
Abstract
With the advance in chromatin immunoprecipitation followed by high-throughput sequencing, there has been a dramatic increase in our understanding of distal enhancer function. In the developing heart, the identification and characterisation of such enhancers have deepened our knowledge of the mechanisms of transcriptional regulation that drives cardiac differentiation. With next-generation sequencing techniques becoming widely accessible, the quantity of data describing the genome-wide distribution of cardiac-specific transcription factor and chromatin modifiers has rapidly increased and it is now becoming clear that the usage of enhancers is highly dynamic and complex, both during the development and in the adult. The identification of those enhancers has revealed new insights into the transcriptional mechanisms of how tissue-specific gene expression patterns are established, maintained, and change dynamically during development and upon physiological stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The development of the four-chambered heart involves coordinate differentiation of multiple cardiac cell types, such changes occurring, whilst the embryonic organ maintains its essential function pumping blood through the developing embryo [1–3]. The transition between different differentiation states of cardiac progenitors during heart morphogenesis is the result of a tightly coordinated process, regulated at the transcriptional level. In that context, the non-coding part of the genome has recently been shown to encode a large collection of enhancers and associated non-coding RNAs (for example micro RNAs or long non-coding RNAs) with regulatory functions within the gene regulatory network driving heart development [4, 5]. Distant enhancers control the dynamic expression of cardiac-specific genes and any disruption of such controls can lead to a large variety of congenital heart defects (CHD). CHD are the most common type of birth defect, affecting 10 % live births [6, 7]. Moreover, mutations within non-coding elements, such as enhancers, have been shown to disrupt heart development [8–10]. Our increased understanding of the transcriptional processes occurring during cardiac development has spurred efforts to identify the mechanisms that would allow the generation of cardiomyocytes in vitro in numbers large enough for therapeutic use. Efforts have focused either on differentiating embryonic stem cells, or using induced pluripotent stem cells, terminally differentiated cells which have been reprogrammed through the introduction of a defined set of genes [11–13].
After describing the common strategies used to identify cardiac enhancers and the difficulty in the design and interpretation of ChIP-seq experiments, we will summarise the state of our knowledge on the dynamics of enhancer usage during the cardiac development and differentiation.
Identification of cardiac enhancers
Gene expression is regulated through the integrated action of different types of cis-regulatory elements, such as distal enhancers [14, 15]. Classically, enhancers are defined as non-coding DNA elements that can increase the transcription of genes located in cis. With a significant role in regulating gene expression, the identification and location in the genome of those enhancers have been the focus of many studies in the cardiovascular development [16].
However, by their nature, the identification of such enhancers is challenging. For example, their location relative to the target gene, or genes, is highly variable and they can function in an orientation-independent manner. Prior to the development of high-throughput sequencing methods, enhancers were largely identified by comparative genomics, with the assumption that non-coding sequence which is conserved between different vertebrate or mammalian species is enriched for enhancers [17, 18]. Using this method in combination with functional assays, it was found that about half of the identified, highly conserved sequences are, indeed, functionally active enhancers. However, this approach has some important limitations. Some conserved sequences have no apparent enhancer function when assayed in transgenic mice. Moreover, it is now clear that a substantial fraction of cardiac enhancers displays modest or no conservation across species [19–22].
With advances in high-throughput sequencing techniques, new approaches have been developed to identify gene enhancers [23]. Two types of strategies have been used to identify cardiac enhancers: genome-wide distribution of key cardiac transcription factors and the identification of appropriate epigenomic marks [16]. Each strategy utilises ChIP-seq (chromatin immunoprecipitation followed by high-throughput sequencing) and is independent of cross-species sequence conservation. Both have proved powerful in identifying cardiac enhancers, but both have important limitations that we will summarise.
Experimental design and data analysis challenges
The genome-wide distribution of post-translational histone modifications of chromatin, (or “chromatin marks”) has widely been used to predict enhancer activity [24]. The identification of de novo mutations in histone-modifying genes in CHD emphasizes the importance of chromatin modification in cardiac differentiation [25]. The protocols used to identify such marks are now well established and multiple different marks have been identified, for example, H3K4me1 correlates with gene promoters, H3K4me3 with active promoters, H3k27me3 with inactive promoters, and H3K27ac with some active enhancers. Importantly, there is no single chromatin mark which can be used to identify all active enhancers. Furthermore, new histone modifications correlating with the presence of new classes of active enhancers are identified regularly [26].
