jphysiol.2010.190033
Volpato AM, Schultz A, Magalhaes-da-Costa E, Correia ML, Aguila MB, Mandarim-de-Lacerda CA (2012) Maternal high-fat diet programs for metabolic disturbances in offspring despite leptin sensitivity. Neuroendocrinology 96(4):272–284. https://doi.org/10.1159/000336377
Desai M, Jellyman JK, Han G, Beall M, Lane RH, Ross MG (2014) Maternal obesity and high-fat diet program offspring metabolic syndrome. Am J Obstet Gynecol 211(3):237. https://doi.org/10.1016/j.ajog.2014.03.025
Bergman RN, Ader M, Huecking K, Van Citters G (2002) Accurate assessment of beta-cell function: the hyperbolic correction. Diabetes 51(Suppl 1):S212–220. https://doi.org/10.2337/diabetes.51.2007.s212
Poy MN, Yang Y, Rezaei K, Fernstrom MA, Lee AD, Kido Y, Erickson SK, Najjar SM (2002) CEACAM1 regulates insulin clearance in liver. Nat Genet 30(3):270–276. https://doi.org/10.1038/ng840
Dumortier O, Blondeau B, Duvillie B, Reusens B, Breant B, Remacle C (2007) Different mechanisms operating during different critical time-windows reduce rat fetal beta cell mass due to a maternal low-protein or low-energy diet. Diabetologia 50(12):2495–2503. https://doi.org/10.1007/s00125-007-0811-0
Petersen MC, Vatner DF, Shulman GI (2017) Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol 13(10):572–587. https://doi.org/10.1038/nrendo.2017.80
DeFronzo RA (2004) Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 88(4):787–835. https://doi.org/10.1016/j.mcna.2004.04.013
Barthel A, Schmoll D (2003) Novel concepts in insulin regulation of hepatic gluconeogenesis. Am J Physiol Endocrinol Metab 285(4):E685–692. https://doi.org/10.1152/ajpendo.00253.2003
Tsuru M, Katagiri H, Asano T, Yamada T, Ohno S, Ogihara T, Oka Y (2002) Role of PKC isoforms in glucose transport in 3T3-L1 adipocytes: insignificance of atypical PKC. Am J Physiol Endocrinol Metab 283(2):E338–345. https://doi.org/10.1152/ajpendo.00457.2001
Leto D, Saltiel AR (2012) Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 13(6):383–396. https://doi.org/10.1038/nrm3351
He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM (2003) Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci USA 100(26):15712–15717. https://doi.org/10.1073/pnas.2536828100
Mooradian AD, Chehade J, Thurman JE (2002) The role of thiazolidinediones in the treatment of patients with type 2 diabetes mellitus. Treat Endocrinol 1(1):13–20. https://doi.org/10.2165/00024677-200201010-00002
Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14(22):2819–2830. https://doi.org/10.1101/gad.844900
Cao G, Liang Y, Broderick CL, Oldham BA, Beyer TP, Schmidt RJ, Zhang Y, Stayrook KR, Suen C, Otto KA, Miller AR, Dai J, Foxworthy P, Gao H, Ryan TP, Jiang XC, Burris TP, Eacho PI, Etgen GJ (2003) Antidiabetic action of a liver x receptor agonist mediated by inhibition of hepatic gluconeogenesis. J Biol Chem 278(2):1131–1136. https://doi.org/10.1074/jbc.M210208200
Westwick JK, Weitzel C, Minden A, Karin M, Brenner DA (1994) Tumor necrosis factor alpha stimulates AP-1 activity through prolonged activation of the c-Jun kinase. J Biol Chem 269(42):26396–26401
Sluss HK, Barrett T, Derijard B, Davis RJ (1994) Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol Cell Biol 14(12):8376–8384. https://doi.org/10.1128/mcb.14.12.8376
Peng K, Pan Y, Li J, Khan Z, Fan M, Yin H, Tong C, Zhao Y, Liang G, Zheng C (2016) 11beta-hydroxysteroid dehydrogenase type 1(11beta-HSD1) mediates insulin resistance through JNK activation in adipocytes. Sci Rep 6:37160. https://doi.org/10.1038/srep37160
