
Abstract
Pulmonary arterial remodeling is a presently irreversible pathologic hallmark of pulmonary arterial hypertension (PAH). This complex disease involves pathogenic dysregulation of all cell types within the small pulmonary arteries contributing to vascular remodeling leading to intimal lesions, resulting in elevated pulmonary vascular resistance and right heart dysfunction. Mutations within the bone morphogenetic protein receptor 2 gene, leading to dysregulated proliferation of pulmonary artery smooth muscle cells, have been identified as being responsible for heritable PAH. Indeed, the disease is characterized by excessive cellular proliferation and resistance to apoptosis of smooth muscle and endothelial cells. Significant gene dysregulation at the transcriptional and signaling level has been identified. MicroRNAs are small non-coding RNA molecules that negatively regulate gene expression and have the ability to target numerous genes, therefore potentially controlling a host of gene regulatory and signaling pathways. The major role of miRNAs in pulmonary arterial remodeling is still relatively unknown although research data is emerging apace. Modulation of miRNAs represents a possible therapeutic target for altering the remodeling phenotype in the pulmonary vasculature. This review will focus on the role of miRNAs in regulating smooth muscle and endothelial cell phenotypes and their influence on pulmonary remodeling in the setting of PAH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung vasculopathy is an irreversible pathologic hallmark of the lung vascular disorder pulmonary arterial hypertension (PAH). PAH is an often fatal and increasingly prevalent disease that is manifested by a maladaptive elevation of pulmonary vascular resistance and pulmonary arterial pressure, consequently leading to right heart failure and eventual death. Clinically, the disease is defined as a mean pulmonary artery pressure of >25 mmHg at rest [1].
There are three main forms of PAH; idiopathic (IPAH), in which the cause is unknown, familial (FPAH), and PAH associated with other risk factors (APAH), such as HIV infection, collagen vascular diseases, and congenital heart disease [2]. The main genetic defect associated with PAH is a mutation in the gene encoding bone morphogenetic protein receptor 2 (BMPR2). Germline mutations in BMPR2 were originally identified in patients with FPAH [3, 4]. In these families, the disease segregates in an autosomal dominant fashion, with markedly reduced penetrance of approximately 20–30 % [5]. As such, many patients who carry the disease gene do not develop clinical PAH. In addition, up to 25 % of patients with apparently sporadic IPAH have been found to harbor similar mutations [6]. A proportion of these mutation carriers are examples of FPAH where the condition has not manifested in relatives due to low penetrance, while others are examples of de novo mutations. The low penetrance of the disease among BMPR2 mutation carriers suggests that other factors are important in the manifestation of clinical PAH and that a “second hit” in addition to a mutation in BMPR2 is required to establish PAH [7].
The incidence of PAH varies from 1.1, 2.0, and 2.4 per million of adult population per year in the UK and Ireland, USA, and France, respectively [8–10]. Recent studies show that females are more susceptible to developing PAH with a female-to-male ratio of 4.3:1 [11] in PAH and 4.1:1 in IPAH [12]. However, severity and survival is worse in males who have developed the disease than in females [13]. This clear difference between the genders is an intriguing phenomenon and much work is underway to identify the role of sex hormones such as estrogen on the development and maintenance of PAH.
Current treatments for PAH include endothelin-1 receptor antagonists, phosphodiesterase type 5 inhibitors, and administration of prostacyclins [14]. Although current therapies do indeed provide a survival benefit, mortality rates still remain high and the treatment does not prevent the aggressive progression of the disease. As a result, newer treatments are required to more effectively manage PAH and regulate the cellular components resulting in pulmonary remodeling. Contributing factors leading to remodeling include vessel injury, hypoxic exposure, and inflammation, resulting in severe remodeling of predominantly the small pulmonary vessels [15]. This remodeling process involves interaction between all cell types present in the distinct layers of the pulmonary arteries causing histological changes to the pulmonary vessel wall [16, 17] (Fig. 1).

Pathogenesis of pulmonary arterial hypertension. Stress to the small pulmonary arteries results in endothelial dysregulation and proliferation in the intima, smooth muscle cell proliferation, and resistance to apoptosis within the medial layer, along with adventitial fibroblast activation. This culminates in vasoconstriction and remodeling of the pulmonary vessels, which can result in plexiform lesions in human PAH
The principal cell type within the adventitia layer of the vessel wall is the fibroblast, and adventitial thickening is observed due to hyperplasia of these cells [18, 19]. Activation of the fibroblasts results in a release of growth factors such as endothelin-1, serotonin, and platelet-derived growth factor (PDGF) [20] causing migration, proliferation, and contraction of fibroblasts and smooth muscle cells (SMC) [21]. An increase in reactive oxygen species (ROS) in the adventitia by NADPH oxidase can lead to activation of fibroblasts. From these activated adventitial fibroblasts, a subset population differentiate into myofibroblasts, characterized by α-SMA expression [22]. Myofibroblasts are mesenchymal cells that can be induced under the control of growth factors such as transforming growth factor (TGF-β1) [23]. These cells have the ability to migrate into the medial layer, increasing medial thickness. Another role of myofibroblasts is the production and deposition of extracellular matrix proteins, such as collagen, elastin, fibronectin, and tenascin-C, which can induce smooth muscle cell (SMC) proliferation and migration within the media [15, 24, 25]. The medial layer within the vessel contains SMCs, which are regulated under the control of many growth factors and cytokines [26]. These factors influence the phenotype of the SMC inducing proliferation, migration, and contraction, while inhibiting apoptosis. Infiltration of the media with myofibroblasts and hypertrophy of SMCs results in muscularization of normally non-muscular or partially muscular distal arteries augmenting medial thickening. Hypertrophy and excessive proliferation of endothelial cells (EC) contributes to increased intimal thickening and endothelial dysfunction, leading to an increase in vasoconstrictor molecules, such as endothelin-1 (ET-1) [27]. It is suggested that EC dysfunction induces release of elastase, which activates matrix metalloproteinases (MMPs) and in turn tenascin-C, therefore further enhancing SMC proliferation in the medial layer [25, 28].
The progression of pulmonary arterial remodeling is through increased proliferation and resistance to apoptosis of ECs and SMCs. In particular, hyperproliferation of ECs can result in a clustered appearance resulting in the formation of complex plexiform lesions [29], occlusion of the blood vessel, and loss of blood flow to the small distal pulmonary arteries [30]. Although pulmonary arterial remodeling is a relatively well-studied condition, the exact cellular and molecular processes that lead to initiation and progression of PAH are still being investigated and are not completely understood. The process is proposed to be due to changes in transcriptional and signaling mechanisms within all three layers of the vessel wall. Thus, suggesting that controlling gene expression and hence protein expression, will allow modulation of the pathways involved in a cell-specific manner and result in more effective treatments for PAH.
miRNA biogenesis
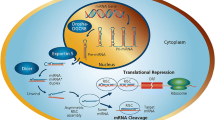
MiRNAs are small non-coding RNA molecules around 22 nucleotides long that target the 3′-untranslated region (UTR) of mRNA to negatively regulate gene expression [31, 32]. Transcription by RNA polymerase II gives rise to the primary miRNA (pri-miRNA) molecule, which can be over 1 kb in size and can produce several mature miRNAs. This primary structure is processed by the RNase III enzyme Drosha in the presence of cofactor DGCR8 to form a ~60-nucleotide stem loop molecule; this is the precursor miRNA (pre-miRNA). Pre-miRNAs are transported out of the nucleus into the cytoplasm by Exportin-5 where they are cleaved by the endonuclease Dicer into a microRNA duplex. This duplex is then bound to an argonaute protein (Ago2) [33, 34] and the mature miRNA is formed by stabilization of the guide strand (identified by the suffix 5p) while the passenger strand (identified by the suffix 3p) was thought to be degraded, however, there is increasing evidence that the function of 3p-derived miRNAs can be as important as the 5p strand [35, 36] (discussed below). The mature miRNA is incorporated into the RNA-induced silencing complex (RISC) where nucleotides 2–8 of the miRNA (“seed” region) bind to the 3′-UTR of the target mRNA sequence. If the complementarity between mRNA and miRNA is complete, cleavage of the mRNA occurs. However, if complementation is incomplete, inhibition of mRNA translation occurs, leading to gene silencing [32, 37, 38] (Fig. 2). This results in miRNAs potentially regulating a multitude of RNA species [39]. Of the non-coding RNA family, miRNAs are one of the largest families that have a relevant and well-studied function. Little is known about the other members of the non-coding RNA family (e.g., long non-coding RNA, lncRNA) however, recent data implicates these lncRNAs in heart development [40] and consequently these molecules are likely to emerge as important regulators of tissue development within different disease models.

