
Abstract
The aggregation and deposition of the amyloid-β peptide (Aβ) in the brain has been linked with neuronal death, which progresses in the diagnostic and pathological signs of Alzheimer’s disease (AD). The transition of an unstructured monomeric peptide into self-assembled and more structured aggregates is the crucial conversion from what appears to be a harmless polypeptide into a malignant form that causes synaptotoxicity and neuronal cell death. Despite efforts to identify the toxic form of Aβ, the development of effective treatments for AD is still limited by the highly transient and dynamic nature of interconverting forms of Aβ. The variability within the in vivo “pool” of different Aβ peptides is another complicating factor. Here we review the dynamical interplay between various components that influence the heterogeneous Aβ system, from intramolecular Aβ flexibility to intermolecular dynamics between various Aβ alloforms and external factors. The complex dynamics of Aβ contributes to the causative role of Aβ in the pathogenesis of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Experimental studies and clinical trials are ongoing in the search for an effective prevention or treatment of Alzheimer’s disease (AD) [1–3]. These studies and trials often target the amyloid-beta peptide (Aβ), which plays a major role in AD pathogenesis [4]. Effective drug development has remained without success and this is thought to originate from the fact that Aβ can appear in many different shapes that can interconvert within a dynamical interplay. This finding triggered a vast exploration of the many conformations the peptide can adopt, as well as the aim to precisely pinpoint which of these conformations can be claimed as “the toxic species”, such that specific drug targeting can be employed. To complicate matters even more, a heterogeneous pool of monomeric Aβ varying in length from 37 to 49 amino acids is produced by proteolytic cleavage from the transmembrane amyloid precursor protein (APP) by β- and γ-secretases [5, 6] (Fig. 1). Most research effort has been focused on the most abundant form Aβ1−40, which comprises 40 amino acids. The longer and less abundant Aβ1−42, C-terminally extended by two residues, has been found to be more aggregation-prone [7]. Nonetheless, it has recently been discovered by us [8–10] and other groups [11–14] that the co-occurrence of peptides varying in length can affect the neurotoxic and aggregation potential of the total Aβ pool. It was also recognized that particularly small aggregated forms of Aβ are potently toxic, rather than the mature amyloid fibrils as observed in the brain of AD patients. Therefore, a lot of research has aimed at understanding the Aβ aggregation mechanism and identifying the intermediate species that occur along the aggregation pathway [15, 16]. The current amyloid cascade hypothesis suggests that AD-related synapto- and neurotoxicity might be mediated by soluble Aβ oligomers [17, 18], which have proven notoriously difficult to study in detail in vivo with the currently available technology. The dynamics, stability, and transient lifetime of potentially toxic species further hamper the possibility to precisely pinpoint the toxic structural aspects of Aβ aggregates. Moreover, the dynamic behavior of aggregation intermediates may actually provide an important source for toxicity of Aβ as isolated Aβ oligomers are only toxic in the presence of Aβ monomers that provide a source for continued growth of oligomers into fibrillar species [13, 19].

Heterogeneity in the Aβ peptide pool. Sequential proteolytic events by the β- and γ-secretase of the amyloid precursor protein (APP) give rise to the carboxy-terminal fragment (CTF), APP intracellular domain (AICD), and the amyloid-β peptide (Aβ). The heterogeneity in the Aβ pool originates from the proteolysis by the γ-secretase, but also post-translational modifications contribute to the formation of various Aβ alloforms. Mutations in Aβ and other exogenous factors can influence the dynamics that are observed within the Aβ system
This review discusses how Aβ peptide dynamics can influence and contribute to Aβ-induced toxicity. Aβ dynamics is mainly considered on two levels. First, we define intramolecular dynamics of Aβ as the intrinsic disorder or polypeptide backbone flexibility that is present in isolated Aβ monomeric peptides or aggregation states. Second, we define intermolecular dynamics as (1) the interplay between different Aβ alloforms present in the in vivo Aβ pool and (2) the dynamical equilibrium that exists between different Aβ species. With the term alloform, we refer to a distinct form of the Aβ peptide that is commonly treated as a single kind of peptide species, like Aβ length variants or side chain modifications. Finally, several external factors and interaction partners that can influence Aβ dynamics are addressed. The potential importance of Aβ dynamics in understanding AD pathology is highlighted with the aim of shaping new research orientations for AD treatment.
Intramolecular dynamics
The Aβ monomer has a high tendency to self-assemble into large aggregates and fibrils. It is increasingly recognized that despite the highly packed and ordered state of these higher-order aggregates, they often do contain a significant portion of flexible and intrinsically disordered regions [20]. The intrinsically disordered nature of the Aβ monomer is fairly well documented, but revealing the structural disorder in oligomers and fibrils has proven more challenging due to the difficulties in studying this phenomenon. In this section, we discuss the intrinsic structural disorder that is present in every Aβ aggregation state, and we illustrate how it contributes to Aβ-induced toxicity.
The intrinsically disordered Aβ monomer
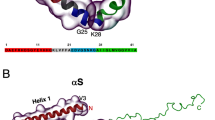
Although the pathological hallmark of AD comprises insoluble Aβ deposits in neuritic plaques in the brain of AD patients, monomeric Aβ peptides have also been purified and characterized from brain tissue [21–24]. Size exclusion chromatography (SEC) experiments suggested that the freshly dissolved peptide eluted as a single low molecular weight species, consistent with a monomer or dimer [25–27]. These low molecular weight Aβ species were competent to deposit onto pre-existing amyloid in preparations of AD cortex, with a first-order kinetic dependence on soluble Aβ concentration [26]. Translational diffusion measurements by nuclear magnetic resonance (NMR) techniques conclusively demonstrated that the form of the peptide active in plaque deposition is a monomer [26]. Further NMR data revealed that monomeric Aβ exists in solution as disordered coils that lack regular α-helical or β-stranded structure [28–30]. Despite the challenging task because of its unstructured and amyloidogenic nature, the Aβ monomer is now well recognized as an intrinsically disordered peptide (IDP). This implies that the monomeric Aβ peptide does not display a unique fold, as would be the case for a typical well-folded protein, but rather comprises a mixture of rapidly interconverting conformations whereby the polypeptide backbone can sample the conformational space without any stable and well-defined conformational ensemble (Fig. 2). Yet, it is possible to bias the ensemble toward distinct secondary structure elements by changing solution conditions and/or the oxidation state of Met35 [30–33].

Various structures of Aβ that correspond to different experimental conditions and phases in the aggregation landscape. a Four representatives of the structural ensemble of monomeric Aβ1−42 under aqueous conditions as derived from a combined molecular dynamics/NMR approach [38]. Extended as well as collapsed coil conformations with secondary structural elements can be observed. b Aβ1−40 in presence of 50 mM NaCl at 15 °C [33] and Aβ1−42 in presence of 30 % hexafluoroisopropanol [32] contain an α-helical segment. c Fibril polymorphism illustrated by fibrillar Aβ1−42 [53], D23N Aβ1−40 [74] and d the ultrastructure of Aβ1−40 [83], and brain-derived Aβ1−40 [89]
Some experimental studies suggested that Aβ is not entirely a “random coil”. Ion mobility mass spectrometry (MS) combined with theoretical modeling showed that Aβ1−42 in aqueous solution adopts both extended chain as well as collapsed-coil structures [34]. Limited proteolysis successfully identified structured and disordered regions within Aβ [35]. This approach revealed a proteolytically resistant decapeptide, Ala21–Ala30, that was found in NMR studies to form a turn-like structure [30]. When the dynamics of monomeric Aβ1−40 in solution was studied using 15N-relaxation experiments, it revealed structural propensities that correlate well with the secondary structure segments of the peptide that are present in the fibrils, and with the α-helical structure in membrane-mimicking systems [32, 36]. NMR studies further revealed subtle differences between Aβ1−40 and Aβ1−42 monomers whereby a modest increase in C-terminal rigidity has been observed in Aβ1−42 versus Aβ1−40 [37]. Various molecular dynamics simulations also hinted that distinct intramolecular interaction patterns occur in Aβ1−42 [28, 38, 39]. Such subtle differences between Aβ1−40 and Aβ1−42 were confirmed by molecular dynamics simulations [40, 41]. Experimental results in combination with computational simulations have thus proven very powerful to shed light on the conformational landscape of IDPs. The emerging picture of Aβ comprises an IDP that can adapt a variety of collapsed and extended monomeric conformations and transiently samples long-range intramolecular interactions without exclusively stabilizing a specific globular fold.