Another difficulty with this approach is the impact of tissue contamination when ChIP-seq is undertaken with cardiac tissue or with the whole organ. The heart contains a large number of different cell types, (cardiomyocytes, fibroblasts, endothelial and smooth muscle cells, and to name a few) and their precise relative contributions have been the subject of numerous debates [27]. Studies of chromatin marks using cardiac tissue will, therefore, identify an assortment of enhancer activities reflecting the contribution of multiple cell types. Single-cell technology offers a potential answer, but there are obvious technical difficulties with the use of such small quantities of starting material and no such study has yet been reported for cardiac cells [28, 29]. Indeed, one limitation of the ChIP-seq technique is the relatively large amount of initial material required, due to low cellular DNA recovery rate. In a single-cell experiment, non-specific binding of the antibody during the immunoprecipitation step leads to experimental noise. New methods are currently being developed to address this specific problem [30]. For instance, microfluidic technologies have been developed to isolate single cells into aqueous droplets, in which the chromatin of individual cells is labelled with a unique oligonucleotide barcode which can subsequently be tracked. The tagged chromatin of hundred of cells is then combined and undergoes the ChIP-seq procedure, allowing reduction in noise associated with small input DNA.
Whilst the presence of chromatin marks has been shown to be a powerful tool to identify enhancers, localisation of key lineage-specific transcription factors has, in some cases, been shown to be a better predictor of enhancer activity [31]. The genome-wide distribution of those transcription factors is not only of particular importance for identifying their direct, downstream transcriptional targets, but it also facilitates the identification of tissue-specific enhancers. One significant advantage of using transcription factor binding is in their potential cell specificity. If the transcription factor is expressed in a cell-type specific manner, for example, only in cardiomyocytes, cell-type contamination will not affect in vivo analysis.
A large number of transcription factors have been shown to be necessary for mouse heart development [32, 33]. Moreover, human mutations for most of those factors have been suggested as the underlying cause of a variety of CHD [34]. Numerous studies have recently addressed the genome-wide distribution of such key, cardiac transcription factors (Table 1) [10, 20, 35–45]. What is striking is that comparison of results reveals wide discrepancies in the number of occupied sites identified in different studies, not only between factors but also for the same factor at different stages (Table 1). It is not yet clear whether such differences are biologically relevant, or simply reflects differences in the methodology used, the data analysis methods chosen, and differences in the precise biological model. What is clear is that a large number of parameters can affect the identification of binding regions [46].
Many technical and methodological factors will affect both the outcome of a ChIP-seq experiment and the extent to which it can be compared with the other studies. The quality of the primary antibody used to recognise the transcription factor has been shown to be crucial, and epitope-tagging has been used to address problems caused by antibody variation and cross reactivity. This can have dramatic results on ChIP-seq results. Comparison of data obtained with an antibody against GATA4 versus a flag-tagged GATA4 shows a dramatic increase in the number of binding events (or “peaks”) from 11,915 to 43,800 [20].
The number of biological replicates can also dramatically affect the number of significant peaks identified. Pioneering studies have largely used only a single biological sample, no doubt partly due to the novelty of the technique and partly the cost of sequencing (Table 1). However, as with all biological analyses, multiple biological replicates have proved to be necessary [46]. Interestingly, increasing from two to three replicates does not appear to have significantly enhanced the quality of results [46].
The number of sequence reads has been shown to be critical for the identification of relevant peaks [47]. The number of identified binding sites increases with sequencing depth, since weaker binding sites become statistically significant with a greater number of reads [48]. Use of an appropriate control data set has also been shown to be critical [49]. This generally comprises “Input”’ DNA, (DNA prepared under the same conditions as the immunoprecipitated DNA) or an “IgG” control, (a ChIP reaction performed with an unrelated antibody). The optimum choice remains under discussion [50].