MiRNA biogenesis. Transcription by RNA polymerase II gives rise to the pri-miRNA, which can produce multiple mature miRNAs. Processing by RNase III enzyme Drosha along with cofactor DGCR8 produces the stem-loop pre-miRNA which is exported out of the nucleus by Exportin-5. In the cytoplasm, cleavage by Dicer results in an miRNA duplex, ~22 nucleotides long. The mature miRNA is incorporated into the RNA-induced silencing complex (RISC) and targets the 3′-UTR of mRNA. Gene silencing is achieved by either mRNA degradation or translational repression
Although the majority of miRNAs follow the canonical biogenesis mentioned above, there are regulatory molecules that aid (or indeed impede) formation of the mature miRNA, thus illustrating how different miRNAs are processed by distinct methods [41]. KH-type splicing regulatory protein (KHSRP) has been found to bind to the terminal stem loop of certain pre-miRNAs (in particular pre-miR-21 and pre-miR-126a) within the nucleus to help recruit Exportin-5 and therefore stimulate miRNA maturation [42]. Similarly, HIV trans-activating response RNA binding protein (TARBP2) can work in concert with Dicer to stabilize the miRNA-Dicer complex as well as ensure efficient recruitment of Ago2 [43]. Conversely, nuclear factor 90 (NF90) and NF45 are found within the nucleus and form a complex that binds to the pri-miRNA, thereby preventing Drosha from cleaving the primary miRNA. This results in reduced levels of pre-miRNA and in turn mature miRNA [44]. It has also been discovered that distinct miRNAs are capable of being processed in a Dicer-independent manner. In particular, miR-451 precursor is a short stem loop sequence and is loaded directly into Ago2 [45, 46]. Of the four argonaute proteins found to be ubiquitously expressed in humans, Ago2 is responsible for the direct cleavage of mRNA when working in concert with RISC. As well as this, Ago2 can also act as an endonuclease and hence cleaves pre-miR-451 independent of Dicer [33, 34, 45, 46].
Until recently, it was thought that the guide strand was always incorporated into the RISC complex and the passenger strand was degraded with no biological role. However, evidence now suggests that both stands of the duplex can have functional roles and both may be important in different signaling cascades [35, 36]. A recent study by Eulalio [35] found that ectopic expression of miR-590-3p and miR-199-3p increased cardiomyocyte proliferation both in vitro and in vivo, illustrating the new emerging role for the 3p strand of the miRNA duplex.
Owing to their pleiotropic vascular effects, miRNAs are thought to function as upstream molecular factors that integratively coordinate pathogenic signaling pathways in PAH. A spectrum of PAH-relevant insults such as hypoxia and inflammation are essential modulators of miRNA expression in vascular cells. Consequently, a number of these dynamically regulated miRNAs are thought to be central regulators in vascular homeostasis essential for thrombosis, metabolism, cellular proliferation, and cell fate. Despite this, only a distinct few miRNAs have been molecularly linked to PAH. Hence, there are undoubtedly a significant number of presently unidentified miRNAs that have an essential role in pulmonary vascular function. Due to the complex molecular pathology of this disease, the combined utilization of bioinformatics and experimental biology may be especially pertinent in deciphering the pervasiveness of miRNA biology in PAH. This review will provide a comprehensive overview concerning miRNA function in pulmonary vascular homeostasis and discuss the contribution of miRNA biology in pulmonary vascular remodeling and disease. We will also speculate on the elucidation of presently unknown miRNAs in PAH and comment on the exciting application of miRNA-based therapies in the treatment of pulmonary vascular disease.
BMP pathway in pulmonary remodeling
There is a large cohort of data implicating the BMP pathway in the etiology of PAH as heterozygous mutations within the gene encoding bone morphogenetic protein type II receptor (BMPR2) have been found in ~70 % of FPAH [3, 4] and ~26 % of IPAH cases [6]. BMPR2 is a serine/threonine receptor kinase that binds the TGF-β superfamily of ligands. The resulting outcome of BMP signaling is cell and site specific. Under normal conditions, BMP-4 signals via a SMAD-dependent pathway to inhibit proliferation in pulmonary artery SMCs (PASMCs) [47]. TGF-β often antagonizes the effect of BMP signaling and is thought to signal through the activin receptor like kinase (ALK5) and Smad-2 and -3 [48]. The BMP signaling pathway is also controlled by a number of negative regulatory molecules, such as Smad-6/-7, which inhibit phosphorylation of Smad-1 and -5 [49, 50] and Smad ubiquitin regulatory factor 1 (Smurf1) resulting in degradation of Smad-1 and -5 [51]. Chan et al. [52] illustrated that certain proteins interact with the terminal domain of BMPR2 (BMPR2-TD), in particular Tribbles 3 (Trb3). Upon BMP4 stimulation of BMPR2, Trb3 dissociates from the terminal domain of the receptor and binds to Smurf1 inducing degradation of Smurf1 via the ubiquitin–proteasome pathway. Reduction in Smurf1 increases signaling through the BMP-Smad-dependent pathway and promotes the contractile phenotype in vascular SMCs [52].
Many mutations have been discovered within the BMPR2 gene encoding region from patients diagnosed with PAH. However, kindred studies have shown that only ~20 % of people with these mutations go on to develop PAH [5], thus indicating that there may be environmental or additional genetic risk factors involved. PAH patients with a heterozygous mutation in the gene encoding BMPR2 demonstrate lowered levels of BMPR2 protein [53] and PASMCs from these patients have an altered response to the BMP growth factors [54]. Similarly, animal models of PAH show a reduction in BMPR2 protein levels in the lungs [55]. Genetic ablation of BMPR2 proves lethal due to lack of mesoderm [56], while BMPR2+/− mice do not spontaneously develop PAH. However, when BMPR2+/− mice are exposed to a secondary insult (e.g., serotonin), increased pulmonary artery pressures and pulmonary remodeling ensues [57]. This strengthens the idea that BMPR2 mutations predispose patients to develop PAH and that other factors are involved in developing the disease.
Screening of miRNAs dysregulated in PAH
The first study to report dysregulation of miRNAs during the development of PAH was by Caruso et al. [58]. Using a microarray followed by validation by quantitative polymerase chain reaction, it was found that miR-22, miR-30, and let-7f were downregulated while miR-322 and miR-451 were upregulated in two commonly used rodent models of PAH (exposure to chronic hypoxia and monocrotaline (MCT) insult in the rat) [58]. Another study showed that in human pulmonary arteries from PAH patients, miR-138, miR-367, miR-27b, miR302b, miR-145, and miR-450a were up-regulated while miR-204 was down-regulated compared to control patient pulmonary arteries [59]. This fundamental work highlighted the possible role of specific miRNAs during disease development.