Even though the physiological function of Aβ remains obscure, the intrinsic structural flexibility offers certain advantages: high specificity and low affinity in the case of binding-induced folding IDPs (mostly exploited in signaling pathways), and high binding promiscuity that is frequently used by hub proteins in large interaction networks [42]. So its IDP nature facilitates the interaction of the peptide with many different binding partners (see “Other players in the game”), including identical peptides and other Aβ alloforms. In addition, the high intramolecular flexibility of Aβ also simplifies post-translational modifications because the involved side chains are readily accessible (see “The in vivo Aβ pool: a cocktail of different interacting species”).
There is a well-established link between intrinsic polypeptide disorder and functional promiscuity. Protein moonlighting, the phenomenon of proteins exhibiting more than one unique biological function, is typically mediated by intrinsically disordered regions in polypeptides [43]. As IDPs can play a role in numerous biological processes, it is not surprising to find some of them involved in human diseases.
Intrinsic fibril flexibility can underlie disease progression and phenotype
Aβ fibrils contain high order and rigidity compared to Aβ monomers, but still retain a considerable amount of disorder in the N-terminal segment [44–47] and they are often polymorphous. The inherent disorder of Aβ fibrils and the associated fibril polymorphism could underlie time-dependent structural changes during aging in AD and differences in disease progression and phenotype.
The molecular dynamic nature of Aβ fibrils
Even though the amyloid fibril state of Aβ has traditionally been viewed as a rigid or semi-rigid state with the typical cross-β X-ray fiber diffraction pattern [48, 49], part of the peptide in this conformation is also flexible. This flexibility has been illustrated first by solid-state and solution NMR [50–54], electron paramagnetic resonance (EPR) [44], site-directed mutagenesis [55], limited proteolysis and hydrogen-deuterium exchange (HDX) evaluated by MS [45–47, 56], and even X-ray crystallography [57]. These studies suggested a hairpin-like arrangement of each Aβ monomer stacked within the fibril, consisting of two semi-rigidly organized β-strands linked by a flexible connecting region (Fig. 2). The hydrophobic C-terminus of Aβ1−42 in the fibril is highly resistant to HDX and forms the fibril core [53, 54]. In contrast, the C-terminus of Aβ1−40 in the fibril contains slightly more disorder [52, 56, 58–61]. The N-terminal segment, which can range from 10 to 19 residues depending on the study, remains intrinsically disordered for both Aβ1−40 and Aβ1−42 fibrils (Table 1). This relatively hydrophilic part of the polypeptide chain is excluded from the H-bonded β-sheet fibril core and remains exposed to the solvent [44–47, 52–54, 58–61]. Recently, differential scanning calorimetry suggested that thermal denaturation of amyloid fibrils can take place and that this process can be considered as a reversible equilibrium under certain experimental conditions, highlighting the dynamic nature of fibrils [62]. These observations illustrate the impact of the various dynamics within the Aβ system.
The inherent flexibility of Aβ fibrils also allows the internal fibril structure to evolve in time. Multidimensional infrared spectroscopy revealed that fresh and 4-year-old fibrils were structurally heterogeneous due to trapped water molecules that perturbed the H-bonding pattern in time [63]. Recently, Nilsson and coworkers [64] revealed conformational rearrangements during aging in plaques in the brains of AD mouse models using different luminescent conjugated polythiophenes.
Although ignored for a long time, structural disorder in fibrils seems to occur in various amyloidogenic proteins (e.g. α-synuclein, tau, and multiple prions) (reviewed in [20]). Structural disorder in fibrils has been suggested to stabilize fibril formation by accommodating destabilizing residues and by limiting the unfavorable entropy associated with the formation of the highly ordered cross-β spine.
Aβ fibrils are polymorphic entities
Overall fibril topology has been studied using cryo-electron microscopy and 3D reconstruction. In general, Aβ fibrils exhibit multiple distinct morphologies that can differ in fibril symmetry, width, twist period, and curvature [65, 66]. This structural diversity is not limited to Aβ fibrils, but appears to be a fundamental property of the amyloid state [67–69]. Inter-sample polymorphism commonly occurs in vitro in different fibril growth conditions and is subject to pH, temperature, agitation, and salt conditions [70, 71]. A Darwinian-type “survival of the fittest” competition allows the type of fibril that is kinetically the most accessible in a given environment to be the most populated [72]. However, Aβ1−40 can also form at least 12 structurally distinct morphotypes under the same solution conditions (intra-sample polymorphism) indicating that this polymorphism arises from an intrinsic variability [73]. Interconversion between fibril polymorphs coexisting in solution can occur, resulting in the thermodynamically more stable polymorph, as was monitored by solid-state NMR over a period of several weeks for Aβ1−40 [74, 75].
Amyloid polymorphism can have several molecular origins that are not mutually exclusive [76–79]. First, mass-per-length values obtained from scanning TEM indicate that fibrils can be composed of one to five protofilaments (the minimal fibrillar entities) [80, 81]. Second, distinct orientations and modes of lateral association of protofilaments by different patterns of inter-residue interactions determine if protofilaments are oriented side-by-side [50, 82], offset from one another [76, 77], or winded around a hollow core [79]. Third, solid-state NMR demonstrated that agitated (striated) and quiescent (twisted) fibrils differ in the residues participating in the β-strands and such variations in the underlying protofilament substructure can contribute to polymorphism [59, 83]. Surprisingly, the Iowa mutant (D23N Aβ1−40) was recently found to form metastable fibrils with an antiparallel cross-β spine, indicating that a familial disease-related mutation can have profound effects on fibril structure [74]. Although the cross-β spine of Aβ fibers is a common feature, fibrils show a great variety of structural complexity that appears inherent to the dynamic nature of the peptide.
Fibril polymorphism could lead to different pathological outcomes
Fibrils can initiate inflammation in brain tissues and cell-cultured microglia and astrocytes. Fibril-induced inflammation then leads to the secretion of pro-inflammatory cytokines and the production of free radicals causing oxidative damage [84, 85]. Substantial evidence provided that different fibril morphologies exert different toxicities in vitro, although toxic activity of oligomeric Aβ was reported to exceed that of the fibrillar form multiple times [53, 59, 86–88]. For example, oligomeric Aβ correlated more strongly to cognitive impairment as compared to fibrillar Aβ of amyloid plaques [86, 87].
Fibril polymorphism could explain the weak correlation between plaque load and cognitive impairment. If plaques are comprised of different fibril polymorphs, different levels of toxicity could be associated to these amyloid deposits. In this case, the structural diversity of fibrils may account for differences in disease progression and phenotype as has been suggested by Tycko and coworkers [89]. They reported that Aβ fibrils seeded from human brain extracts differed between patients with different clinical history and neuropathology [89]. Moreover, fibril polymorphism has been linked previously to different phenotypes for hereditary transthyretin amyloidosis [90]. In this regard, the different architectures of wild-type Aβ and Iowa D23N fibrils, comprising respectively parallel and antiparallel β-sheet orientations, could underlie the different pathological outcomes: sporadic AD versus early onset AD associated with cerebral amyloid angiopathy (CAA).