With the identification of thousands of putative enhancers, limitations enforced by the need for experimental validation of their function. Transgenic mouse reporter assays are the most commonly used approach to evaluate the function of enhancers in vivo. Such experiments consist in delivering a linear plasmid containing a reporter gene (for example, LacZ or GFP) linked to the putative enhancer into the mouse zygote, through pronuclear injection [51]. The transgenic embryos generated, generally in a transient manner, are assessed for the spatial expression of the reporter gene in the whole embryo. However, if that technique is a powerful way to identify qualitative enhancer activity, copy number, and position effect due to random transgene insertion impede reliable quantitative analysis. Moreover, mouse transgenic experiments are clearly not an appropriate technique for high-throughput testing of enhancers identified with ChIP-seq, due to their relatively high cost. Furthermore, in the context of dynamic studies, in which an enhancer has to be tested at different stages of development and/or different conditions of biological stress, this approach becomes extremely time-consuming. Stable transgenic lines would most likely need to be generated and studied in different conditions, significantly increasing the time of study as well as escalating an already high cost.
In summary, despite technological improvements which have facilitated wider adoption of the technique, design and analysis of ChIP-seq experiments remain challenging and different choices can often lead to conflicting results [52].
Dynamic of enhancers during cardiac development
If the number of studies describing the identification of cardiac enhancers has been relatively high, the number of studies describing the dynamics of those enhancers—their differential usage between stages of development and/or in disease conditions—has been relatively poor.
Early studies established that even if evolutionary sequence conservation is a powerful tool to identify enhancers, fewer than 2 % identified in this way are active in the heart [17, 18]. Location of the transcriptional co-activator p300 proved to be to be a powerful predictor of enhancer activity in brain and limb [53] and this was, therefore, used in the mouse heart at embryonic day 11 (E11.5) [19]. Results demonstrated that most candidate cardiac enhancers identified by p300 binding are less conserved in vertebrate evolution than those in limb or brain [19].
Comparison of p300 binding between the foetal and adult human heart shows that a large proportion of the enhancers identified in this way are highly dynamic during heart development [21]. Indeed, 48 % of human cardiac enhancers active in the adult are also identified in foetal hearts, whilst only 21 % of foetal cardiac enhancers are identified with adult tissue [21]. Similar results are obtained when comparing the embryonic (E14.5) and the adult mouse heart, using the chromatin marks H3K4me1 or H3K27ac [54]. Such dynamic enhancer usage appears to reflect distinct biological function; enhancers specific to embryonic stages are associated with genes expressed during cardiac differentiation, whilst adult enhancers are associated with genes important for adult heart function [54].
Interestingly, comparison of enhancers (identified through p300 binding) between the postnatal day 2 mouse heart and the approximately equivalent 16-week-old human foetal heart shows only a 21 % overlap. This suggests a considerable and, perhaps, unexpected degree of species specificity for cardiac enhancers [21]. Since even poorly conserved human enhancers are able to drive cardiac expression in a mouse cardiac transgenic assay, even if enhancer usage between species is different, the cardiac transcription factors that regulate those enhancers are most probably the same [21].
Similarly dynamic activity of cardiac enhancers is found comparing the chromatin mark H3K27ac at different stages of mouse heart development (E11.5, E14.5, E17.5, P0, P7, P21, and P56) [22]. Indeed, only 3 % of cardiac enhancers identified in this way are predicted to be active at all stages examined. That extensive and fast turnover is illustrated with only 45 % identified actives in at most two consecutive time points [22]. The large majority of cardiac enhancers identified in this way are predicted to have a highly restricted, temporal window of activity during development, which, in turn, suggests that dynamic developmental processes are regulated by the transient activities of such enhancers.
Interestingly, substantial differences are observed in sequence conservation of putative enhancers within a given tissue across time points and also across tissues at the same timepoint [22]. As mentioned above, heart enhancers show relatively weak conservation compared with enhancers identified in either liver or more dramatically, forebrain. However, it is interesting to note that this conservation is not constant during development; enhancer conservation is maximal at early stage of heart development when compared with enhancers active in the adult heart [22].
As with heart development, enhancer usage during the differentiation of ESCs into cardiomyocytes is also highly dynamic [55]. Moreover, the distribution of four different chromatin marks at four different stages of ESC differentiation (ESC, mesoderm, cardiac progenitor, and cardiomyocytes) shows that genes with similar expression patterns can show substantial variation in chromatin states during cardiac differentiation. These results suggest that histone mark profile is not very useful for predicting dynamics of gene expression [55].