In contrast, others have used a candidate approach to identify miRNAs involved in the remodeling process. MiRNAs which affect signaling pathways involved in the phenotypic changes during remodeling are studied along with their target genes, focusing specifically on the changes in expression during disease initiation and progression [60].
miR-204
An elegant study by Courboulin et al. [59] was the first to confirm a mechanistic link between miRNAs and cellular pathways affecting pulmonary arterial remodeling. MiR-204 is located within and transcribed from intron 6 of the transient receptor potential melastatin 3 (TRPM3) gene on human chromosome 9. MiR-204 and TRPM3 are transcribed as a single unit [61] however, the effects of miR-204 on signaling pathways involved in PAH do not appear to be mediated via TRPM3 [59]. MiR-204 has been implicated in the development of several tissues (e.g., maintaining epithelial barrier function in eye development [62]) and disease states. The expression of miR-204 has been linked with survival rate in neuroblastoma [63] and miR-204 is found to be down-regulated in tumor tissue from gastric cancer patients [64]. Both studies found that miR-204 directly targets Bcl-2 (B cell lymphoma 2), an inhibitor of apoptosis and with suppressed levels of miR-204, Bcl-2 levels were de-repressed therefore promoting inhibition of apoptosis [63, 64].
With regard to pulmonary remodeling, miR-204 was first observed to be dysregulated in the MCT and hypoxic rat model of PAH with levels of miR-204 down-regulated in the lungs from the disease model [58] as well as in the hypoxic mouse lung [59]. In situ hybridization demonstrated localization of miR-204 within the PASMC compartment and miR-204 expression was reduced in PASMCs from IPAH patients, accompanied by increased levels of proliferation and lower levels of apoptosis compared to control PASMCs. Additionally, treatment of MCT rats with synthetic miR-204 mimic significantly lowered pulmonary artery pressure and medial wall thickness in small pulmonary arteries [59]. It will be important to define, for potential therapeutic purposes, the optimal mimic delivery strategy that leads to effective and safe mimic administration to the pulmonary vascular compartment in vivo.
The effect of miR-204 on proliferation and apoptosis is proposed to be via the Src-STAT3-NFAT/BMPR2 pathway (Fig. 3). During the initiation of PAH, circulating vasoconstrictor molecules such as endothelin-1 and PDGF activate STAT3, which can directly bind to miR-204 leading to down-regulation of this miRNA. This causes an increase in miR-204 target gene SHP2, a tyrosine phosphatase involved in growth factor signaling, leading to activation of Src and increasing STAT3 activation [59]. This positive feedback of STAT3 may indicate why the progression of pulmonary arterial remodeling is so severe. STAT3 has been shown to directly activate the serine/threonine protein kinase Pim1 which in turn triggers activation and nuclear translocation of NFATc2. In vivo inhibition of Pim1was able to reverse MCT-induced PAH in rats while Pim1−/− mice were resistant to PAH development [65]. NFATc2 is up-regulated in pulmonary arteries from PAH patients [66] and animal models of PAH have illustrated a down-regulation of voltage-gated potassium channels, in particular Kv1.5 [67]. A key study by Bonnet et al. [66] found that NFATc2 activation decreased the expression of Kv1.5 resulting in PASMC depolarization and opening of the voltage gated calcium channels, hence increasing intracellular levels of potassium ([K+]i) and calcium ([Ca2+]i). Increased [Ca2+]i promotes further NFATc2 activity and leads to vasoconstriction and PASMC proliferation [68]. NFATc2 also increased anti-apoptotic Bcl-2 leading to mitochondrial hyperpolarization and resistance to apoptosis [66]. Treatment of PAH PASMCs with VIVIT (a direct inhibitor of NFAT) or cyclosporine A (an indirect inhibitor of NFAT), lowered levels of nuclear activated NFATc2, reduced [K+]i and [Ca2+]i and depolarized PASMC mitochondria to levels comparable to normal PASMCs. Similar results were obtained in vivo using cyclosporine A, with inhibition of NFATc2 reversing MCT-induced PAH by increasing expression of Kv1.5, reducing Bcl-2 levels and promoting mitochondrial-dependent apoptosis. This culminated in reduced right ventricular hypertrophy (RVH) and medial hypertrophy of the small pulmonary arteries [66].

MiRNA regulatory pathways. Summary schematic illustrating the complex pathways controlled by miRNAs. Dysregulation of endothelial cells (EC) and smooth muscle cells (SMC) can occur in response to stress or injury to the vessel leading to vasoconstriction and pulmonary arterial remodeling
Administration of phenamil, a derivative of the diuretic amiloride, to the hypoxic rat model of PAH caused a reduction in PAH development and pulmonary remodeling. This is thought to be due to phenamil inducing Trb3 therefore increasing signaling through the BMP pathway and resulting in pro-proliferative and anti-apoptotic PASMCs [52, 69]. These findings highlight an essential role of miR-204 in controlling PASMC proliferation and apoptosis via NFAT regulation. There are many molecules involved in the NFAT signaling pathway and modulating either miR-204 or down-stream effectors may provide a target for future therapy of pulmonary remodeling. Consideration has to be given to the method by which these pathways are modulated. Over-expression of miR-204 via synthetic miR mimics would require optimization of mimic delivery to target specifically the pulmonary vasculature therefore limiting off-target effects. Although much work is required to further the present data and before a viable therapeutic agent is produced, the work shows promise.
miR143/145 cluster
Smooth muscle cells (SMCs), one of the key cellular components of pulmonary vascular remodeling, are able to “phenotypically switch” between the quiescent “contractile” phenotype and the migratory and proliferative “synthetic” phenotype in response to different growth factors to induce repair during vascular injury [70, 71]. Persistent activation of the highly proliferative synthetic state can be detrimental and lead to disease progression [72]. Two of the key miRNAs involved in regulation of the phenotype of SMCs are miR-143 and miR-145. MiR-145 is transcribed bicistronically along with miR-143 from human chromosome 5 [73]. Expression of the miR-143/miR145 cluster is high in cardiomyocytes during heart development, after which expression is exclusively localized within the SMCs [73–75]. Activation of miR143/miR145 is highly regulated through a DNA element called CArG box, contained within the promoter region of the pri-miRNA cluster, which is activated by binding of serum response factor (SRF) and its cofactors myocardin (Myocd) and myocardin-related transcription factors (MRTF) [73]. Growth factors TGF-β and BMP4 have been shown to activate miR-143/miR-145 via distinct mechanisms. TGF-β induces Myocd expression while BMP4 stimulation leads to MRTF-A activation, both pathways resulting in activation of miR-143/miR-145 cluster via the CArG box [70, 76, 77].
With relevance to PAH, miR-143 and miR-145 can potentially target many of the same genes, leading to co-operativity of this cluster in translational regulation. Down-regulation of target genes klf4 and klf5 by activation of miR-143/miR-145 (via TGF-β and BMP4) increases expression of smooth muscle-specific genes such as SMA, calponin, and SM22-α, promoting differentiation and repression of proliferation in vascular SMCs [75, 76, 78, 79]. It has also been shown that klf4 down-regulates Myocd and MRTF-A, two of the critical signaling molecules involved in promoting the contractile phenotype via miR-143/miR-145 activation [76, 78]. Another transcription factor, klf2, has been shown to bind to a promoter region within the miR-143/miR-145 cluster up-regulating expression of this miRNA cluster [80] and stimulating cell–cell communication between ECs and SMCs. Klf2 has been shown to generate the atheroprotective effects in human ECs in response to shear stress, one of the major contributing factors of atherosclerosis [80, 81]. A recent study revealed that over-expression of klf2 in human umbilical vein endothelial cells (HUVECs) led to an up-regulation of miR-143 and miR-145 within the HUVECs but also within the extracellular vesicles released by these cells. These vesicles were then able to control the phenotype of co-cultured SMCs by down-regulating target genes of miR-143/miR-145, therefore promoting the contractile phenotype [80]. Further to this, intravenous injection of extracellular vesicles from ECs known to have elevated Klf2 expression reduced atherosclerotic lesion formation in the ApoE−/− mouse [80].