Aβ oligomers: a mishmash of conformations and sizes
Since the Aβ plaque load and AD severity could not be correlated [86, 87], growing evidence has revealed that soluble oligomers, either on- or off-pathway to fibrils (see “The in vivo Aβ pool: a cocktail of different interacting species”), play a primary role in AD. Soluble oligomers have commonly been associated with disease severity, the loss of synapses and neuronal damage (reviewed in [18]). The low abundance, heterogeneity, low solubility, and transient nature of Aβ oligomers have hindered structural studies. It now becomes clear that Aβ oligomers exist in a broad range of interconverting assemblies varying in size, conformation, and associated toxicity (reviewed in [91, 92]).
Aβ oligomers can cause toxicity by a variety of mechanisms (reviewed in [93]). To enable drug design, it is essential to establish the key determinants of oligomer toxicity. Several studies report that neurotoxic activity varies with Aβ oligomer size with small oligomers (n < 14) being most toxic [94, 95]. However, oligomer size is not sufficient to define toxicity as Aβ oligomers with similar size have been shown to exert different toxicities [96–98]. The underlying peptide conformation also needs to be taken into account as the interplay between Aβ oligomer size and conformation plays an important role in toxicity (reviewed in [92]). The design of a well-controlled study to investigate size and conformational impact on toxicity is notoriously difficult as different oligomer conformations and sizes are in continuous exchange. However, studies in which different conformations or sizes have been enriched or stabilized by means of crosslinking have been performed and careful conclusions can be drawn from such studies. For example, different Aβ oligomer conformations have been shown to induce neurotoxicity by distinct mechanisms in human cortical neurons [99]. One possibility to classify oligomers according to their underlying structure is based on recognition by conformation-dependent antibodies [100–103]. Surprisingly, soluble oligomers of a wide variety of amyloidogenic polypeptides (Aβ, α-synuclein, islet amyloid polypeptide, polyglutamine, lysozyme, human insulin and prion peptide) react with the oligomer-specific A11 antibody developed in the laboratory of Charles Glabe, suggesting that there has to be a common denominator to their toxic origin. Interestingly, pre-incubation of mouse hippocampal neurons with the A11 antibody, before treatment with Aβ, rescues them from the neurotoxic effects induced by Aβ [8]. It has been suggested that A11 positive oligomers are composed of antiparallel β-sheets, based on Fourier transform infrared (FTIR) spectroscopy. This antiparallel signature might represent a critical step in perturbation or permeabilization of cell membranes leading to cell toxicity [104]. Later studies using FTIR, EPR, and X-ray crystallography have confirmed that oligomeric species can be characterized by an antiparallel β-sheet orientation, while most fibrils consist of in-register, parallel β-sheets [105–109]. Moreover, antiparallel oligomers displayed a lower content in secondary structure and faster HDX kinetics compared to fibrils, suggesting a higher intrinsic flexibility [104].
Apart from size and peptide conformation, this intrinsic flexibility of the Aβ oligomer can also be a key determinant of Aβ-induced toxicity. Several studies have shown that the N-terminus retains a degree of flexibility upon oligomerization and is exposed to the solvent [41, 109–111]. Ahmed and coworkers reported solution NMR measurements of Aβ1−42 pentamers [111]. The authors found that the loosely packed N-terminal segment of Aβ was defined by HDX ratios approaching 1 for residues Asp1-Gly9, indicating high solvent accessibility and nearly complete exchange within the acquisition time (<1.5 h). In contrast, Val40-Ala42 were less solvent accessible and most likely buried within the center of the oligomer. Similar results were obtained for packing of the Aβ peptide within Aβ1−42 dodecamers. Site-directed spin labeling of Aβ1−42 combined with EPR spectroscopy showed that the N-terminus was loosely packed within the dodecamer, while residues Ile32-Val40 formed a tight core [109]. Increased structural disorder and solvent exposure of hydrophobic segments of the oligomer have been suggested to be a common feature of highly toxic, soluble aggregates [96–98, 112]. Recent work has shown that the most cytotoxic, oligomeric species of the E22G (arctic) variant of Aβ1−42 interacted more strongly with 1-anilinoaphthalene 8-sulfonate (ANS), a dye sensitive to exposed hydrophobic patches [112]. A higher degree of solvent-exposed, hydrophobic regions was further shown to lead to a disturbed cellular calcium homeostasis, likely due to disruption of the cell membrane [98]. Moreover, oligomers have been shown to bind with higher affinity and cause more disruption of synthetic membranes as compared to the higher-ordered fibrils [113]. These data emphasize the importance of intrinsic disorder and molecular flexibility of Aβ oligomers for the toxicity mechanism.
In conclusion, a re-evaluation of the oligomer cascade hypothesis is needed (reviewed in [114]). Whereas earlier hypotheses held one single oligomer of a predefined size responsible for toxicity [23, 115], it is obvious that a diverse “Aβ oligomeric soup” exists, consisting of a large variety of rapidly exchangeable polymorphs that differ in size, conformation, hydrophobicity, solvent exposure, intrinsic disorder (or internal flexibility), and toxicity. The oligomer cascade hypothesis should take into account that it is likely that the entire dynamic Aβ oligomeric soup contributes to the heterogeneity of AD progression and phenotype, via various toxic mechanisms.
Intermolecular dynamics
As the in vivo Aβ pool is a mix of species influencing one another, one must also consider the dynamics between different Aβ species when regarding Aβ-related toxicity. First, Aβ peptides of various lengths are produced due to the heterogeneous cleavage pattern of APP by γ-secretase [5, 6]. This gives rise to the production of Aβ1−40, smaller amounts of Aβ1−42, and trace amounts of peptides ranging in length from 37 to 49 amino acids [116–118]. Second, a dynamical equilibrium exists between different aggregation states during Aβ aggregation. Studying the behavior of Aβ peptide mixtures and revealing the dynamics of interconversion among different aggregate species will be crucial in understanding the AD-related toxic effects of Aβ.
The in vivo Aβ pool: a cocktail of different interacting species
The large majority of biophysical and cell biological studies investigating the role of Aβ in AD have focused either on pure Aβ1−40 or on pure Aβ1−42, the two predominant Aβ alloforms present in the brain [7, 119]. The in vivo Aβ pool not only contains different Aβ peptide lengths but also comprises post-translationally modified Aβ [120] (Fig. 1). Aβ peptides can undergo racemization [121, 122], isomerization [123], phosphorylation [124, 125], oxidation [126, 127], non-enzymatic glycation [128], and pyroglutamylation [129]. Post-translational oxidation of Met35 affects fibril flexibility within Aβ plaques [127]. Met35 oxidation also has been shown to impede the rate of Aβ aggregation in vitro [30], possibly by decreasing the β-strand content of the C-terminal region [130]. Furthermore, proteins can become modified by non-enzymatic glycation upon aging. Advanced glycation end products (AGEs), found in Aβ plaques and in neurons, and their receptor RAGE play an important role in AD by contributing to oxidative stress and by triggering inflammation signaling pathways [128, 131, 132]. For other modifications, it remains largely unknown how they can affect Aβ aggregation dynamics.
Various forms of Aβ co-exist and co-deposit in amyloid fibrils and plaques [23, 128]. It has become clear that biologically relevant mixtures of Aβ alloforms behave in a more complex manner in vitro than anticipated from their behavior in isolation, in terms of aggregation properties and toxicity [8–12]. For example, Aβ1−38 and Aβ1−40 exerted little toxicity in isolation, but were highly toxic to a neuroblastoma cell line when tested in a mixture, whereas addition of Aβ1−38 to Aβ1−42 had a protective effect [10].