Saegusa H, Nakagawa Y, Liu Y-J, Ohzeki T (1999) Influence of placental 11β-hydroxysteroid dehydrogenase (11β-HSD) inhibition on glucose metabolism and 11β-HSD regulation in adult offspring of rats. Metabol Clin Exp 48(12):1584–1588
Lindsay R, Lindsay R, Waddell B, Seckl J (1996) Prenatal glucocorticoid exposure leads to offspring hyperglycaemia in the rat: studies with the 11 b-hydroxysteroid dehydrogenase inhibitor carbenoxolone. Diabetologia 39(11):1299–1305
Thompson MD, Cismowski MJ, Trask AJ, Lallier SW, Graf AE, Rogers LK, Lucchesi PA, Brigstock DR (2016) Enhanced steatosis and fibrosis in liver of adult offspring exposed to maternal high-fat diet. Gene Expr 17(1):47–59. https://doi.org/10.3727/105221616x692135
Qasem RJ, Cherala G, D'Mello AP (2010) Maternal protein restriction during pregnancy and lactation in rats imprints long-term reduction in hepatic lipid content selectively in the male offspring. Nutr Res 30(6):410–417. https://doi.org/10.1016/j.nutres.2010.05.008
Wattez JS, Delahaye F, Barella LF, Dickes-Coopman A, Montel V, Breton C, Mathias P, Foligne B, Lesage J, Vieau D (2014) Short- and long-term effects of maternal perinatal undernutrition are lowered by cross-fostering during lactation in the male rat. J Dev Orig Health Dis 5(2):109–120. https://doi.org/10.1017/S2040174413000548
Tapanainen PJ, Bang P, Wilson K, Unterman TG, Vreman HJ, Rosenfeld RG (1994) Maternal hypoxia as a model for intrauterine growth retardation: effects on insulin-like growth factors and their binding proteins. Pediatr Res 36(2):152–158. https://doi.org/10.1203/00006450-199408000-00004
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D (1999) Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA 96(13):7324–7329. https://doi.org/10.1073/pnas.96.13.7324
Sferruzzi-Perri AN, Vaughan OR, Forhead AJ, Fowden AL (2013) Hormonal and nutritional drivers of intrauterine growth. Curr Opin Clin Nutr Metab Care 16(3):298–309. https://doi.org/10.1097/MCO.0b013e32835e3643
Aguirre GA, De Ita JR, de la Garza RG, Castilla-Cortazar I (2016) Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med 14:3. https://doi.org/10.1186/s12967-015-0762-z
Lewitt MS, Dent MS, Hall K (2014) The insulin-like growth factor system in obesity, insulin resistance and type 2 diabetes mellitus. J Clin Med 3(4):1561–1574. https://doi.org/10.3390/jcm3041561
Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, Spiegelman BM (2002) C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev 16(1):22–26. https://doi.org/10.1101/gad.948702
Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM (2000) Transcriptional regulation of adipogenesis. Genes Dev 14(11):1293–1307
Ouchi N, Parker JL, Lugus JJ, Walsh K (2011) Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11(2):85–97. https://doi.org/10.1038/nri2921
Bird A (2007) Perceptions of epigenetics. Nature 447(7143):396–398. https://doi.org/10.1038/nature05913
Claycombe KJ, Uthus EO, Roemmich JN, Johnson LK, Johnson WT (2013) Prenatal low-protein and postnatal high-fat diets induce rapid adipose tissue growth by inducing Igf2 expression in Sprague Dawley rat offspring. J Nutr 143(10):1533–1539. https://doi.org/10.3945/jn.113.178038
Zeng Y, Gu P, Liu K, Huang P (2013) Maternal protein restriction in rats leads to reduced PGC-1alpha expression via altered DNA methylation in skeletal muscle. Mol Med Rep 7(1):306–312. https://doi.org/10.3892/mmr.2012.1134
Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdge GC (2008) Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br J Nutr 100(2):278–282. https://doi.org/10.1017/s0007114507894438