Recent studies have demonstrated an SRF-independent pathway of miR-143/miR-145 activation [82]. Jag-1 activates translocation of notch intracellular domain, which can then interact with CBF1 regulators. This complex binds to the miR-143/miR-145 promoter region (independent of the CArG box) resulting in up-regulation of SMC contractile genes [82]. These critical studies emphasize the importance of miR-143/miR-145 in regulating and maintaining vascular SMC phenotype under normal conditions.
The SMC phenotypic switch is clearly integral to the pathogenesis of vascular diseases and hence many studies have reported the involvement of miR-143/miR-145 in this setting. Expression of miR-143/miR-145 was significantly reduced in animal models characterized by neointimal formation, including carotid artery ligation, carotid artery balloon injury, transverse aortic constriction (TAC), and ApoE knockout mice [75, 79, 83]. Additionally, over-expression of miR-145 in models of vascular disease has resulted in reduced neo-intimal formation and increased expression of SMC genes, indicative of the contractile phenotype [79, 83]. Use of miR-143/miR-145 knockout mice supports these results with increased neo-intimal formation compared with wild-type mice [74, 79]. Xin et al. [73] found opposing results with miR-143 and miR-145 knockout mice displaying reduced neo-intimal formation in response to carotid artery ligation. However, this result is thought to be due to maladaption of the knockout mice to injury. While these studies highlight the importance and implication of this axis in vascular pathology, they do not directly assess the influence of the miRNAs in PAH.
In the setting of PAH, recent work established that miR-145 was up-regulated in pulmonary arteries from IPAH patients [59], PAH-PASMCs containing a known BMPR2 mutation, and in lung tissue from IPAH and FPAH patients [84]. Expression of miR-145 was noted within the muscular regions of the lesions [84] with greater expression of miR-145 in the concentric lesions compared to plexiform lesions [85]. MiR-145 expression was also significantly increased in the hypoxic mouse model of PAH, both in the lungs and the right ventricle. Furthermore, knockout of miR-145 by both the use of miR-145−/− mice and mice treated with antimiR-145 significantly reduced the systolic right ventricular pressure (RVP) and number of remodeled vessels compared to control hypoxic animals. Target analysis provides evidence that members of the Wnt signaling pathway are targets for miR-145. WIF1, FRZB, and DAB2, which are part of the Wnt signaling family, were increased in hypoxic miR-145−/− mice [84]. In addition to this, other members of the Wnt signaling pathway have previously been shown to regulate PASMC proliferation [86], thus illustrating that the protective remodeling effect observed in miR-145−/− mice may be due to inhibition of Wnt/β-Catenin canonical signaling pathway. Loss of miR-143 did not illustrate the same protective effect as miR-145 [84] however, further studies are required to ascertain the exact role of miR-143 in the pulmonary vasculature and the remodeling process. Taken together, in vitro studies have demonstrated the essential role for miR-143/miR-145 for controlling SMC phenotype. Studies in vivo using rodent models of PAH indicate that specifically knocking down miR-145 expression appears to confer protection against development of PAH. Consequently, miR-145 may be targeted as treatment for pulmonary diseases once more is known about the exact pathways activated within the pulmonary vasculature in response to modulation of miR-145.
miR-17/92 cluster
As well as regulating the NFAT pathway, miR-204 has also been implicated in regulating the BMPR2 pathway indirectly as miR-204 has been shown to regulate the cytokine interleukin-6 (IL-6). IL-6 has been shown to be involved in PAH development as levels are increased in PAH patients and over-expression of IL-6 induces PAH in mice [87]. In human pulmonary artery endothelial cells (PAEC), IL-6 induces expression of the miR-17/92 cluster via activation of STAT3, which can directly bind to the promoter region of the miR-17/92 cluster (specifically miR-20a). BMPR2 is a target for this miR cluster and accordingly, BMPR2 protein levels are down-regulated in response to IL-6 [88]. Both miR-204 and miR-17/92 cluster are activated through different mechanisms but there are common molecules to both signaling pathways, indicating the importance of miRNAs working in concert with one another to affect cellular phenotype.
MiR-17/92 is a polycistronic cluster located on human chromosome 13, which gives rise to six mature miRNAs; miR17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92-1 [89]. Although all six miRNAs are transcribed together, each miRNA will regulate its own set of targets. Hence, regulation of the miR-17/92 cluster has the potential to control a great deal of target genes. The miR-17/92 cluster was first identified to be involved in tumor development due to up-regulation in tumor cells [90]. O’Donnell et al. [91] demonstrated that activation of the miR-17/92 cluster was via the transcription factor c-Myc. MiR-17-5p and miR-20a down-regulate target gene E2F1, which is involved in control of cell cycle and can induce c-Myc in a feedback loop. The miR-17/92 cluster is highly involved in controlling tumor growth via regulation of proliferation and apoptosis [92] and another cell cycle regulator, cyclin-dependent kinase inhibitor 1A (p21), is also targeted by miR-17. Inhibition of miR-17 in the hypoxic mouse and MCT rat model of PAH caused a reduction in systolic RVP and pulmonary vascular remodeling with an increase in p21 expression [93]. The exact mechanisms through which this miRNA cluster are acting are still relatively unknown, therefore further knowledge must be gained before we can understand the true role of this cluster in the pulmonary vasculature.
miR-424/503
Recent work has highlighted the role of miR-424/503 in PAH. MiR-424 and miR-503 are located within 250 base pairs of each other on the human X chromosome and are transcribed together [94]. The influence of these miRNAs on pulmonary remodeling has been suggested due to their activation by Apelin. Apelin is highly expressed in pulmonary endothelial cells and expression is decreased in lung ECs and in serum from IPAH and FPAH patients [94, 95]. Apelin−/− mice were shown to develop exaggerated PAH when exposed to hypoxia with obliteration of the small pulmonary arteries, compared to wild-type mice [95]. This finding was proposed to be via Apelin targeting AMP-activated kinase, which in turn activates klf2 to regulate endothelial nitric oxide synthase (eNOS) production [95]. Kim et al. [94] then found that decreased apelin expression in PAECs resulted in up-regulation of fibroblast growth factor 2 (FGF2) and increased proliferation. This study postulates that apelin can target miR-424 and miR-503 in PAECs, which can then directly repress FGF2 and FGFR1. Over-expression of these two miRNAs resulted in down-regulation of FGF2 and FGFR1, reducing phosphorylation of ERK1/2 and inhibition of proliferation [94, 96]. Furthermore, PAECs regulate PASMCs in a paracrine fashion as exposure of PASMCs to media conditioned by PAH PAECs led to hyperproliferation of PASMCs due to increased FGF2 expression [97]. Lentiviral over-expression of miR-424 and miR-503 in the lung of animal models of PAH reduced systolic RVP, RVH, muscularization of small pulmonary arteries and decreased proliferation, highlighting the protective effect of miR-424 and miR-503 in the pulmonary vasculature [94]. From these studies, apelin appears to have a critical function in controlling both PAEC and PASMC phenotype via down-stream signaling.