Dynamic enhancer usage is also observed when the binding of cardiac transcription factors, such as GATA4, is used to identify heart enhancers [20]. 66.5 % of the adult GATA4-bound regions are occupied by GATA4 in the E12.5 foetal heart, whilst 80.1 % of foetal GATA4 regions are not bound by GATA4 in the adult heart. Those proportions are similar to those found by assaying chromatin marks, with again, a much larger number of enhancers predicted to be active during development than in the adult. Furthermore, the authors also noted that GATA4 binding, which is mainly distal to the transcription start site (TSS) in the embryonic mouse heart, is shifted more proximal to the TSS in the adult heart.
Foetal-specific, shared, and adult-specific GATA4 regions were associated with different co-enriched-binding sites, with, for example, MEF2 and TEAD1 motifs enriched in foetal-specific regions, whilst adult regions were enriched for the EGR-1 motif. The functional consequence of such association remains undocumented.
GATA4 binding is not only changed dynamically between embryonic development and the adult heart, but also in the pathological state of cardiac hypertrophy [20], a condition in which GATA4 is well known to play a role [56]. When hypertrophy of the heart is induced by pressure overload, only 49 % of the GATA4 bound regions are found shared between ascending aortic band and the sham condition [20]. In pathological stress, such as pressure overload, reactivation of a foetal gene expression programme has been suggested to occur [57]. However, if there is clearly a redistribution of GATA4 binding under pathological stress, the redistribution to foetal-specific enhancers is relatively small. Indeed, only 20.5 % of stress-induced GATA4 binding was on foetal regions, whilst 40 % was to regions bound in neither foetal nor the adult heart [20]. These results suggest that under pathological hypertrophy, rather than reallocation to foetal enhancers, GATA4 binds to a set of stress-specific, cardiac enhancers. Similar results are found with the regulation of Nppa–Nppb gene cluster during cardiac development and hypertrophy [58]. Those two genes are induced by stress and are well-described clinical markers of heart failure. In that study, the distribution of H3K27ac and the association of Pol2 across the locus reveal that foetal expression and stress-induced Nppa expression is dependent on different enhancers, again suggesting that the foetal and stress transcriptional regulatory networks are different [58].
As observed in vivo, the binding pattern of GATA4, NKX2–5, and TBX5 is all highly dynamic during ESC differentiation into cardiomyocytes [40]. During the transition of cardiac progenitors to cardiomyocytes in vitro, more than 50 % of the binding events occur at a single, specific stage [40]. Moreover, the same study highlights the extensive co-binding of these cardiac transcription factors on common enhancers regulating cardiac gene expression. Interestingly, in a null background for NKX2–5, TBX5, or both, a significant ectopic binding of the remaining factors is observed [40]. This suggests that interdependent binding may also be necessary to prevent transcription factors from distributing ectopically and activating lineage-inappropriate genes. Such a mechanism of ectopic reallocation of transcription factor binding is also observed when the genome distribution of the transcription factor NKX2–5 is compared with that of a CHD-associated, mutated form of the protein [35]. If NKX2–5 mutants’ proteins fail to bind the majority of its wild-type targets in HL-1 cells, they are still able to recognise a large number of them along with a unique set of ectopic sites [35]. The significance of those ectopic sites of binding in the genome is not yet clear, but in the context of CHD, they could affect transcription in either a positive or dominant negative manner [35].
Extensive co-binding of NKX2–5 has also been observed with both TBX5 and SHOX2 [45]. In the E12.5 mouse heart, nearly 80 % of NKX2–5-bound regions overlap with SHOX2 binding regions [45]. Extending that co-occupancy analysis with a TBX5 data set published previously [37], the authors found that 67 % of the SHOX2–NKX2–5 co-occupied sites are also occupied by TBX5 [45]. Such significant co-occupancy of enhancers is consistent with observations indicating an antagonising role of SHOX2 on the transcriptional output of NKX2–5 in the pulmonary vein of the developing heart [45].
Our own efforts to identify active cardiac enhancers in the E11.5 mouse embryonic heart demonstrate that, at least at this stage, this is achieved more effectively using NKX2–5 binding than by p300 binding [19, 36]. 83 % of enhancers enriched for NKX2–5 binding versus 53 % for those enriched for p300 drive expression in the mouse heart. This increase in efficiency is mainly due to a reduction in the number of false positives. This suggests that binding of a critical cardiac transcription factor is an efficient predictor of cardiac enhancer activity, an observation consistent with the previous findings in equivalent studies using blood cells [31].