Recently, it has been demonstrated that minor shifts in the Aβ1−42:Aβ1−40 ratio can modulate neurotoxicity [8]. The aggregation of samples of Aβ lengths in various compositions were monitored by NMR allowing simultaneous investigation of both Aβ1−42 and Aβ1−40 in the same sample by combining 15N-isotope-labeling of one Aβ alloform with 15N-edited filter experiments [9]. It was revealed that Aβ1−42 and Aβ1−40 directly interact and influence oligomer formation and aggregation kinetics. Moreover, cross-seeding data revealed structural differences between the different ratios at the level of the oligomeric state. A subtle change in the Aβ1−42:Aβ1−40 ratio was suggested to induce differences in conformational plasticity of the oligomeric peptide mixtures [9]. High molecular weight (HMW) mass spectra further showed that a continuous range of oligomeric intermediates were formed upon incubation of Aβ through a monomer addition process for the time frame within which toxicity exists [8, 9]. This observation is in agreement with the “coalescence and reorganization model of amyloid formation” [133], but also with the principle of a template-dependent dock-and-lock-and-block mechanism whereby the locking of a peptide cannot efficiently occur unless the previously loaded peptide has assembled into the correct position [134]. This can be envisaged in the following way: intrinsically disordered Aβ monomers diffuse freely and can attach individually to each other, to a pre-existing oligomer or to the fiber surface, especially through the distal ends. The crucial step occurs when the incoming monomer collides with the docking surface. In the case of a productive association, a permanent attachment can then take place, perhaps accompanied by a minor structural rearrangement. The conformational constraints of the monomers will therefore influence the efficiency and kinetics of the aggregation as well as the architecture of Aβ fibrils [135] (see “Aβ fibrils are polymorphic entities”). Alloform differences of the monomeric conformation are essential at this point to interpret productive or non-productive interactions [38, 40, 41], particularly in the complex in vivo pool of peptides. Aβ1−42 has the tendency to sample more fibril-like conformations compared to Aβ1−40 and as such can simply dock to fibril-like oligomers leading to highly productive (on-pathway) interactions. The more rigid and less flexible C-terminus of Aβ1−42 was suggested to enable the formation of a larger number of intramolecular contacts than Aβ1−40 [37, 40] and therefore provide a more extensive hydrophobic surface for intermolecular interactions. Experiments using amino acid substitutions in the C-terminal part of Aβ1−40 and Aβ1−42 confirmed that (i) the stability of the β-hairpin structure was increased by reducing the backbone flexibility and strengthening the hydrophobic interactions between the putative β-strands, (ii) destabilizing mutations in the C-terminal part of Aβ1−42 lead to a more Aβ1−40-like behavior, and (iii) stabilizing mutations in the C-terminus of Aβ1−40 lead to a more Aβ1−42-like behavior [143]. The conformational search of the incoming peptide for binding on the docking surface and for the proper orientation to lock-in-place could explain the complex aggregation behavior of Aβ alloform mixtures. The balance between productive and non-productive interactions in the transient encounter states is essential to guide the kinetics of aggregation, which in turn will define the time window within which the toxic species exist. Now that it becomes evident from independent research groups that the pattern of oligomer formation is mainly influenced by (patho)physiologically relevant Aβ1−42:Aβ1−40 ratios [136], it is also important to realize that independent (on- and off-) pathways exist for oligomerization and fibrillization of Aβ [137, 138].
Experimental approaches to obtain insight into complex Aβ dynamics
It seems logical that the assembly and disassembly of toxic species is a dynamic and continuous process, at least in the initial stages, that is directed by the Aβ pool composition. However, the possibility that toxicity is present over a series of conformers or sizes should not be disregarded [91, 92, 94, 139, 140]. The question is thus how biophysical parameters influence this process in vivo and affect the relative distribution of Aβ species over toxic and non-toxic conformations over time. Given the complexity of the biophysical environment in which Aβ aggregation occurs in vivo, such a question is extremely difficult to address. Nevertheless, it is possible to analyze the dynamic features of this process in simplified and controlled conditions in vitro, and to evaluate the effect of the relative concentrations of Aβ1−40 and Aβ1−42 (and other alloforms) to the generation of neurotoxic species over time.
The combination of high-resolution NMR and HMW MS is perfectly suited to investigate the individual aggregation behavior of the diverse Aβ alloforms in complex and heterogeneous sample compositions. This can yield a comprehensive aggregation fingerprint that allows us to understand how the different compositions of the Aβ peptide pool influence the overall aggregation behavior. This aggregation fingerprint can be related to cytotoxicity, membrane integrity, apoptotic responses, and functional read-outs such as microelectrode arrays (MEA), in which synaptic activities at different timeframes and under various conditions are monitored in response to Aβ [8, 141]. Such a fingerprint also opens perspectives to the diagnostics and therapeutics field when it can be correlated to biomarkers. Patient-specific treatment (personalized medicine) could be based on the detailed characterization of the composition of the Aβ pool. It will be essential to correlate the aggregation fingerprint of such compositions with disease severity and the (ir)reversibility of the disease “progress”. It is also important to cover the overall dynamics in these pools rather than focusing on particular “toxic” intermediates that are only transient in the aggregation process. This will allow tackling the source of toxicity and limiting the time frame in which the toxic assemblies can exist. Aggregation fingerprints will thus be essential to better understand the Aβ-induced pathogenesis of AD and the biophysical processes that underlie the cell biological responses.
The interaction between different species present during Aβ aggregation
NMR relaxation measurements showed that monomers are constantly binding to and being released from oligomers in vitro [142, 143]. Estimates showed that approximately 3 % of the peptide within the oligomer undergoes exchange with free monomer in pseudo-equilibrium conditions, suggesting that exchange occurs predominantly from the oligomer surface. A large part of the hydrophobic C-terminal region is involved in the association of monomer onto the oligomer surface [142]. In a next aggregation phase, protofibrils are formed that are also in constant exchange with monomers through the same surface region [144]. An elegant combination of 19F-NMR and other biophysical techniques revealed a heterogeneous mixture of small Aβ oligomers that exist in pseudo-equilibrium with protofibrils and fibrils during the early stages of aggregation [145].
Protofibrils self-associate and give rise to mature fibrils that can thermodynamically be considered as the most stable aggregation state due to the high density of intermolecular hydrogen bonding and steric zipper interactions [146]. However, fibrils are not static and irreversible end species, as was the traditional view, but were shown to continuously dissociate and reassociate through both fibril ends [147]. Aβ1−40 fibrils recycle to a greater extent than Aβ1−42 fibrils, which could be attributed to a difference in fibril dissolution rate. These findings are consistent with a dynamical model for interpreting plaque morphology, in which aggregation and disaggregation were proposed to be in steady-state equilibrium [148]. The species involved in the fibril recycling process are still a matter of debate. Differential solution NMR isotope labeling experiments revealed that Aβ1−40 monomers can replace Aβ1−42 on Aβ1−42 aggregates, recycling Aβ1−42 monomers back into solution [14]. Later reports confirmed the constant recycling of Aβ1−40 and Aβ1−42 monomers and competition of binding for the ends of protofibrillar and fibrillar aggregates [13]. Alternatively, the accumulation of fibrils could be associated with the generation of diffusible lower molecular weight aggregates. This idea is consistent with the observation of a halo of oligomeric Aβ surrounding senile plaques when analyzed by array tomography [149]. Recently, Knowles and coworkers demonstrated that the secondary nucleation pathway can be a major source of oligomers once the critical concentration of amyloid fibrils (in the order of 10 nM) has formed [150]. Hereby, the surfaces of existing fibrils catalyze the nucleation of new aggregates from the monomeric state, with a rate dependent on both the concentration of the monomers and that of the existing fibrils. As the critical fibril concentration is lower than the aggregate loads present in brains of AD patients, this pathway is likely to be active in the brain [150].