Gong L, Pan YX, Chen H (2010) Gestational low protein diet in the rat mediates Igf2 gene expression in male offspring via altered hepatic DNA methylation. Epigenetics 5(7):619–626. https://doi.org/10.4161/epi.5.7.12882
van Straten EM, Bloks VW, Huijkman NC, Baller JF, van Meer H, Lutjohann D, Kuipers F, Plosch T (2010) The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am J Physiol Regul Integr Comp Physiol 298(2):R275–282. https://doi.org/10.1152/ajpregu.00413.2009
Jousse C, Parry L, Lambert-Langlais S, Maurin AC, Averous J, Bruhat A, Carraro V, Tost J, Letteron P, Chen P, Jockers R, Launay JM, Mallet J, Fafournoux P (2011) Perinatal undernutrition affects the methylation and expression of the leptin gene in adults: implication for the understanding of metabolic syndrome. Faseb J 25(9):3271–3278. https://doi.org/10.1096/fj.11-181792
Yu HL, Dong S, Gao LF, Li L, Xi YD, Ma WW, Yuan LH, Xiao R (2015) Global DNA methylation was changed by a maternal high-lipid, high-energy diet during gestation and lactation in male adult mice liver. Br J Nutr 113(7):1032–1039. https://doi.org/10.1017/s0007114515000252
Liang X, Yang Q, Fu X, Rogers CJ, Wang B, Pan H, Zhu MJ, Nathanielsz PW, Du M (2016) Maternal obesity epigenetically alters visceral fat progenitor cell properties in male offspring mice. J Physiol 594(15):4453–4466. https://doi.org/10.1113/jp272123
Jiao F, Yan X, Yu Y, Zhu X, Ma Y, Yue Z, Ou H, Yan Z (2016) Protective effects of maternal methyl donor supplementation on adult offspring of high fat diet-fed dams. J Nutr Biochem 34:42–51. https://doi.org/10.1016/j.jnutbio.2016.04.005
Moody L, Wang H, Jung PM, Chen H, Pan YX (2019) Maternal and post-weaning high-fat diets produce distinct DNA methylation patterns in hepatic metabolic pathways within specific genomic contexts. Int J Mol Sci. https://doi.org/10.3390/ijms20133229
Simmons RA, Templeton LJ, Gertz SJ (2001) Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50(10):2279–2286. https://doi.org/10.2337/diabetes.50.10.2279
Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM (2010) Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J Biol Chem 285(20):15111–15118. https://doi.org/10.1074/jbc.M109.095133
Keshet I, Lieman-Hurwitz J, Cedar H (1986) DNA methylation affects the formation of active chromatin. Cell 44(4):535–543. https://doi.org/10.1016/0092-8674(86)90263-1
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462(7271):315–322. https://doi.org/10.1038/nature08514
Bestor T, Laudano A, Mattaliano R, Ingram V (1988) Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol 203(4):971–983. https://doi.org/10.1016/0022-2836(88)90122-2
Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19(3):219–220. https://doi.org/10.1038/890
Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA, Lillycrop KA (2007) Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br J Nutr 97(3):435–439. https://doi.org/10.1017/s0007114507352392
Khalyfa A, Cortese R, Qiao Z, Ye H, Bao R, Andrade J, Gozal D (2017) Late gestational intermittent hypoxia induces metabolic and epigenetic changes in male adult offspring mice. J Physiol 595(8):2551–2568. https://doi.org/10.1113/jp273570
Giudicelli F, Brabant AL, Grit I, Parnet P, Amarger V (2013) Excess of methyl donor in the perinatal period reduces postnatal leptin secretion in rat and interacts with the effect of protein content in diet. PLoS ONE 8(7):e68268. https://doi.org/10.1371/journal.pone.0068268