The role of miR-21 in PAH
MiR-21 is located on human chromosome 17 within the coding gene for transmembrane protein 49 (TMEM49), however miR-21 has its own promoter region and is therefore transcribed independently [98]. The role of miR-21 in pulmonary arterial remodeling has been investigated in many cell types and in vivo models. As stated before, hypoxia is one of the main triggers for pulmonary remodeling and miR-21 expression is up-regulated in human PASMCs [99], human PAECs [60], and mouse lungs [93, 100] exposed to hypoxia. Increased PASMC proliferation and migration in hypoxic PASMCs or control PASMCs over-expressing miR-21 has been observed, with reversal of phenotype with treatment of antimiR-21 [99, 100]. Down-regulation of miR-21 target genes PDCD4, SPRY2, and PPARα may be responsible for the increased proliferation observed [99, 101] and reduction in PDCD4 has recently been proposed to have anti-apoptotic effects [102].
Bone morphogenetic protein (BMP) signaling also plays a part in modulating miR-21 activation once again in a ligand-specific manner. BMP4 stimulates SMAD-1 and -5, while TGF-β stimulates SMAD-3, to bind to the RNA helicase p68, which is part of the Drosha complex. This complex promotes processing of pri-miR-21 into pre-miR-21, therefore increasing mature-miR-21 expression, which can down-regulate target genes such as PDCD4, resulting in increased smooth muscle contractile gene expression [101]. Upon induction of miR-21 by BMP4, members of the dedicator of cytokinesis (DOCK) 180-related protein family (DOCK-4, -5 and -7) were down-regulated. A reduction in DOCK protein leads to an inhibition of cell migration and promotion of the contractile SMC phenotype [103], thus illustrating another miRNA that can modulate PASMC phenotype via BMP signaling.
Tissues taken from patients with heart failure and animal models of cardiac disease show an up-regulation of miR-21 [104, 105]. However, two separate groups have found opposing results regarding therapeutically regulating miR-21 in the heart. Thum et al. [104] found that the significant up-regulation of miR-21 in cardiac fibroblasts after TAC lead to repression of its target Sprouty homologue 1 (Spry1), resulting in activation of ERK-MAP kinase signaling pathway and increased cell survival. Treatment with antagomiR-21 was able to reverse these changes, leading to a reduction in fibrosis [104]. On the contrary, Patrick et al. [105] found that a reduction in miR-21 did not influence cardiac hypertrophy and fibrosis. In response to various cardiac injuries, including TAC, infusion of AngII and coronary artery ligation, miR-21−/− mice showed comparable phenotypes to the wild-type mice with increased remodeling and fibrosis, with similar results obtained using an LNA-antimiR targeting miR-21 [105]. The differences observed between these two studies may be due to differences in chemistry between the oligonucleotides used for miR-21 knock down. These studies indicate the possible challenges faced when using knock down of a miRNA as a target for prevention and reversal of the diseased state.
Similar to the controversy surrounding miR-21 in cardiac disease, conflicting results have also been obtained from in vivo experiments regarding the role of miR-21 in PAH. Knock down of miR-21 in hypoxic mice using antimiR-21 resulted in a reduction in systolic RVP [93] and lowered muscularization of distal pulmonary arteries, thought to be brought about by a reduction in fibronectin, endothelin-1, α-SMA, Cald1 and SM22-α [100]. On the contrary, Parikh et al. [60] observed an exaggerated PAH phenotype in mice void of miR-21 expression in the pulmonary vasculature after exposure to Sugen-5416 (vascular endothelial growth factor receptor inhibitor) coupled with chronic hypoxia with an increase in systolic RVP and increased pulmonary arterial remodeling shown by increased muscularization in the arterial wall. This amplified response may be due to repression by RhoB, a target of miR-21. RhoB expression induces Rho-kinase activation, which attenuates endothelial nitric oxide synthase, leading to pulmonary vasoconstriction [60]. To support this data, RhoB knockout mice have reduced pulmonary remodeling and RVH when exposed to hypoxia [106].
Conversely, another study has reported that expression of miR-21 was decreased in male rats exposed to MCT with expression unchanged in hypoxic conditions and down-regulation of miR-21 also observed in lung samples from IPAH patients [58]. The variation between the studies mentioned may be due to multiple experimental variations, including differences in species and distinct methods used to modulate miR-21 levels as mentioned above. Another possibility that may lead to differences in findings could be gender. Both males and females are known to have different susceptibility to developing PAH and therefore the miRNA profile may be modulated differently between the sexes. Taken together, these findings highlight the importance of miR-21 in pulmonary remodeling, however, the exact mechanism of action of miR-21 appears to be dependent on various experimental factors.
Other pathogenic mechanisms linking miRNAs and PAH
Resistance to apoptosis is a major component leading to the development of pulmonary remodeling and recent work has highlighted the role of p53 and miR-34a [107]. MiR-34a is located on human chromosome 1 and pri-miR-34a is a direct target for the tumor suppressor protein p53. Thus, miR-34a is thought to be involved in various cancer pathways [108, 109]. Over-expression of miR-34a leads to a reduction in proliferation and augmented apoptosis [110–112]. A study by Mizuno et al. [107] demonstrated that hypoxia up-regulates p53 and in turn miR-34a. Accordingly, p53 knockout mice display an exaggerated PAH phenotype with increased systolic RVP, RVH, and medial wall thickening in small pulmonary arteries when exposed to hypoxia compared with hypoxic wild-type mice [107]. Notch1 has been verified as a direct target for miR-34a [113], thus over-expression of miR-34a down-regulates Notch1 and its downstream targets Bcl-2 and Survivin [110, 112], both of which are anti-apoptotic. Although the majority of studies reporting the p53/miR-34a/Notch1 pathway have examined the role in cancer, the pathogenesis of cancer and remodeling share several common features, however, further studies will confirm if the same response is initiated in pulmonary artery cells.
MiR-328 is primarily localized within the PASMC and is found to be down-regulated in pulmonary arteries from hypoxic rats and PAH patients. Over-expression of miR-328 in rat PASMC displayed an increase in cellular apoptosis and insulin growth factor 1 receptor (IGF-1R) was identified as a target for miR-328 [114]. This data implicates miR-328 in remodeling by regulating the apoptotic pathway, although the exact molecules and targets involved in the apoptosis response via miR-328 need to be further clarified.
Another miRNA thought to be involved in remodeling is miR-206. MiR-206 is down-regulated in PASMCs from hypoxic mice and hPASMC proliferation and migration is reduced with over-expression of miR-206, along with increases in smooth muscle cell markers therefore promoting the contractile phenotype. Furthermore, hPASMCs over-expressing miR-206 demonstrate increased apoptotic activity, as shown by TUNEL staining and caspase 3 activity [115], indicating that lowered expression of miR-206, as in the case during PAH and hypoxia, promotes excessive proliferation and resistance to apoptosis in PASMCs. The mechanism of action is thought to be due to repression of notch3 by miR-206 as notch3 has been shown to increase SMC growth and prevent SMC apoptosis [116, 117]. Little is known about miR-206 in disease development with previous studies showing its involvement in regulation of breast cancer cell lines [118]. With further in vivo work focusing on up-regulation of miR-206, the pathways and molecules involved in miR-206 modulation can be identified.
What causes miRNA dysregulation?
The focus of this review has primarily been on the events occurring downstream of miRNA and the ensuing changes in gene regulation and phenotype. However, it is fundamentally important to understand the mechanisms by which these miRNAs are initially regulated during disease. Hypoxia is one of the major rodent models used to study PAH and in addition to this, hypoxia can occur in site-specific regions in response to inflammation or vascular injury. Therefore, one of the key triggers for miRNA dysregulation may be activation of transcription factors, such as hypoxia inducible factors (HIF), which promote regulation of many pathways in response to hypoxic conditions. Mice heterozygous for HIF1α had reduced pulmonary remodeling compared to wild-type mice [119], indicating that HIF1 plays a vital role in controlling vascular remodeling in response to hypoxia. Numerous miRNAs have been identified as being dysregulated in hypoxic conditions in a HIF-dependent manner [120]. Recent studies have found that miR-210 expression is induced by HIF1α in pancreatic cancer cells [121] and endothelial cells [122] therefore providing a possible explanation as to why HIF1α activation can result in gene silencing. As well as inducing miRNA expression, HIF1α can also be targeted by miRNAs to provide a complex and interactive signaling pathway. One such example is miR-155, which has been shown to provide a negative feedback loop involving HIF1α in hypoxic conditions [123].