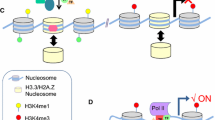
Sequence analysis of NKX2–5-bound regions suggests that NKX2–5 and MEIS1 share an overlapping-binding site which is present in a number of cardiac enhancers in vivo (Fig. 1). Interestingly, these two transcription factors are sequentially expressed in the differentiating cardiac progenitors of the embryo. As cardiac progenitor mature, they experience successively high levels of MEIS1 in cardiac progenitors, followed by high levels of NKX2–5 in differentiated cardiomyocytes. This suggests a simple mechanism of transcriptional regulation, in which one factor successively replaces the other at cardiac enhancer harbouring the shared-binding site (Fig. 1).

Mechanism of transcriptional regulation by successive binding of MEIS1 and NKX2–5 on cardiac enhancers. NKX2–5 and MEIS1 are able to bind in vitro on a DNA motif (GTGNTGACAG) which is an overlapping-binding site for the two transcription factors. As cardiac progenitors differentiate in the mouse embryo, they successively experience high levels of MEIS1 expression in the secondary heart field (grey) followed by high levels of NKX2–5 expression in the heart tube (red). Cardiac enhancers with an overlapping-binding site for MEIS1 and NKX2–5 are bound successively by those factors
The mesodermal core of the branchial arches contains a population of cardiac progenitor cells which populate the outflow tract of the developing heart [59]. Strikingly, nearly 30 % of NKX2–5-bound regions are also bound by MEIS in the first branchial arch [60], suggesting that this mechanism can be extended to an entire subset of cardiac enhancers. A similar mechanism of transcriptional regulation through an overlapping-binding site has been described for the gene Fgf10 [61]. A cardiac enhancer of that gene is successively bound by ISL1 and NKX2–5 [61]. That Fgf10 enhancer is activated in cardiac progenitors by high levels of ISL1 and is repressed in the myocardium by high levels of NKX2–5, which is likely to act both directly and indirectly through Isl1 suppression [61]. Taken together, these findings suggest that overlapping transcription factor-binding sites in cardiac enhancers might be of broad significance during the differentiation of cardiac progenitors. Moreover, such a mechanism of replacement of one transcription factor by another has been found in other systems. When ESCs become specified to form neural precursors and subsequently differentiate into neurons, an ordered and sequential binding of Sox2, Sox3, and Sox11 to target enhancers drive neurogenesis. In this case, Sox2 binding would first preselect neural genes in ES cells, ensuring their proper activation in neural precursors and then inducing neuronal differentiation [62].
Conclusion
Recent progress in ChIP-seq techniques has allowed great advances in the systemic identification of cardiac enhancers active during cardiac development, as well as in the normal and pathological adult heart. With the decrease in cost and improvement in new technologies, such as single-cell studies, identification of such enhancers in different pathological conditions as well as a more detailed spatial resolution in the mouse embryonic heart should lead to a better understanding of the gene regulatory network responsible for cardiovascular development and differentiation.