Altobelli G, Bogdarina IG, Stupka E, Clark AJ, Langley-Evans S (2013) Genome-wide methylation and gene expression changes in newborn rats following maternal protein restriction and reversal by folic acid. PLoS ONE 8(12):e82989. https://doi.org/10.1371/journal.pone.0082989
Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, Uribe-Lewis S, Ito Y, Jones RH, Marquez VE, Cairns W, Tadayyon M, O'Neill LP, Murrell A, Ling C, Constancia M, Ozanne SE (2011) Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc Natl Acad Sci USA 108(13):5449–5454. https://doi.org/10.1073/pnas.1019007108
Claycombe KJ, Vomhof-DeKrey EE, Garcia R, Johnson WT, Uthus E, Roemmich JN (2016) Decreased beige adipocyte number and mitochondrial respiration coincide with increased histone methyl transferase (G9a) and reduced FGF21 gene expression in Sprague-Dawley rats fed prenatal low protein and postnatal high-fat diets. J Nutr Biochem 31:113–121. https://doi.org/10.1016/j.jnutbio.2016.01.008
Zheng S, Rollet M, Pan YX (2011) Maternal protein restriction during pregnancy induces CCAAT/enhancer-binding protein (C/EBPbeta) expression through the regulation of histone modification at its promoter region in female offspring rat skeletal muscle. Epigenetics 6(2):161–170. https://doi.org/10.4161/epi.6.2.13472
Strakovsky RS, Zhang X, Zhou D, Pan YX (2011) Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J Physiol 589(Pt 11):2707–2717. https://doi.org/10.1113/jphysiol.2010.203950
Zhou D, Wang H, Cui H, Chen H, Pan YX (2015) Early-life exposure to high-fat diet may predispose rats to gender-specific hepatic fat accumulation by programming Pepck expression. J Nutr Biochem 26(5):433–440. https://doi.org/10.1016/j.jnutbio.2014.10.009
Panchenko PE, Voisin S, Jouin M, Jouneau L, Prezelin A, Lecoutre S, Breton C, Jammes H, Junien C, Gabory A (2016) Expression of epigenetic machinery genes is sensitive to maternal obesity and weight loss in relation to fetal growth in mice. Clin Epigenet 8:22. https://doi.org/10.1186/s13148-016-0188-3
Masuyama H, Hiramatsu Y (2012) Treatment with constitutive androstane receptor ligand during pregnancy prevents insulin resistance in offspring from high-fat diet-induced obese pregnant mice. Am J Physiol Endocrinol Metab 303(2):E293–300. https://doi.org/10.1152/ajpendo.00167.2012
Masuyama H, Mitsui T, Nobumoto E, Hiramatsu Y (2015) The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology 156(7):2482–2491. https://doi.org/10.1210/en.2014-2020
Pinney SE, Jaeckle Santos LJ, Han Y, Stoffers DA, Simmons RA (2011) Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia 54(10):2606–2614. https://doi.org/10.1007/s00125-011-2250-1
Marino-Ramirez L, Kann MG, Shoemaker BA, Landsman D (2005) Histone structure and nucleosome stability. Expert Rev Proteom 2(5):719–729. https://doi.org/10.1586/14789450.2.5.719
Greer EL, Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13(5):343–357. https://doi.org/10.1038/nrg3173
Sakabe K, Wang Z, Hart GW (2010) Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci USA 107(46):19915–19920. https://doi.org/10.1073/pnas.1009023107
Lee DY, Hayes JJ, Pruss D, Wolffe AP (1993) A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72(1):73–84. https://doi.org/10.1016/0092-8674(93)90051-q
Strahl BD, Ohba R, Cook RG, Allis CD (1999) Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci USA 96(26):14967–14972. https://doi.org/10.1073/pnas.96.26.14967
Zhang T, Cooper S, Brockdorff N (2015) The interplay of histone modifications—writers that read. EMBO Rep 16(11):1467–1481. https://doi.org/10.15252/embr.201540945