The dynamical equilibrium potentially contributes to Aβ-associated toxicity
The co-existence of different Aβ aggregate species should be taken into account when analyzing Aβ toxicity studies. For example, fibrils act as a reservoir of soluble aggregates that can diffuse and induce toxic effects. The halo of oligomers surrounding senile plaques co-localizes with loss of excitatory synapses and spine collapse [149] and the disruption of dendritic spines in the vicinity of plaques is dependent on their distance from these plaques [151]. Moreover, fibrils can be destabilized by brain lipids and reverted into neurotoxic soluble protofibrils [139]. Amyloid fibrils can thus be toxic per se (see “Fibril polymorphism could lead to different pathological outcomes”) or can function as a potential source of neurotoxic oligomeric species [152, 153]. It has also been suggested that the ongoing polymerization process, rather than the formation of one stable aggregate, is responsible for Aβ-related toxicity [19, 154]. In accordance with this hypothesis, crude Aβ1−42 preparations containing a monomeric and heterogeneous mixture of Aβ1−42 oligomers and protofibrils were more toxic than purified monomeric, protofibrillar fractions or fibrils. The toxicity of protofibrils was directly linked with their interactions with monomeric Aβ1−42 and strongly dependent on their ability to convert into fibrils. Moreover, the ongoing Aβ aggregation process, rather than distinct aggregation states, elicited alterations in astrocyte metabolic phenotypes [19]. Therefore, insight into the dynamic equilibrium is required to fully understand Aβ toxicity.
Other players in the game
The modulation of Aβ production, aggregation, and degradation by environmental factors [155–157], genetic risk factors [158–161], post-translational modifications [127], and an individual’s lifestyle [162–169] has been extensively reviewed before and does not lie in the scope of this review. Only a few reports discuss the influence of these factors on Aβ dynamics.
Metals have been shown to affect Aβ intramolecular dynamics. Binding of zinc to the N-terminus of the Aβ monomer leads to a decrease in the intrinsic mobility of this region and the formation of a turn-like conformation in residues Val24-Lys28 promoting aggregation, as shown by 15N relaxation measurements [170]. Copper can also bind to the N-terminus, causing a structural ordering in this region [171], but slowing down aggregation [110].
There is evidence that membrane composition and properties, in turn, play a critical role in Aβ cytotoxicity associated with its conformational changes and aggregation into oligomers and fibrils ([172–174], reviewed in [175]). Moreover, interaction with lipid membranes can modulate Aβ peptide conformation and aggregation properties (reviewed in [175, 176]).
Genetic evidence suggested a role for chaperones in AD [177] and abundant chaperone levels block formation of Aβ aggregates as was demonstrated in a Caenorhabditis elegans disease model [178]. In vitro results indicated a role for heat shock proteins in the early aggregation events by interfering with the dynamical aggregation process [179]. The BRICHOS domain, a chaperone-like domain found in lung surfactant protein C, is reported to be a potent in vitro inhibitor of Aβ aggregation [180]. The contribution of chaperones in the context of AD is reviewed in [181].
Interactions of Aβ with small molecules designed to target Aβ toxicity and/or Aβ aggregation have also been extensively studied. These ligands are not only interesting in light of drug development, but also provide a tool for addressing the modulation of Aβ dynamics upon ligand interaction [182–184].
As the Aβ monomer concentration affects the dynamical equilibrium between monomers, oligomers, protofibrils, and fibrillar Aβ, it is also worthwhile to consider factors that modulate Aβ metabolism. Aluminium is known to increase the Aβ brain burden in experimental animals and this might be due to a direct influence upon Aβ anabolism or to direct or indirect effects on Aβ catabolism [185]. Holtzman and coworkers reported that human cerebrospinal fluid Aβ levels undergo diurnal fluctuations and that this cycle is disturbed following plaque formation before the appearance of any cognitive symptoms [186]. Aβ fluctuations were affected by perturbation of the orexin signaling pathway and the sleep-wake cycle and this suggested that sleep abnormalities in earlier life might predispose an individual to AD [187]. Cholesterol has been suggested to provide stability to membrane-adjacent lipid rafts and therefore facilitate the Aβ cleavage from APP [168]. Recent evidence showed that the γ-secretase subunit composition defines the Aβ profile and affects the ratio between alloforms [6]. This implies that external factors influencing the γ-secretase subunit composition will have a profound effect on Aβ toxicity.
Conclusions
Understanding the intrinsic molecular flexibility, dynamics of interactions, and the structural behavior of the various Aβ peptides is crucial to comprehend the molecular mechanisms underlying the pathophysiology of Alzheimer’s disease. This will allow a more rational design of therapeutic intervention strategies to halt the disease progress and neutralize the malignant action of Aβ aggregation. To gain understanding of these events is difficult if not impossible to follow in real-time in the human brain. Therefore, these events are often mimicked in the test tube in research laboratories where information on Aβ behavior can be followed in molecular detail using advanced biophysical and biochemical assays in the course of seconds to hours or days, which happen in patients over a range of years.
The intrinsically disordered nature of Aβ raises the question of whether this peptide may act as signaling peptide, which is known to require a high degree of flexibility. It is striking to observe that many proteins involved in human diseases are in fact classified as IDPs (alpha-synuclein, tau, multiple prions) [188, 189]. This raises the question as to whether protein flexibility may act as a disease-contributing factor as opposed to the generally accepted idea that specific sizes or conformations of oligomeric forms of these peptides induce pathogenesis. In this review we state that different types of dynamics can be distinguished varying from inter- to intramolecular factors as well as external factors and that recent observations strongly indicate that indeed the contribution of dynamics to pathogenesis warrants further investigation. As the dynamic nature of Aβ and its ability to undergo conformational changes and aggregation has hampered its study, promising new experimental approaches and chemical tools [182] are being developed to address Aβ dynamics, having the major advantage that they can be used directly without the need for modification of Aβ with additional amino acids or fluorophores [110, 190]. While a lot has been learned in the past from the behavior of the Aβ system, it is clear that the picture is still incomplete and extremely complex. Variability in terms of space (intra- and extracellular space, brain compartments, patient-to-patient differences, etc.) and time (circadian rhythm, aging, lifestyle, etc.) imposes additional dynamical factors, emphasizing the importance to better understand the fluctuating microenvironment. Therefore, it is opportune to compare the Aβ system to a complex ecosystem or society, where minor perturbations might have profound effects that can result in cataclysmic events. Various Aβ alloforms interact and mutually influence each other’s behavior, but they also interact with the complex biological cell surface where they might exert a toxic effect by interfering with its normal functionality. Therefore, a holistic view of the dynamical Aβ ecosystem would enable us to initiate a successful ecosystem management strategy to prevent or remediate the AD pathobiology.
We summarized the evidence supporting the role of structural flexibility and in particular of the intrinsic protein disorder in the Aβ system to AD pathogenesis. A more systematic approach to the study of molecular flexibility in the Aβ system is required. This knowledge should then be integrated into future research efforts to optimize the clinical outcomes of drug trials.