References
Buckingham M, Meilhac S, Zaffran S (2005) Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 6(11):826–835. doi:10.1038/nrg1710
Miquerol L, Kelly RG (2013) Organogenesis of the vertebrate heart. Wiley Interdiscip Rev Dev Biol 2(1):17–29. doi:10.1002/wdev.68
Moorman AF, Christoffels VM (2003) Cardiac chamber formation: development, genes, and evolution. Physiol Rev 83(4):1223–1267. doi:10.1152/physrev.00006.2003
Thum T, Condorelli G (2015) Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res 116(4):751–762. doi:10.1161/CIRCRESAHA.116.303549
Wronska A, Kurkowska-Jastrzebska I, Santulli G (2015) Application of microRNAs in diagnosis and treatment of cardiovascular disease. Acta Physiol (Oxf) 213(1):60–83. doi:10.1111/apha.12416
Fahed AC, Gelb BD, Seidman JG, Seidman CE (2013) Genetics of congenital heart disease: the glass half empty. Circ Res 112(4):707–720. doi:10.1161/CIRCRESAHA.112.300853
Nichols M, Townsend N, Scarborough P, Rayner M (2014) Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J 35(42):2950–2959. doi:10.1093/eurheartj/ehu299
Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA (2012) Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet 21(14):3255–3263. doi:10.1093/hmg/dds165
van den Boogaard M, Smemo S, Burnicka-Turek O, Arnolds DE, van de Werken HJ, Klous P, McKean D, Muehlschlegel JD, Moosmann J, Toka O, Yang XH, Koopmann TT, Adriaens ME, Bezzina CR, de Laat W, Seidman C, Seidman JG, Christoffels VM, Nobrega MA, Barnett P, Moskowitz IP (2014) A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Investig 124(4):1844–1852. doi:10.1172/JCI73140
van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, t Hoen PA, Bakkers J, Barnett P, Christoffels VM (2012) Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest 122(7):2519–2530. doi:10.1172/JCI62613
Madonna R, Van Laake LW, Davidson SM, Engel FB, Hausenloy DJ, Lecour S, Leor J, Perrino C, Schulz R, Ytrehus K, Landmesser U, Mummery CL, Janssens S, Willerson J, Eschenhagen T, Ferdinandy P, Sluijter JP (2016) Position Paper of the European Society of Cardiology Working Group Cellular Biology of the Heart: cell-based therapies for myocardial repair and regeneration in ischemic heart disease and heart failure. Eur Heart J. doi:10.1093/eurheartj/ehw113
Sadahiro T, Yamanaka S, Ieda M (2015) Direct cardiac reprogramming: progress and challenges in basic biology and clinical applications. Circ Res 116(8):1378–1391. doi:10.1161/CIRCRESAHA.116.305374
Srivastava D, Yu P (2015) Recent advances in direct cardiac reprogramming. Curr Opin Genet Dev 34:77–81. doi:10.1016/j.gde.2015.09.004
Davidson EH (2010) Emerging properties of animal gene regulatory networks. Nature 468(7326):911–920. doi:10.1038/nature09645
Spitz F, Furlong EE (2012) Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13(9):613–626. doi:10.1038/nrg3207
Kathiriya IS, Nora EP, Bruneau BG (2015) Investigating the transcriptional control of cardiovascular development. Circ Res 116(4):700–714. doi:10.1161/CIRCRESAHA.116.302832
Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, Plajzer-Frick I, Akiyama J, De Val S, Afzal V, Black BL, Couronne O, Eisen MB, Visel A, Rubin EM (2006) In vivo enhancer analysis of human conserved non-coding sequences. Nature 444(7118):499–502. doi:10.1038/nature05295
Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Afzal V, Rubin EM, Pennacchio LA (2008) Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet 40(2):158–160. doi:10.1038/ng.2007.55
Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Bristow J, Ren B, Black BL, Rubin EM, Visel A, Pennacchio LA (2010) ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42(9):806–810. doi:10.1038/ng.650
He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA, Visel A, Pennacchio LA, Pu WT (2014) Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun 5:4907. doi:10.1038/ncomms5907
May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Afzal V, Simpson PC, Rubin EM, Black BL, Bristow J, Pennacchio LA, Visel A (2012) Large-scale discovery of enhancers from human heart tissue. Nat Genet 44(1):89–93. doi:10.1038/ng.1006
Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, Rubenstein JL, Rubin EM, Pennacchio LA, Visel A (2013) Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155(7):1521–1531. doi:10.1016/j.cell.2013.11.033