Dai Y, Ghosh S, Shin BC, Devaskar SU (2019) Role of microRNA-122 in hepatic lipid metabolism of the weanling female rat offspring exposed to prenatal and postnatal caloric restriction. J Nutr Biochem 73:108220. https://doi.org/10.1016/j.jnutbio.2019.108220
Dumortier O, Hinault C, Gautier N, Patouraux S, Casamento V, Van Obberghen E (2014) Maternal protein restriction leads to pancreatic failure in offspring: role of misexpressed microRNA-375. Diabetes 63(10):3416–3427. https://doi.org/10.2337/db13-1431
Ferland-McCollough D, Fernandez-Twinn DS, Cannell IG, David H, Warner M, Vaag AA, Bork-Jensen J, Brons C, Gant TW, Willis AE, Siddle K, Bushell M, Ozanne SE (2012) Programming of adipose tissue miR-483-3p and GDF-3 expression by maternal diet in type 2 diabetes. Cell Death Differ 19(6):1003–1012. https://doi.org/10.1038/cdd.2011.183
Sanchez-Parra C, Jacovetti C, Dumortier O, Lee K, Peyot ML, Guay C, Prentki M, Laybutt DR, Van Obberghen E, Regazzi R (2018) Contribution of the long noncoding RNA H19 to beta-cell mass expansion in neonatal and adult rodents. Diabetes 67(11):2254–2267. https://doi.org/10.2337/db18-0201
Zheng J, Xiao X, Zhang Q, Wang T, Yu M, Xu J (2017) Maternal low-protein diet modulates glucose metabolism and hepatic MicroRNAs expression in the early life of offspring dagger. Nutrients. https://doi.org/10.3390/nu9030205
Fernandez-Twinn DS, Alfaradhi MZ, Martin-Gronert MS, Duque-Guimaraes DE, Piekarz A, Ferland-McCollough D, Bushell M, Ozanne SE (2014) Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol Metab 3(3):325–333. https://doi.org/10.1016/j.molmet.2014.01.007
Zhang J, Wang C, Terroni PL, Cagampang FR, Hanson M, Byrne CD (2005) High-unsaturated-fat, high-protein, and low-carbohydrate diet during pregnancy and lactation modulates hepatic lipid metabolism in female adult offspring. Am J Physiol Regul Integr Comp Physiol 288(1):R112–118. https://doi.org/10.1152/ajpregu.00351.2004
Zhang J, Zhang F, Didelot X, Bruce KD, Cagampang FR, Vatish M, Hanson M, Lehnert H, Ceriello A, Byrne CD (2009) Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genom 10:478. https://doi.org/10.1186/1471-2164-10-478
Benatti RO, Melo AM, Borges FO, Ignacio-Souza LM, Simino LA, Milanski M, Velloso LA, Torsoni MA, Torsoni AS (2014) Maternal high-fat diet consumption modulates hepatic lipid metabolism and microRNA-122 (miR-122) and microRNA-370 (miR-370) expression in offspring. Br J Nutr 111(12):2112–2122. https://doi.org/10.1017/s0007114514000579
de Paula Simino LA, de Fante T, Figueiredo Fontana M, Oliveira Borges F, Torsoni MA, Milanski M, Velloso LA, Souza Torsoni A (2017) Lipid overload during gestation and lactation can independently alter lipid homeostasis in offspring and promote metabolic impairment after new challenge to high-fat diet. Nutr Metab (Lond) 14:16. https://doi.org/10.1186/s12986-017-0168-4
Zheng J, Zhang Q, Mul JD, Yu M, Xu J, Qi C, Wang T, Xiao X (2016) Maternal high-calorie diet is associated with altered hepatic microRNA expression and impaired metabolic health in offspring at weaning age. Endocrine 54(1):70–80. https://doi.org/10.1007/s12020-016-0959-9
Wang H, Chen Y, Mao X, Du M (2019) Maternal obesity impairs fetal mitochondriogenesis and brown adipose tissue development partially via upregulation of miR-204-5p. Biochim Biophys Acta Mol Basis Dis 1865(10):2706–2715. https://doi.org/10.1016/j.bbadis.2019.07.012
Wei JW, Huang K, Yang C, Kang CS (2017) Non-coding RNAs as regulators in epigenetics (Review). Oncol Rep 37(1):3–9. https://doi.org/10.3892/or.2016.5236