References
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M (2010) Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 9(7):702–716
Huang Y, Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148(6):1204–1222
Hamaguchi T, Ono K, Yamada M (2006) Anti-amyloidogenic therapies: strategies for prevention and treatment of Alzheimer’s disease. Cell Mol Life Sci 63(13):1538–1552
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12(10):383–388
Takami M et al (2009) Gamma-secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci 29(41):13042–13052
Acx H et al (2013) Signature Aβ profiles are produced by different γ-secretase complexes. J Biol Chem 289(7):4346–4355
Finder VH, Vodopivec I, Nitsch RM, Glockshuber R (2010) The recombinant amyloid-beta peptide Abeta1-42 aggregates faster and is more neurotoxic than synthetic Abeta1-42. J Mol Biol 396(1):9–18
Kuperstein I et al (2010) Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J 29(19):3408–3420
Pauwels K et al (2012) Structural basis for increased toxicity of pathological aβ42:aβ40 ratios in Alzheimer disease. J Biol Chem 287(8):5650–5660
Vandersteen A et al (2012) Molecular plasticity regulates oligomerization and cytotoxicity of the multipeptide-length amyloid-β peptide pool. J Biol Chem 287(44):36732–36743
Yoshiike Y, Chui DH, Akagi T, Tanaka N, Takashima A (2003) Specific compositions of amyloid-beta peptides as the determinant of toxic beta-aggregation. J Biol Chem 278(26):23648–23655
Snyder SW et al (1994) Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys J 67(3):1216–1228
Jan A, Gokce O, Luthi-Carter R, Lashuel HA (2008) The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J Biol Chem 283(42):28176–28189
Yan Y, Wang C (2007) Abeta40 protects non-toxic Abeta42 monomer from aggregation. J Mol Biol 369(4):909–916
Morris AM, Watzky MA, Finke RG (2009) Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim Biophys Acta 1794(3):375–397
Finder VH, Glockshuber R (2007) Amyloid-beta aggregation. Neurodegener Dis 4(1):13–27
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10(9):698–712
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8(2):101–112
Jan A et al (2011) Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J Biol Chem 286(10):8585–8596
Tompa P (2009) Structural disorder in amyloid fibrils: its implication in dynamic interactions of proteins. FEBS J 276(19):5406–5415
Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890
Klyubin I et al (2008) Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci 28(16):4231–4237
Shankar GM et al (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8):837–842
Jin M et al (2011) Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA 108(14):5819–5824
Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K (1991) Aggregation and secondary structure of synthetic amyloid beta A4 peptides of Alzheimer’s disease. J Mol Biol 218(1):149–163
Tseng BP et al (1999) Deposition of monomeric, not oligomeric, Abeta mediates growth of Alzheimer’s disease amyloid plaques in human brain preparations. Biochemistry 38(32):10424–10431
Jan A, Hartley DM, Lashuel HA (2010) Preparation and characterization of toxic Abeta aggregates for structural and functional studies in Alzheimer’s disease research. Nat Protoc 5(6):1186–1209
Zhang S et al (2000) The Alzheimer’s peptide a beta adopts a collapsed coil structure in water. J Struct Biol 130(2–3):130–141
Riek R, Güntert P, Döbeli H, Wipf B, Wüthrich K (2001) NMR studies in aqueous solution fail to identify significant conformational differences between the monomeric forms of two Alzheimer peptides with widely different plaque-competence, A beta(1-40)(ox) and A beta(1-42)(ox). Eur J Biochem 268(22):5930–5936
Hou L et al (2004) Solution NMR studies of the A beta(1-40) and A beta(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc 126(7):1992–2005
Shao H, Jao S, Ma K, Zagorski MG (1999) Solution structures of micelle-bound amyloid beta-(1-40) and beta-(1-42) peptides of Alzheimer’s disease. J Mol Biol 285(2):755–773
Tomaselli S et al (2006) The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1-42) peptide in aqueous media is reversible: a step-by-step conformational analysis suggests the location of beta conformation seeding. Chem Bio Chem 7(2):257–267
Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A (2011) A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem Biophys Res Commun 411(2):312–316
Baumketner A et al (2006) Structure of the 21-30 fragment of amyloid beta-protein. Protein Sci 15(6):1239–1247
Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB (2005) On the nucleation of amyloid beta-protein monomer folding. Protein Sci 14(6):1581–1596
Danielsson J, Andersson A, Jarvet J, Gräslund A (2006) 15N relaxation study of the amyloid beta-peptide: structural propensities and persistence length. Magn Reson Chem 44 Spec No:S114–121
Yan Y, Wang C (2006) Abeta42 is more rigid than Abeta40 at the C terminus: implications for Abeta aggregation and toxicity. J Mol Biol 364(5):853–862
Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE (2007) The Alzheimer’s peptides Abeta40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J Mol Biol 368(5):1448–1457
Sgourakis NG et al (2011) Atomic-level characterization of the ensemble of the Aβ(1−42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms. J Mol Biol 405(2):570–583
Yang M, Teplow DB (2008) Amyloid beta-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J Mol Biol 384(2):450–464
Rosenman DJ, Connors CR, Chen W, Wang C, García AE (2013) Aβ monomers transiently sample oligomer and fibril-like configurations: ensemble characterization using a combined MD/NMR approach. J Mol Biol 425(18):3338–3359
Wright PE, Dyson HJ (2009) Linking folding and binding. Curr Opin Struct Biol 19(1):31–38
Tompa P, Szász C, Buday L (2005) Structural disorder throws new light on moonlighting. Trends Biochem Sci 30(9):484–489
Török M et al (2002) Structural and dynamic features of Alzheimer’s Abeta peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem 277(43):40810–40815
Kheterpal I, Zhou S, Cook KD, Wetzel R (2000) Abeta amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc Natl Acad Sci USA 97(25):13597–13601
Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R (2001) Structural features of the Abeta amyloid fibril elucidated by limited proteolysis. Biochemistry 40(39):11757–11767
Wang SS, Tobler SA, Good TA, Fernandez EJ (2003) Hydrogen exchange-mass spectrometry analysis of beta-amyloid peptide structure. Biochemistry 42(31):9507–9514
Sunde M et al (1997) Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol 273(3):729–739
Jahn TR et al (2010) The common architecture of cross-beta amyloid. J Mol Biol 395(4):717–727
Petkova AT et al (2002) A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA 99(26):16742–16747
Bertini I, Gonnelli L, Luchinat C, Mao J, Nesi A (2011) A new structural model of Aβ40 fibrils. J Am Chem Soc 133(40):16013–16022
Whittemore NA et al (2005) Hydrogen-deuterium (H/D) exchange mapping of Abeta(1-40) amyloid fibril secondary structure using nuclear magnetic resonance spectroscopy. Biochemistry 44(11):4434–4441
Luhrs T et al (2005) 3D structure of Alzheimer’s amyloid-beta(1−42) fibrils. Proc Natl Acad Sci USA 102(48):17342–17347
Olofsson A, Sauer-Eriksson AE, Ohman A (2006) The solvent protection of alzheimer amyloid-beta-(1-42) fibrils as determined by solution NMR spectroscopy. J Biol Chem 281(1):477–483
Morimoto A et al (2004) Analysis of the secondary structure of beta-amyloid (Abeta42) fibrils by systematic proline replacement. J Biol Chem 279(50):52781–52788
Kheterpal I, Chen M, Cook KD, Wetzel R (2006) Structural differences in Abeta amyloid protofibrils and fibrils mapped by hydrogen exchange–mass spectrometry with on-line proteolytic fragmentation. J Mol Biol 361(4):785–795
Sawaya MR et al (2007) Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 447(7143):453–457
Williams AD et al (2004) Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol 335(3):833–842
Petkova AT et al (2005) Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 307(5707):262–265
Scheidt HA, Morgado I, Rothemund S, Huster D, Fändrich M (2011) Solid-state NMR spectroscopic investigation of Aβ protofibrils: implication of a β-sheet remodeling upon maturation into terminal amyloid fibrils. Angew Chem Int Ed Engl 50(12):2837–2840
Scheidt HA, Morgado I, Rothemund S, Huster D (2012) Dynamics of amyloid β fibrils revealed by solid-state NMR. J Biol Chem 287(3):2017–2021
Morel B, Varela L, Conejero-Lara F (2010) The thermodynamic stability of amyloid fibrils studied by differential scanning calorimetry. J Phys Chem B 114(11):4010–4019