The cellular response to hypoxia is only one aspect that may promote changes in miRNA expression during disease. Another factor thought to be involved in miRNA dysregulation is inflammation. Inflammation is an important process thought to contribute to PAH phenotype due to the release of cytokines, chemokines, and various growth factors which can result in cellular proliferation, migration and survival. Due to their extensive gene regulatory effects, miRNAs may be involved in the regulation of inflammation, and as a result have an impact on the pathogenesis of PAH. In support of this, Kuhn et al. [124] observed that miR-25 was down-regulated in human airway SMCs in response to pro-inflammatory stimuli including IL-1β, TNF-α, and IFN-γ, while Klf4, a target gene of miR-25, was increased. On the other hand, several miRNAs including miR-155 [125, 126] and miR-146 [126] are induced by various inflammatory mediators. MiR-146a is induced in response to pro-inflammatory stimuli via NF-κB and targets TRAF6 and IRAK1which are molecules involved in NF-κB activation, thus providing a negative feedback loop [126]. Various studies have found an up-regulation of miR-21 expression after exposure to inflammatory stimuli including heat-inactivated bacterium [127], LPS exposure [128], and in a model of allergic airway inflammation [129]. In particular, transcription of pri-miR-21 has been shown to be induced by IL-6, a pro-inflammatory cytokine, in a STAT3-dependent manner [130] and it is believed that initial IL-6 release is due to NF-κB nuclear translocation in response to tissue damage [131]. The fact that miRNAs can be modulated by inflammation as well as target inflammatory mediators adds to the complexity of this regulatory pathway, making it a challenging task to study in a disease model.
Another possible mechanism through which miRNAs are modulated may be through tyrosine kinase pathways. The majority of growth factors (such as PDGF and vascular endothelial growth factor, VEGF) signal through tyrosine kinase receptors that can activate STAT proteins. Recent research has highlighted the function of miRNAs in this pathway as several miRNAs have been shown to be regulated by tyrosine kinase Src and STAT3, in particular miR-204 [59] and miR-17/92 [88] (see individual sections above and Fig. 3). There is strong evidence linking tyrosine kinase dysregulation and signaling dysfunction in disease as the tyrosine kinase inhibitor imatinib was developed as a treatment for chronic myeloid leukemia [132]. Many of the pathologies associated with PAH share similarities with cancer and as a result, imatinib has been the subject of a phase 3 trial as an “add-on” therapy for PAH [133]. Imatinib treatment caused an increase in exercise capacity and improved hemodynamic parameters, however, adverse effects were recorded that limit clinical application in the setting of PAH. Hence, more knowledge is required regarding the exact mechanisms by which tyrosine kinases exert their effect within the pulmonary circulation and miRNAs may provide a crucial link into the downstream effects of this pathway.
One way to characterize the exact processes that are initiating miRNA dysregulation in PAH would be to use a bioinformatics approach, as has been utilized previously [60]. This would allow pathways activated during PAH to be identified and assess the precise mechanisms by which miRNAs are affected. The information gathered from a screening approach would add to the knowledge already known about the downstream effectors of these miRNAs and create a better picture of PAH disease initiation and progression through miRNA pathways.
miRNAs as biomarkers for disease
The studies presented within this review demonstrate that expression of miRNAs are tissue and cell specific and can be dysregulated during disease. More importantly, many miRNA expression levels correlate with disease severity. This provides a unique signature of miRNAs which are expressed in disease and it is believed that circulating miRNAs can be used as diagnostic biomarkers for early detection of disease. Identifying miRNA profiles in the serum/plasma from certain diseases is ideal, as it is a non-invasive method and miRNAs are highly stable in the blood. It was originally thought that the majority of miRNAs within the circulation were enclosed within exosomes [134, 135] or apoptotic bodies [136], which were released from cells into the circulation, allowing cell to cell communication and miRNA function in cells distinct from where they were synthesized [136]. However, Arroyo et al. [137] used differential centrifugation and size-exclusion chromatography to establish that almost 90 % of circulating miRNAs exist in a protein complex (primarily with Ago2, a key molecule in miRNA biosynthesis). Biomarkers of disease have already been suggested for a number of diseases [138] including heart failure, with miR-423-5p distinguishing diseased state [139] and type II diabetes mellitus characterized with a loss of endothelial miR-126 [140]. There are of course difficulties when analyzing miRNA expression from either serum or plasma, for instance lack of a standard molecule of reference, and further challenges faced in isolating and accurately quantifying circulating miRNAs are discussed in a review by Zampetaki and Mayr [141]. In addition, questions still need to be answered on how the circulating miRNAs are associated with disease initiation and progression and how early these miRNA profiles arise in humans.
Conclusions
MiRNA research is a relatively recent field and there is still much to discover about the intricate mechanisms involved in the signaling pathways involving miRNAs. It is apparent that miRNAs play a pivotal role in regulating pulmonary arterial remodeling via distinct pathways (Fig. 3). One of the biggest challenges in miRNA research is validating direct targets of specific miRNAs and hence understanding the regulation by which miRNAs exert their effect. Gain- and loss-of-function experiments highlight the beneficial effects of regulating miRNAs and identification of genes involved in the miRNA pathway represent attractive therapeutic targets. The most effective therapies will most likely target the pathways directly involved in controlling pro-proliferation and anti-apoptosis of cells within the vessel wall and reversing these effects. The modulation of miRNAs using either synthetic miRNAs or antimiRs individually or in combination, is highly studied in animal models of PAH, however, there are problems with this treatment. The role of miRNAs throughout the body is broad and the advantage of therapeutically targeting miRNAs is the ability to hit many different pathways involved in disease development and progression. However, this can also be a significant drawback as targeting a miRNA may produce off target effects via modulation of pathways not involved in the diseased state. To overcome this problem, highlighting particular targets of selected miRNAs may prove more successful in reducing disease severity while limiting the effects to pathways involved in PAH. Direct targeting of the miRNA via local delivery to the lung is essential to minimize off-target effects. In addition, directing the miRNA treatment to particular cellular compartments will further reduce unwanted effects. Altogether, the potential to target miRNAs involved in pulmonary remodeling is high and will potentially lead to the generation of novel therapeutic agents.