Weinberg RA, Penman S (1968) Small molecular weight monodisperse nuclear RNA. J Mol Biol 38(3):289–304. https://doi.org/10.1016/0022-2836(68)90387-2
Knight E Jr, Darnell JE (1967) Distribution of 5 s RNA in HeLa cells. J Mol Biol 28(3):491–502. https://doi.org/10.1016/s0022-2836(67)80099-8
Diamantopoulos MA, Tsiakanikas P, Scorilas A (2018) Non-coding RNAs: the riddle of the transcriptome and their perspectives in cancer. Ann Transl Med 6(12):241. https://doi.org/10.21037/atm.2018.06.10
Holoch D, Moazed D (2015) RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16(2):71–84. https://doi.org/10.1038/nrg3863
Bartolomei MS, Webber AL, Brunkow ME, Tilghman SM (1993) Epigenetic mechanisms underlying the imprinting of the mouse H19 gene. Genes Dev 7(9):1663–1673. https://doi.org/10.1101/gad.7.9.1663
Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, Lu L, Liu C, Yi JS, Zhang H, Min W, Bennett AM, Gregory RI, Ding Y, Huang Y (2013) The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell 52(1):101–112. https://doi.org/10.1016/j.molcel.2013.08.027
Goyal N, Kesharwani D, Datta M (2018) Lnc-ing non-coding RNAs with metabolism and diabetes: roles of lncRNAs. Cell Mol Life Sci 75(10):1827–1837. https://doi.org/10.1007/s00018-018-2760-9
Guay C, Regazzi R (2013) Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat Rev Endocrinol 9(9):513–521. https://doi.org/10.1038/nrendo.2013.86
Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M (2004) A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432(7014):226–230. https://doi.org/10.1038/nature03076
Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, Zavolan M, Stoffel M (2009) miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci USA 106(14):5813–5818. https://doi.org/10.1073/pnas.0810550106
Erener S, Mojibian M, Fox JK, Denroche HC, Kieffer TJ (2013) Circulating miR-375 as a biomarker of beta-cell death and diabetes in mice. Endocrinology 154(2):603–608. https://doi.org/10.1210/en.2012-1744
Zhu X, Wu YB, Zhou J, Kang DM (2016) Upregulation of lncRNA MEG3 promotes hepatic insulin resistance via increasing FoxO1 expression. Biochem Biophys Res Commun 469(2):319–325. https://doi.org/10.1016/j.bbrc.2015.11.048
Yan C, Chen J, Chen N (2016) Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP-1c protein stability. Sci Rep 6:22640. https://doi.org/10.1038/srep22640
Sferruzzi-Perri AN, Camm EJ (2016) The programming power of the placenta. Front Physiol 7:33. https://doi.org/10.3389/fphys.2016.00033
Burton GJ, Fowden AL, Thornburg KL (2016) Placental origins of chronic disease. Physiol Rev 96(4):1509–1565. https://doi.org/10.1152/physrev.00029.2015
Myatt L, Thornburg KL (2018) Effects of prenatal nutrition and the role of the placenta in health and disease. Methods Mol Biol 1735:19–46. https://doi.org/10.1007/978-1-4939-7614-0_2
Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ (2010) Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 467(7318):963–966. https://doi.org/10.1038/nature09491
Watkins AJ, Dias I, Tsuro H, Allen D, Emes RD, Moreton J, Wilson R, Ingram RJM, Sinclair KD (2018) Paternal diet programs offspring health through sperm- and seminal plasma-specific pathways in mice. Proc Natl Acad Sci USA 115(40):10064–10069. https://doi.org/10.1073/pnas.1806333115
Chang RC, Wang H, Bedi Y, Golding MC (2019) Preconception paternal alcohol exposure exerts sex-specific effects on offspring growth and long-term metabolic programming. Epigenet Chromatin 12(1):9. https://doi.org/10.1186/s13072-019-0254-0