Ma J et al (2013) Intrinsic structural heterogeneity and long-term maturation of amyloid β peptide fibrils. ACS Chem Neurosci 4(8):1236–1243
Nyström S et al (2013) Evidence for age-dependent in vivo conformational rearrangement within Aβ amyloid deposits. ACS Chem Biol 8(6):1128–1133
Fändrich M, Meinhardt J, Grigorieff N (2009) Structural polymorphism of Alzheimer Abeta and other amyloid fibrils. Prion 3(2):89–93
Tycko R, Wickner RB (2013) Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc Chem Res 46(7):1487–1496
Crowther RA, Goedert M (2000) Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol 130(2–3):271–279
Jiménez JL, Tennent G, Pepys M, Saibil HR (2001) Structural diversity of ex vivo amyloid fibrils studied by cryo-electron microscopy. J Mol Biol 311(2):241–247
Bousset L et al (2013) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4:2575
Kodali R, Williams AD, Chemuru S, Wetzel R (2010) Abeta(1-40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated. J Mol Biol 401(3):503–517
Klement K et al (2007) Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer’s abeta(1-40) amyloid fibrils. J Mol Biol 373(5):1321–1333
Pedersen JS, Otzen DE (2008) Amyloid-a state in many guises: survival of the fittest fibril fold. Protein Sci 17(1):2–10
Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, Fändrich M (2009) Abeta(1-40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J Mol Biol 386(3):869–877
Qiang W, Yau WM, Luo Y, Mattson MP, Tycko R (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc Natl Acad Sci USA 109(12):4443–4448
Qiang W, Kelley K, Tycko R (2013) Polymorph-specific kinetics and thermodynamics of β-amyloid fibril growth. J Am Chem Soc 135(18):6860–6871
Sachse C et al (2006) Quaternary structure of a mature amyloid fibril from Alzheimer’s Abeta(1-40) peptide. J Mol Biol 362(2):347–354
Sachse C, Fändrich M, Grigorieff N (2008) Paired beta-sheet structure of an Abeta(1-40) amyloid fibril revealed by electron microscopy. Proc Natl Acad Sci USA 105(21):7462–7466
Schmidt M et al (2009) Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci USA 106(47):19813–19818
Zhang R et al (2009) Interprotofilament interactions between Alzheimer’s Abeta1-42 peptides in amyloid fibrils revealed by cryoEM. Proc Natl Acad Sci USA 106(12):4653–4658
Goldsbury CS et al (2000) Studies on the in vitro assembly of a beta 1-40: implications for the search for a beta fibril formation inhibitors. J Struct Biol 130(2–3):217–231
Goldsbury C, Frey P, Olivieri V, Aebi U, Müller SA (2005) Multiple assembly pathways underlie amyloid-beta fibril polymorphisms. J Mol Biol 352(2):282–298
Petkova AT, Yau WM, Tycko R (2006) Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry 45(2):498–512
Paravastu AK, Leapman RD, Yau WM, Tycko R (2008) Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci USA 105(47):18349–18354
Cameron B, Landreth GE (2010) Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis 37(3):503–509
Ill-Raga G et al (2010) Amyloid-β peptide fibrils induce nitro-oxidative stress in neuronal cells. J Alzheimers Dis 22(2):641–652
McKee AC, Kosik KS, Kowall NW (1991) Neuritic pathology and dementia in Alzheimer’s disease. Ann Neurol 30(2):156–165
Berg L et al (1998) Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55(3):326–335
Seilheimer B et al (1997) The toxicity of the Alzheimer’s beta-amyloid peptide correlates with a distinct fiber morphology. J Struct Biol 119(1):59–71
Lu JX et al (2013) Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154(6):1257–1268
Ihse E et al (2008) Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J Pathol 216(2):253–261
Benilova I, Karran E, De Strooper B (2012) The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15(3):349–357
Broersen K, Rousseau F, Schymkowitz J (2010) The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: oligomer size or conformation? Alzheimers Res Ther 2(4):12
Kayed R, Lasagna-Reeves CA (2013) Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis 33(Suppl 1):S67–S78
Ono K, Condron MM, Teplow DB (2009) Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci USA 106(35):14745–14750
Cizas P et al (2010) Size-dependent neurotoxicity of beta-amyloid oligomers. Arch Biochem Biophys 496(2):84–92
Campioni S et al (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol 6(2):140–147
Nekooki-Machida Y et al (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci USA 106(24):9679–9684
Ladiwala AR et al (2012) Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J Biol Chem 287(29):24765–24773
Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26(22):6011–6018
Kayed R et al (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300(5618):486–489
Glabe CG (2008) Structural classification of toxic amyloid oligomers. J Biol Chem 283(44):29639–29643
Kayed R et al (2009) Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J Biol Chem 284(7):4230–4237
Kayed R et al (2010) Conformation-dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol Neurodegener 5:57
Cerf E et al (2009) Antiparallel beta-sheet: a signature structure of the oligomeric amyloid beta-peptide. Biochem J 421(3):415–423
Sarroukh R et al (2011) Transformation of amyloid β(1-40) oligomers into fibrils is characterized by a major change in secondary structure. Cell Mol Life Sci 68(8):1429–1438
Celej MS et al (2012) Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem J 443(3):719–726
Vandersteen A et al (2012) A comparative analysis of the aggregation behavior of amyloid-β peptide variants. FEBS Lett 586(23):4088–4093
Laganowsky A et al (2012) Atomic view of a toxic amyloid small oligomer. Science 335(6073):1228–1231
Gu L, Liu C, Guo Z (2013) Structural insights into Aβ42 oligomers using site-directed spin labeling. J Biol Chem 288(26):18673–18683
Zhang Y et al (2013) Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc Natl Acad Sci USA 110(36):14604–14609
Ahmed M et al (2010) Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat Struct Mol Biol 17(5):561–567
Bolognesi B et al (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chem Biol 5(8):735–740
Williams TL et al (2011) Aβ42 oligomers, but not fibrils, simultaneously bind to and cause damage to ganglioside-containing lipid membranes. Biochem J 439(1):67–77
Teplow DB (2013) On the subject of rigor in the study of amyloid β-protein assembly. Alzheimers Res Ther 5(4):39
Lesné S et al (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440(7082):352–357
Qi-Takahara Y et al (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci 25(2):436–445
Wiltfang J et al (2002) Highly conserved and disease-specific patterns of carboxyterminally truncated Abeta peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J Neurochem 81(3):481–496
Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB (1993) Characterization of beta-amyloid peptide from human cerebrospinal fluid. J Neurochem 61(5):1965–1968
Masters CL, Selkoe DJ (2012) Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2(6):a006262
Moro ML, Collins MJ, Cappellini E (2010) Alzheimer’s disease and amyloid beta-peptide deposition in the brain: a matter of ‘aging’? Biochem Soc Trans 38(2):539–544
Mori H et al (1994) Racemization: its biological significance on neuropathogenesis of Alzheimer’s disease. Tohoku J Exp Med 174(3):251–262
Kubo T, Kumagae Y, Miller CA, Kaneko I (2003) Beta-amyloid racemized at the Ser26 residue in the brains of patients with Alzheimer disease: implications in the pathogenesis of Alzheimer disease. J Neuropathol Exp Neurol 62(3):248–259
Kuo YM, Webster S, Emmerling MR, De Lima N, Roher AE (1998) Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of A beta peptides of Alzheimer’s disease. Biochim Biophys Acta 1406(3):291–298
Milton NG (2001) Phosphorylation of amyloid-beta at the serine 26 residue by human cdc2 kinase. Neuroreport 12(17):3839–3844
Kumar S et al (2011) Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J 30(11):2255–2265
Dong J et al (2003) Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42(10):2768–2773
Hou L et al (2013) Modification of amyloid-β1-42 fibril structure by methionine-35 oxidation. J Alzheimers Dis 37(1):9–18
Vitek MP et al (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA 91(11):4766–4770
Jawhar S, Wirths O, Bayer TA (2011) Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. J Biol Chem 286(45):38825–38832
Brown AM, Lemkul JA, Schaum N, Bevan DR (2014) Simulations of monomeric amyloid β-peptide (1-40) with varying solution conditions and oxidation state of Met35: implications for aggregation. Arch Biochem Biophys 545:44–52