References
Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, Gaine S (2004) Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 43 (12 Suppl S):40S–47S
Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, Krowka MJ, Langleben D, Nakanishi N, Souza R (2009) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54(1 Suppl):S43–S54
Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA III, Loyd JE, Nichols WC, Trembath RC (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 26(1):81–84
Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, Gruenig E, Janssen B, Koehler R, Seeger W, Eickelberg O, Olschewski H, Elliott CG, Glissmeyer E, Carlquist J, Kim M, Torbicki A, Fijalkowska A, Szewczyk G, Parma J, Abramowicz MJ, Galie N, Morisaki H, Kyotani S, Nakanishi N, Morisaki T, Humbert M, Simonneau G, Sitbon O, Soubrier F, Coulet F, Morrell NW, Trembath RC (2006) Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat 27(2):121–132
Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA, Loyd JE (2001) Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 345(5):319–324
Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JS, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC (2000) Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet 37(10):741–745
Maclean MR, Dempsie Y (2010) The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol 661:309–322
Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, Pepke-Zaba J, Sheares KK, Corris PA, Fisher AJ, Lordan JL, Gaine S, Coghlan JG, Wort SJ, Gatzoulis MA, Peacock AJ (2012) Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 186(8):790–796
Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, Krichman A, Liou TG, Raskob GE, Wason P, Feldkircher K, Turner M, McGoon MD (2011) The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US Contemporary Registries. Chest 139(1):128–137
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G (2006) Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 173(9):1023–1030
Walker AM, Langleben D, Korelitz JJ, Rich S, Rubin LJ, Strom BL, Gonin R, Keast S, Badesch D, Barst RJ, Bourge RC, Channick R, Frost A, Gaine S, McGoon M, McLaughlin V, Murali S, Oudiz RJ, Robbins IM, Tapson V, Abenhaim L, Constantine G (2006) Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J 152(3):521–526
Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP, McGoon MD (2010) Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 137(2):376–387
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jais X, Montani D, Souza R, Simonneau G (2010) Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 122(2):156–163
Frumkin LR (2012) The pharmacological treatment of pulmonary arterial hypertension. Pharmacol Rev 64(3):583–620
Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M (2006) Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 21:134–145
Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M (2010) Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121(25):2747–2754
Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM (2004) Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 43(12 Suppl S):25S–32S
Jelaska A, Strehlow D, Korn JH (1999) Fibroblast heterogeneity in physiological conditions and fibrotic disease. Springer Semin Immunopathol 21(4):385–395
Das M, Dempsey EC, Reeves JT, Stenmark KR (2002) Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am J Physiol Lung Cell Mol Physiol 282(5):L976–L986
Liao DF, Jin ZG, Baas AS, Daum G, Gygi SP, Aebersold R, Berk BC (2000) Purification and identification of secreted oxidative stress-induced factors from vascular smooth muscle cells. J Biol Chem 275(1):189–196
Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM (2003) Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 92(6):683–691
Stenmark KR, Gerasimovskaya E, Nemenoff RA, Das M (2002) Hypoxic activation of adventitial fibroblasts: role in vascular remodeling. Chest 122(6 Suppl):326S–334S
Jiang YL, Dai AG, Li QF, Hu RC (2006) Transforming growth factor-beta1 induces transdifferentiation of fibroblasts into myofibroblasts in hypoxic pulmonary vascular remodeling. Acta Biochim Biophys Sin (Shanghai) 38(1):29–36
Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J (2009) Fibronectin is an important regulator of flow-induced vascular remodeling. Arter Thromb Vasc Biol 29(7):1074–1079
Rabinovitch M (2007) Pathobiology of pulmonary hypertension. Annu Rev Pathol 2:369–399
Rakesh K, Agrawal DK (2005) Cytokines and growth factors involved in apoptosis and proliferation of vascular smooth muscle cells. Int Immunopharmacol 5(10):1487–1506
Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M (2004) Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43(12):13S–24S
Cowan KN, Jones PL, Rabinovitch M (2000) Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest 105(1):21–34
Sakao S, Tatsumi K, Voelkel NF (2010) Reversible or irreversible remodeling in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 43(6):629–634
Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, Tuder RM (1999) Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol 155(2):411–419
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Van Rooij E, Olson EN (2007) MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Investig 117(9):2369–2376
Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T (2004) Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 15(2):185–197
Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ (2004) Argonaute2 is the catalytic engine of mammalian RNAi. Science 305(5689):1437–1441
Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M (2012) Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492(7429):376–381
Kos A, Olde Loohuis NF, Wieczorek ML, Glennon JC, Martens GJ, Kolk SM, Aschrafi A (2012) A potential regulatory role for intronic microRNA-338-3p for its host gene encoding apoptosis-associated tyrosine kinase. PLoS ONE 7(2):e31022
Flynt AS, Lai EC (2008) Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet 9(11):831–842
Liu N, Olson EN (2010) MicroRNA regulatory networks in cardiovascular development. Dev Cell 18(4):510–525
Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA-target recognition. PLoS Biol 3(3):e85
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, Abo R, Tabebordbar M, Lee RT, Burge CB, Boyer LA (2013) Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152(3):570–583
Volk N, Shomron N (2011) Versatility of MicroRNA biogenesis. PLoS ONE 6(5):e19391
Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG (2009) The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459(7249):1010–1014
Chakravarthy S, Sternberg SH, Kellenberger CA, Doudna JA (2010) Substrate-specific kinetics of Dicer-catalyzed RNA processing. J Mol Biol 404(3):392–402
Sakamoto S, Aoki K, Higuchi T, Todaka H, Morisawa K, Tamaki N, Hatano E, Fukushima A, Taniguchi T, Agata Y (2009) The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol Cell Biol 29(13):3754–3769
Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, Wolfe SA, Giraldez AJ (2010) A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328(5986):1694–1698
Yang J-S, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC (2010) Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci USA 107(34):15163–15168
Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW (2005) Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 96(10):1053–1063
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P (2002) Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 21(7):1743–1753
Afrakhte M, Moren A, Jossan S, Itoh S, Sampath K, Westermark B, Heldin CH, Heldin NE, ten Dijke P (1998) Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem Biophys Res Commun 249(2):505–511
Ishisaki A, Yamato K, Hashimoto S, Nakao A, Tamaki K, Nonaka K, ten Dijke P, Sugino H, Nishihara T (1999) Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J Biol Chem 274(19):13637–13642
Shi W, Chen H, Sun J, Chen C, Zhao J, Wang YL, Anderson KD, Warburton D (2004) Overexpression of Smurf1 negatively regulates mouse embryonic lung branching morphogenesis by specifically reducing Smad1 and Smad5 proteins. Am J Physiol Lung Cell Mol Physiol 286(2):L293–L300
Chan MC, Nguyen PH, Davis BN, Ohoka N, Hayashi H, Du K, Lagna G, Hata A (2007) A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol Cell Biol 27(16):5776–5789
Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW (2002) Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 105(14):1672–1678
Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC (2001) Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation 104(7):790–795
Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, Fukuchi Y (2006) Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 290(3):L450–L458
Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O, Noda T, Miyazono K (2000) BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol 221(1):249–258
Long L, MacLean MR, Jeffery TK, Morecroft I, Yang XD, Rudarakanchana N, Southwood M, James V, Trembath RC, Morrell NW (2006) Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res 98(6):818–827
Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R, Denby L, Dempsie Y, Long L, Morrell NW, Baker AH (2010) Dynamic changes in lung MicroRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arter Thromb Vasc Biol 30(4):716–U182
Courboulin A, Paulin R, Giguere NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Cote J, Simard MJ, Bonnet S (2011) Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 208(3):535–548
Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, Shaik RS, Waxman AB, Zhang YY, Maron BA, Hartner JC, Fujiwara Y, Orkin SH, Haley KJ, Barabasi AL, Loscalzo J, Chan SY (2012) MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 125(12):1520–1532
Deo M, Yu JY, Chung KH, Tippens M, Turner DL (2006) Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev Dyn 235(9):2538–2548
Wang FE, Zhang C, Maminishkis A, Dong L, Zhi C, Li R, Zhao J, Majerciak V, Gaur AB, Chen S, Miller SS (2010) MicroRNA-204/211 alters epithelial physiology. FASEB J 24(5):1552–1571
Ryan J, Tivnan A, Fay J, Bryan K, Meehan M, Creevey L, Lynch J, Bray IM, O’Meara A, Davidoff AM, Stallings RL (2012) MicroRNA-204 increases sensitivity of neuroblastoma cells to cisplatin and is associated with a favourable clinical outcome. Br J Cancer 107(6):967–976