Levo M, Segal E (2014) In pursuit of design principles of regulatory sequences. Nat Rev Genet 15(7):453–468. doi:10.1038/nrg3684
Wamstad JA, Wang X, Demuren OO, Boyer LA (2014) Distal enhancers: new insights into heart development and disease. Trends Cell Biol 24(5):294–302. doi:10.1016/j.tcb.2013.10.008
Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung YH, Deanfield J, DePalma S, Fakhro KA, Glessner J, Hakonarson H, Italia MJ, Kaltman JR, Kaski J, Kim R, Kline JK, Lee T, Leipzig J, Lopez A, Mane SM, Mitchell LE, Newburger JW, Parfenov M, Pe’er I, Porter G, Roberts AE, Sachidanandam R, Sanders SJ, Seiden HS, State MW, Subramanian S, Tikhonova IR, Wang W, Warburton D, White PS, Williams IA, Zhao H, Seidman JG, Brueckner M, Chung WK, Gelb BD, Goldmuntz E, Seidman CE, Lifton RP (2013) De novo mutations in histone-modifying genes in congenital heart disease. Nature 498(7453):220–223. doi:10.1038/nature12141
Pradeepa MM, Grimes GR, Kumar Y, Olley G, Taylor GC, Schneider R, Bickmore WA (2016) Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet. doi:10.1038/ng.3550
Zhou P, Pu WT (2016) Recounting cardiac cellular composition. Circ Res 118(3):368–370. doi:10.1161/CIRCRESAHA.116.308139
Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren G, Ni B, Sklar J, Przytycka TM, Childs R, Levens D, Zhao K (2015) Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528(7580):142–146. doi:10.1038/nature15740
Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, Bernstein BE (2015) Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 33(11):1165–1172. doi:10.1038/nbt.3383
Hyun BR, McElwee JL, Soloway PD (2015) Single molecule and single cell epigenomics. Methods 72:41–50. doi:10.1016/j.ymeth.2014.08.015
Dogan N, Wu W, Morrissey CS, Chen KB, Stonestrom A, Long M, Keller CA, Cheng Y, Jain D, Visel A, Pennacchio LA, Weiss MJ, Blobel GA, Hardison RC (2015) Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility. Epigenet Chromatin 8:16. doi:10.1186/s13072-015-0009-5
McCulley DJ, Black BL (2012) Transcription factor pathways and congenital heart disease. Curr Top Dev Biol 100:253–277. doi:10.1016/B978-0-12-387786-4.00008-7
Bruneau BG (2008) The developmental genetics of congenital heart disease. Nature 451(7181):943–948. doi:10.1038/nature06801
Andersen TA, Troelsen Kde L, Larsen LA (2014) Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci 71(8):1327–1352. doi:10.1007/s00018-013-1430-1
Bouveret R, Waardenberg AJ, Schonrock N, Ramialison M, Doan T, de Jong D, Bondue A, Kaur G, Mohamed S, Fonoudi H, Chen CM, Wouters MA, Bhattacharya S, Plachta N, Dunwoodie SL, Chapman G, Blanpain C, Harvey RP (2015) NKX2–5 mutations causative for congenital heart disease retain functionality and are directed to hundreds of targets. Elife 4:e06942. doi:10.7554/eLife.06942. https://elifesciences.org/content/4/e06942
Dupays L, Shang C, Wilson R, Kotecha S, Wood S, Towers N, Mohun T (2015) Sequential binding of MEIS1 and NKX2–5 on the Popdc2 gene: a mechanism for spatiotemporal regulation of enhancers during cardiogenesis. Cell Rep 13(1):183–195. doi:10.1016/j.celrep.2015.08.065
He A, Kong SW, Ma Q, Pu WT (2011) Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci USA 108(14):5632–5637. doi:10.1073/pnas.1016959108
Jain R, Li D, Gupta M, Manderfield LJ, Ifkovits JL, Wang Q, Liu F, Liu Y, Poleshko A, Padmanabhan A, Raum JC, Li L, Morrisey EE, Lu MM, Won KJ, Epstein JA (2015) HEART DEVELOPMENT. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science 348(6242):aaa6071. doi:10.1126/science.aaa6071
Liang X, Zhang Q, Cattaneo P, Zhuang S, Gong X, Spann NJ, Jiang C, Cao X, Zhao X, Zhang X, Bu L, Wang G, Chen HS, Zhuang T, Yan J, Geng P, Luo L, Banerjee I, Chen Y, Glass CK, Zambon AC, Chen J, Sun Y, Evans SM (2015) Transcription factor ISL1 is essential for pacemaker development and function. J Clin Investig 125(8):3256–3268. doi:10.1172/JCI68257
Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, Nagy A, Holloway AK, Pollard KS, Muller CW, Bruneau BG (2016) Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell 164(5):999–1014. doi:10.1016/j.cell.2016.01.004
Shen T, Aneas I, Sakabe N, Dirschinger RJ, Wang G, Smemo S, Westlund JM, Cheng H, Dalton N, Gu Y, Boogerd CJ, Cai CL, Peterson K, Chen J, Nobrega MA, Evans SM (2011) Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J Clin Investig 121(12):4640–4654. doi:10.1172/JCI59472