Sharp GC, Lawlor DA (2019) Paternal impact on the life course development of obesity and type 2 diabetes in the offspring. Diabetologia 62(10):1802–1810. https://doi.org/10.1007/s00125-019-4919-9
Soubry A, Hoyo C, Jirtle RL, Murphy SK (2014) A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. BioEssays 36(4):359–371. https://doi.org/10.1002/bies.201300113
Dupont C, Kappeler L, Saget S, Grandjean V, Levy R (2019) Role of miRNA in the transmission of metabolic diseases associated with paternal diet-induced obesity. Front Genet 10:337. https://doi.org/10.3389/fgene.2019.00337
Rando OJ (2012) Daddy issues: paternal effects on phenotype. Cell 151(4):702–708. https://doi.org/10.1016/j.cell.2012.10.020
McPherson NO, Fullston T, Aitken RJ, Lane M (2014) Paternal obesity, interventions, and mechanistic pathways to impaired health in offspring. Ann Nutr Metab 64(3–4):231–238. https://doi.org/10.1159/000365026
Sales VM, Ferguson-Smith AC, Patti ME (2017) Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab 25(3):559–571. https://doi.org/10.1016/j.cmet.2017.02.016
Blake GE, Watson ED (2016) Unravelling the complex mechanisms of transgenerational epigenetic inheritance. Curr Opin Chem Biol 33:101–107. https://doi.org/10.1016/j.cbpa.2016.06.008
Roseboom TJ, Watson ED (2012) The next generation of disease risk: are the effects of prenatal nutrition transmitted across generations? Evidence from animal and human studies. Placenta 33(Suppl 2):e40–44. https://doi.org/10.1016/j.placenta.2012.07.018
Khorram O, Keen-Rinehart E, Chuang TD, Ross MG, Desai M (2015) Maternal undernutrition induces premature reproductive senescence in adult female rat offspring. Fertil Steril 103(1):291–298. https://doi.org/10.1016/j.fertnstert.2014.09.026
Leonhardt M, Lesage J, Croix D, Dutriez-Casteloot I, Beauvillain JC, Dupouy JP (2003) Effects of perinatal maternal food restriction on pituitary-gonadal axis and plasma leptin level in rat pup at birth and weaning and on timing of puberty. Biol Reprod 68(2):390–400. https://doi.org/10.1095/biolreprod.102.003269
Sloboda DM, Howie GJ, Pleasants A, Gluckman PD, Vickers MH (2009) Pre- and postnatal nutritional histories influence reproductive maturation and ovarian function in the rat. PLoS ONE 4(8):e6744. https://doi.org/10.1371/journal.pone.0006744
Sathishkumar K, Elkins R, Yallampalli U, Yallampalli C (2009) Protein restriction during pregnancy induces hypertension and impairs endothelium-dependent vascular function in adult female offspring. J Vasc Res 46(3):229–239. https://doi.org/10.1159/000166390
Molendi-Coste O, Grumolato L, Laborie C, Lesage J, Maubert E, Ghzili H, Vaudry H, Anouar Y, Breton C, Vieau D (2006) Maternal perinatal undernutrition alters neuronal and neuroendocrine differentiation in the rat adrenal medulla at weaning. Endocrinology 147(6):3050–3059. https://doi.org/10.1210/en.2005-1331
Li Y (2018) Epigenetic mechanisms link maternal diets and gut microbiome to obesity in the offspring. Front Genet 9:342. https://doi.org/10.3389/fgene.2018.00342
Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, Wen L (2015) Maternal antibiotic treatment protects offspring from diabetes development in nonobese diabetic mice by generation of tolerogenic APCs. J Immunol 195(9):4176–4184. https://doi.org/10.4049/jimmunol.1500884
Turner N, Robker RL (2015) Developmental programming of obesity and insulin resistance: does mitochondrial dysfunction in oocytes play a role? Mol Hum Reprod 21(1):23–30. https://doi.org/10.1093/molehr/gau042