Srikanth V et al (2011) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32(5):763–777
Gu L, Guo Z (2013) Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J Neurochem 126(3):305–311
Serio TR et al (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289(5483):1317–1321
Esler WP et al (2000) Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 39(21):6288–6295
Brännström K, Ohman A, Olofsson A (2011) Aβ peptide fibrillar architectures controlled by conformational constraints of the monomer. PLoS One 6(9):e25157
Johnson RD et al (2013) Single-molecule imaging reveals aβ42:aβ40 ratio-dependent oligomer growth on neuronal processes. Biophys J 104(4):894–903
Necula M, Kayed R, Milton S, Glabe CG (2007) Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282(14):10311–10324
Necula M et al (2007) Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry 46(30):8850–8860
Martins IC et al (2008) Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J 27(1):224–233
Hepler RW et al (2006) Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry 45(51):15157–15167
Chong SA et al (2011) Synaptic dysfunction in hippocampus of transgenic mouse models of Alzheimer’s disease: a multi-electrode array study. Neurobiol Dis 44(3):284–291
Fawzi NL, Ying J, Torchia DA, Clore GM (2010) Kinetics of amyloid beta monomer-to-oligomer exchange by NMR relaxation. J Am Chem Soc 132(29):9948–9951
Krishnamoorthy J, Brender JR, Vivekanandan S, Jahr N, Ramamoorthy A (2012) Side-chain dynamics reveals transient association of Aβ(1-40) monomers with amyloid fibers. J Phys Chem B 116(46):13618–13623
Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM (2011) Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 480(7376):268–272
Suzuki Y et al (2013) Resolution of oligomeric species during the aggregation of Aβ1-40 using (19)F NMR. Biochemistry 52(11):1903–1912
Hartl FU, Hayer-Hartl M (2009) Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 16(6):574–581
Carulla N, Zhou M, Giralt E, Robinson CV, Dobson CM (2010) Structure and intermolecular dynamics of aggregates populated during amyloid fibril formation studied by hydrogen/deuterium exchange. Acc Chem Res 43(8):1072–1079
Cruz L et al (1997) Aggregation and disaggregation of senile plaques in Alzheimer disease. Proc Natl Acad Sci USA 94(14):7612–7616
Koffie RM et al (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA 106(10):4012–4017
Cohen SI et al (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA 110(24):9758–9763
Spires-Jones TL et al (2009) Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neurobiol Dis 33(2):213–220
Xue WF, Hellewell AL, Hewitt EW, Radford SE (2010) Fibril fragmentation in amyloid assembly and cytotoxicity: when size matters. Prion 4(1):20–25
Cremades N et al (2012) Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149(5):1048–1059
Wogulis M et al (2005) Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neurosci 25(5):1071–1080
Tiiman A, Palumaa P, Tõugu V (2013) The missing link in the amyloid cascade of Alzheimer’s disease-metal ions. Neurochem Int 62(4):367–378
Pithadia AS, Lim MH (2012) Metal-associated amyloid-β species in Alzheimer’s disease. Curr Opin Chem Biol 16(1–2):67–73
Watt AD, Villemagne VL, Barnham KJ (2013) Metals, membranes, and amyloid-β oligomers: key pieces in the Alzheimer’s disease puzzle? J Alzheimers Dis 33(Suppl 1):S283–S293
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10(5):333–344
Holtzman DM, Herz J, Bu G (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2(3):a006312
Mahley RW, Weisgraber KH, Huang Y (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci USA 103(15):5644–5651
Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein E in Alzheimer’s disease. Neuron 63(3):287–303
Ramesh BN, Rao TS, Prakasam A, Sambamurti K, Rao KS (2010) Neuronutrition and Alzheimer’s disease. J Alzheimers Dis 19(4):1123–1139
Smid SD, Maag JL, Musgrave IF (2012) Dietary polyphenol-derived protection against neurotoxic β-amyloid protein: from molecular to clinical. Food Funct 3(12):1242–1250
Radak Z et al (2010) Exercise plays a preventive role against Alzheimer’s disease. J Alzheimers Dis 20(3):777–783
Farina N, Rusted J, Tabet N (2013) The effect of exercise interventions on cognitive outcome in Alzheimer’s disease: a systematic review. Int Psychogeriatr 1–10
Hoffmann K et al (2013) preserving cognition, quality of life, physical health and functional ability in Alzheimer’s disease: the effect of physical exercise (ADEX trial): rationale and design. Neuroepidemiology 41(3–4):198–207
Li S et al (2013) Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron 77(5):929–941
Silva T, Teixeira J, Remião F, Borges F (2013) Alzheimer’s disease, cholesterol, and statins: the junctions of important metabolic pathways. Angew Chem Int Ed Engl 52(4):1110–1121
Ju YE, Lucey BP, Holtzman DM (2013) Sleep and Alzheimer disease pathology-a bidirectional relationship. Nat Rev Neurol
Rezaei-Ghaleh N, Giller K, Becker S, Zweckstetter M (2011) Effect of zinc binding on β-amyloid structure and dynamics: implications for Aβ aggregation. Biophys J 101(5):1202–1211
Syme CD, Nadal RC, Rigby SE, Viles JH (2004) Copper binding to the amyloid-beta (Abeta) peptide associated with Alzheimer’s disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta-(1-28): insights from a range of complementary spectroscopic techniques. J Biol Chem 279(18):18169–18177
Peters I et al (2009) The interaction of beta-amyloid protein with cellular membranes stimulates its own production. Biochim Biophys Acta 1788(5):964–972
Williams TL, Serpell LC (2011) Membrane and surface interactions of Alzheimer’s Aβ peptide–insights into the mechanism of cytotoxicity. FEBS J 278(20):3905–3917
Sciacca MF et al (2012) Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys J 103(4):702–710
Kotler SA, Walsh P, Brender JR, Ramamoorthy A (2014) Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem Soc Rev. doi:10.1039/C3CS60431D
Burke KA, Yates EA, Legleiter J (2013) Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front Neurol 4:17
Magrané J, Smith RC, Walsh K, Querfurth HW (2004) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci 24(7):1700–1706
Fonte V et al (2002) Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci USA 99(14):9439–9444
Evans CG, Wisén S, Gestwicki JE (2006) Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. J Biol Chem 281(44):33182–33191
Knight SD, Presto J, Linse S, Johansson J (2013) The BRICHOS domain, amyloid fibril formation, and their relationship. Biochemistry 52(43):7523–7531
Koren J et al (2009) Chaperone signalling complexes in Alzheimer’s disease. J Cell Mol Med 13(4):619–630
Lee S et al (2014) Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer’s disease. J Am Chem Soc 136(1):299–310
Ramamoorthy A, Lim MH (2013) Structural characterization and inhibition of toxic amyloid-β oligomeric intermediates. Biophys J 105(2):287–288
DeToma AS, Salamekh S, Ramamoorthy A, Lim MH (2012) Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem Soc Rev 41(2):608–621
Exley C (2005) The aluminium-amyloid cascade hypothesis and Alzheimer’s disease. Subcell Biochem 38:225–234
Roh JH et al (2012) Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med 4(150):150ra122
Kang JE et al (2009) Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326(5955):1005–1007
Deleersnijder A, Gerard M, Debyser Z, Baekelandt V (2013) The remarkable conformational plasticity of alpha-synuclein: blessing or curse? Trends Mol Med 19(6):368–377
Uversky VN (2014) The triple power of D3: protein intrinsic disorder in degenerative diseases. Front Biosci (Landmark Ed) 19:181–258
Santi S et al (2013) Real-time amyloid aggregation monitoring with a photonic crystal-based approach. Chem Phys Chem 14(15):3476–3482
Kheterpal I, Wetzel R (2006) Hydrogen/deuterium exchange mass spectrometry: a window into amyloid structure. Acc Chem Res 39(9):584–593
Acknowledgments
EH is supported by a FWO doctoral fellowship. NvN is supported by the VIB and the Flemish Hercules Foundation. KB is supported by a grant from the Internationale Stichting Alzheimer Onderzoek (ISAO), an Odysseus II award from FWO and a UTWIST fellowship. KP is the recipient of a FWO Pegasus long-term postdoctoral fellowship and is supported by the Stichting Alzheimer Onderzoek (SAO-FRA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Hubin, E., van Nuland, N.A.J., Broersen, K. et al. Transient dynamics of Aβ contribute to toxicity in Alzheimer’s disease. Cell. Mol. Life Sci. 71, 3507–3521 (2014). https://doi.org/10.1007/s00018-014-1634-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1634-z