Tao Y, Zhang M, Li L, Bai Y, Zhou Y, Moon AM, Kaminski HJ, Martin JF (2014) Pitx2, an atrial fibrillation predisposition gene, directly regulates ion transport and intercalated disc genes. Circ Cardiovasc Genet 7(1):23–32. doi:10.1161/CIRCGENETICS.113.000259
Weber D, Heisig J, Kneitz S, Wolf E, Eilers M, Gessler M (2015) Mechanisms of epigenetic and cell-type specific regulation of Hey target genes in ES cells and cardiomyocytes. J Mol Cell Cardiol 79:79–88. doi:10.1016/j.yjmcc.2014.11.004
Wu SP, Cheng CM, Lanz RB, Wang T, Respress JL, Ather S, Chen W, Tsai SJ, Wehrens XH, Tsai MJ, Tsai SY (2013) Atrial identity is determined by a COUP-TFII regulatory network. Dev Cell 25(4):417–426. doi:10.1016/j.devcel.2013.04.017
Ye W, Wang J, Song Y, Yu D, Sun C, Liu C, Chen F, Zhang Y, Wang F, Harvey RP, Schrader L, Martin JF, Chen Y (2015) A common Shox2–Nk2–5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinoatrial node. Development 142(14):2521–2532. doi:10.1242/dev.120220
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22(9):1813–1831. doi:10.1101/gr.136184.111
Consortium EP (2011) A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9(4):e1001046. doi:10.1371/journal.pbio.1001046
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP (2014) Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 15(2):121–132. doi:10.1038/nrg3642
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9(9):R137. doi:10.1186/gb-2008-9-9-r137
de Boer BA, van Duijvenboden K, van den Boogaard M, Christoffels VM, Barnett P, Ruijter JM (2014) OccuPeak: ChIP-Seq peak calling based on internal background modelling. PLoS One 9(6):e99844. doi:10.1371/journal.pone.0099844
Palmiter RD, Brinster RL (1986) Germ-line transformation of mice. Annu Rev Genet 20:465–499. doi:10.1146/annurev.ge.20.120186.002341
Sweet DJ (2016) Foxd3: a repressor, an activator, or both? Cell Stem Cell 18(1):1–2. doi:10.1016/j.stem.2015.12.009
Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Ren B, Rubin EM, Pennacchio LA (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457(7231):854–858. doi:10.1038/nature07730
Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B (2012) A map of the cis-regulatory sequences in the mouse genome. Nature 488(7409):116–120. doi:10.1038/nature11243
Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG (2012) Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151(1):206–220. doi:10.1016/j.cell.2012.07.035
Akazawa H, Komuro I (2003) Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res 92(10):1079–1088. doi:10.1161/01.RES.0000072977.86706.23
Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H (2007) Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev 12(3–4):331–343. doi:10.1007/s10741-007-9034-1
Sergeeva IA, Hooijkaas IB, Ruijter JM, van der Made I, de Groot NE, van de Werken HJ, Creemers EE, Christoffels VM (2016) Identification of a regulatory domain controlling the Nppa-Nppb gene cluster during heart development and stress. Development 143(12):2135–2146. doi:10.1242/dev.132019
Lescroart F, Kelly RG, Le Garrec JF, Nicolas JF, Meilhac SM, Buckingham M (2010) Clonal analysis reveals common lineage relationships between head muscles and second heart field derivatives in the mouse embryo. Development 137(19):3269–3279. doi:10.1242/dev.050674
Amin S, Donaldson IJ, Zannino DA, Hensman J, Rattray M, Losa M, Spitz F, Ladam F, Sagerstrom C, Bobola N (2015) Hoxa2 selectively enhances Meis binding to change a branchial arch ground state. Dev Cell 32(3):265–277. doi:10.1016/j.devcel.2014.12.024
Watanabe Y, Zaffran S, Kuroiwa A, Higuchi H, Ogura T, Harvey RP, Kelly RG, Buckingham M (2012) Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nk2–5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc Natl Acad Sci USA 109(45):18273–18280. doi:10.1073/pnas.1215360109
Bergsland M, Ramskold D, Zaouter C, Klum S, Sandberg R, Muhr J (2011) Sequentially acting Sox transcription factors in neural lineage development. Genes Dev 25(23):2453–2464. doi:10.1101/gad.176008.111
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dupays, L., Mohun, T. Spatiotemporal regulation of enhancers during cardiogenesis. Cell. Mol. Life Sci. 74, 257–265 (2017). https://doi.org/10.1007/s00018-016-2322-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2322-y