Jia Y, Li R, Cong R, Yang X, Sun Q, Parvizi N, Zhao R (2013) Maternal low-protein diet affects epigenetic regulation of hepatic mitochondrial DNA transcription in a sex-specific manner in newborn piglets associated with GR binding to its promoter. PLoS ONE 8(5):e63855. https://doi.org/10.1371/journal.pone.0063855
Calatayud M, Koren O, Collado MC (2019) Maternal microbiome and metabolic health program microbiome development and health of the offspring. Trends Endocrinol Metab 30(10):735–744. https://doi.org/10.1016/j.tem.2019.07.021
McMullen S, Langley-Evans SC, Gambling L, Lang C, Swali A, McArdle HJ (2012) A common cause for a common phenotype: the gatekeeper hypothesis in fetal programming. Med Hypotheses 78(1):88–94. https://doi.org/10.1016/j.mehy.2011.09.047
Tsunoda Y, Tokunaga T, Sugie T (1985) Altered sex ratio of live young after transfer of fast-and slow-developing mouse embryos. Gamete Res 12(3):301–304
Howerton CL, Morgan CP, Fischer DB, Bale TL (2013) O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci USA 110(13):5169–5174. https://doi.org/10.1073/pnas.1300065110
Montano MM, Wang MH, vom Saal FS (1993) Sex differences in plasma corticosterone in mouse fetuses are mediated by differential placental transport from the mother and eliminated by maternal adrenalectomy or stress. J Reprod Fertil 99(2):283–290. https://doi.org/10.1530/jrf.0.0990283
Gasparini SJ, Swarbrick MM, Kim S, Thai LJ, Henneicke H, Cavanagh LL, Tu J, Weber MC, Zhou H, Seibel MJ (2019) Androgens sensitise mice to glucocorticoid-induced insulin resistance and fat accumulation. Diabetologia 62(8):1463–1477. https://doi.org/10.1007/s00125-019-4887-0
Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, Onishi T, Sugiura T, Shizuta Y (2003) Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol 176(2):237–246. https://doi.org/10.1677/joe.0.1760237
Weinberg J, Sliwowska JH, Lan N, Hellemans KG (2008) Prenatal alcohol exposure: foetal programming, the hypothalamic-pituitary-adrenal axis and sex differences in outcome. J Neuroendocrinol 20(4):470–488. https://doi.org/10.1111/j.1365-2826.2008.01669.x
Aiken CE, Ozanne SE (2013) Sex differences in developmental programming models. Reproduction 145(1):R1–13. https://doi.org/10.1530/rep-11-0489
O'Dowd R, Kent JC, Moseley JM, Wlodek ME (2008) Effects of uteroplacental insufficiency and reducing litter size on maternal mammary function and postnatal offspring growth. Am J Physiol Regul Integr Comp Physiol 294(2):R539–548. https://doi.org/10.1152/ajpregu.00628.2007
Briffa JF, O'Dowd R, Romano T, Muhlhausler BS, Moritz KM, Wlodek ME (2019) Reducing pup litter size alters early postnatal calcium homeostasis and programs adverse adult cardiovascular and bone health in male rats. Nutrients. https://doi.org/10.3390/nu11010118
Leiter EH (2009) Selecting the "right" mouse model for metabolic syndrome and type 2 diabetes research. Methods Mol Biol 560:1–17. https://doi.org/10.1007/978-1-59745-448-3_1
Acknowledgements
E. R. C. is funded by a Cambridge Rutherford PhD Scholarship from the Cambridge Trust and Rutherford Foundation and A. N. S.-P is supported by a Royal Society Dorothy Hodgkin Research Fellowship (RG74249), Lister Institute Research Prize (RG93692) and Medical Research Council New Investigator Grant (RG93186).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Christoforou, E.R., Sferruzzi-Perri, A.N. Molecular mechanisms governing offspring metabolic programming in rodent models of in utero stress. Cell. Mol. Life Sci. 77, 4861–4898 (2020). https://doi.org/10.1007/s00018-020-03566-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03566-z