Sacconi A, Biagioni F, Canu V, Mori F, Di Benedetto A, Lorenzon L, Ercolani C, Di Agostino S, Cambria AM, Germoni S, Grasso G, Blandino R, Panebianco V, Ziparo V, Federici O, Muti P, Strano S, Carboni F, Mottolese M, Diodoro M, Pescarmona E, Garofalo A, Blandino G (2012) miR-204 targets Bcl-2 expression and enhances responsiveness of gastric cancer. Cell Death Dis 3:e423
Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, Cote J, Provencher S, Sussman MA, Bonnet S (2011) Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 123(11):1205–1215
Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED (2007) The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA 104(27):11418–11423
Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL (2002) Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 105(2):244–250
Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX (2000) Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 279(5):C1540–C1549
Chan MC, Weisman AS, Kang H, Nguyen PH, Hickman T, Mecker SV, Hill NS, Lagna G, Hata A (2011) The amiloride derivative phenamil attenuates pulmonary vascular remodeling by activating NFAT and the bone morphogenetic protein signaling pathway. Mol Cell Biol 31(3):517–530
Lagna G, Ku MM, Nguyen PH, Neuman NA, Davis BN, Hata A (2007) Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J Biol Chem 282(51):37244–37255
Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L (2011) miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet 4(2):197–205
Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84(3):767–801
Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN (2009) MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 23(18):2166–2178
Boettger T, Beetz N, Kostin S, Schneider J, Krueger M, Hein L, Braun T (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Investig 119(9):2634–2647
Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460(7256):705–710
Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A (2011) Down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem 286(32):28097–28110
Long X, Miano JM (2011) Transforming growth factor-beta1 (TGF-beta1) utilizes distinct pathways for the transcriptional activation of microRNA 143/145 in human coronary artery smooth muscle cells. J Biol Chem 286(34):30119–30129
Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK (2005) Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem 280(10):9719–9727
Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C (2009) MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res 105(2):158–166
Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S (2012) Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol 14(3):249–256
Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJ (2002) Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood 100(5):1689–1698
Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L (2011) The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. J Biol Chem 286(32):28312–28321
Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G (2009) The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ 16(12):1590–1598
Caruso P, Dempsie Y, Stevens HC, McDonald RA, Long L, Lu R, White K, Mair KM, McClure JD, Southwood M, Upton P, Xin M, van Rooij E, Olson EN, Morrell NW, MacLean MR, Baker AH (2012) A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res 111(3):290–300
Bockmeyer CL, Maegel L, Janciauskiene S, Rische J, Lehmann U, Maus UA, Nickel N, Haverich A, Hoeper MM, Golpon HA, Kreipe H, Laenger F, Jonigk D (2012) Plexiform vasculopathy of severe pulmonary arterial hypertension and microRNA expression. J Heart Lung Transpl 31(7):764–772
Sklepkiewicz P, Schermuly RT, Tian X, Ghofrani HA, Weissmann N, Sedding D, Kashour T, Seeger W, Grimminger F, Pullamsetti SS (2011) Glycogen synthase kinase 3beta contributes to proliferation of arterial smooth muscle cells in pulmonary hypertension. PLoS ONE 6(4):e18883
Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB (2009) Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 104(2):236–244 (228 p following 244)
Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC (2009) Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 104(10):1184–1191
Tanzer A, Stadler PF (2004) Molecular evolution of a microRNA cluster. J Mol Biol 339(2):327–335
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM (2005) A microRNA polycistron as a potential human oncogene. Nature 435(7043):828–833
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435(7043):839–843
Li C, Hashimi SM, Good DA, Cao S, Duan W, Plummer PN, Mellick AS, Wei MQ (2012) Apoptosis and microRNA aberrations in cancer. Clin Exp Pharmacol Physiol 39(8):739–746
Pullamsetti SS, Doebele C, Fischer A, Savai R, Kojonazarov B, Dahal BK, Ghofrani HA, Weissmann N, Grimminger F, Bonauer A, Seeger W, Zeiher AM, Dimmeler S, Schermuly RT (2012) Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am J Respir Crit Care Med 185(4):409–419
Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, Erzurum SC, Chun HJ (2013) An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 19(1):74–82
Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V, Zamanian RT, Quertermous T, Chun HJ (2011) Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arter Thromb Vasc Biol 31(4):814–820
Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G, Humbert M, Eddahibi S, Guignabert C (2011) Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am J Respir Cell Mol Biol 45(2):311–322
Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, Raffestin B, Eddahibi S (2009) Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest 119(3):512–523
Kumarswamy R, Volkmann I, Thum T (2011) Regulation and function of miRNA-21 in health and disease. RNA Biol 8(5):706–713
Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU (2010) MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 299(6):L861–L871
Yang S, Banerjee S, Freitas A, Cui H, Xie N, Abraham E, Liu G (2012) miR-21 regulates chronic hypoxia-induced pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 302(6):L521–L529
Davis BN, Hilyard AC, Lagna G, Hata A (2008) SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454(7200):56–61
Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C (2010) Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res 87(3):431–439
Kang H, Davis-Dusenbery BN, Nguyen PH, Lal A, Lieberman J, Van Aelst L, Lagna G, Hata A (2012) Bone morphogenetic protein 4 promotes vascular smooth muscle contractility by activating microRNA-21 (miR-21), which down-regulates expression of family of dedicator of cytokinesis (DOCK) proteins. J Biol Chem 287(6):3976–3986
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S (2008) MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456(7224):980–984
Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, van Rooij E, Olson EN (2010) Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest 120(11):3912–3916
Wojciak-Stothard B, Zhao L, Oliver E, Dubois O, Wu Y, Kardassis D, Vasilaki E, Huang M, Mitchell JA, Harrington LS, Prendergast GC, Wilkins MR (2012) Role of RhoB in the regulation of pulmonary endothelial and smooth muscle cell responses to hypoxia. Circ Res 110(11):1423–1434
Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Voelkel NF, Ishizaki T (2011) p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am J Physiol Lung Cell Mol Physiol 300(5):L753–L761
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ (2007) A microRNA component of the p53 tumour suppressor network. Nature 447(7148):1130–1134
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H (2007) Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 104(39):15472–15477
Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, Maitra A, Mendell JT (2007) Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26(5):745–752
Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M (2007) Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 26(5):731–743
Ji X, Wang Z, Geamanu A, Goja A, Sarkar FH, Gupta SV (2012) Delta-tocotrienol suppresses Notch-1 pathway by upregulating miR-34a in nonsmall cell lung cancer cells. Int J Cancer 131(11):2668–2677
Li WB, Ma MW, Dong LJ, Wang F, Chen LX, Li XR (2011) MicroRNA-34a targets notch1 and inhibits cell proliferation in glioblastoma multiforme. Cancer Biol Ther 12(6):477–483
Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S, Tian H, Jiang C, Zhu D (2012) The microRNA-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and L-type calcium channel-alpha1C. Hypertension 59(5):1006–1013
Jalali S, Ramanathan GK, Parthasarathy PT, Aljubran S, Galam L, Yunus A, Garcia S, Cox RR Jr, Lockey RF, Kolliputi N (2012) Mir-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS ONE 7(10):e46808
Sweeney C, Morrow D, Birney YA, Coyle S, Hennessy C, Scheller A, Cummins PM, Walls D, Redmond EM, Cahill PA (2004) Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jk-dependent pathway. FASEB J 18(12):1421–1423
Song G, Zhang Y, Wang L (2009) MicroRNA-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. J Biol Chem 284(46):31921–31927
Adams BD, Furneaux H, White BA (2007) The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol 21(5):1132–1147
Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL (1999) Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 103(5):691–696
Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M (2007) A microRNA signature of hypoxia. Mol Cell Biol 27(5):1859–1867
Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, Story M, Le QT, Giaccia AJ (2009) Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell 35(6):856–867
Fasanaro P, D’Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F (2008) MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem 283(23):15878–15883
Bruning U, Cerone L, Neufeld Z, Fitzpatrick SF, Cheong A, Scholz CC, Simpson DA, Leonard MO, Tambuwala MM, Cummins EP, Taylor CT (2011) MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol Cell Biol 31(19):4087–4096
Kuhn AR, Schlauch K, Lao R, Halayko AJ, Gerthoffer WT, Singer CA (2010) MicroRNA expression in human airway smooth muscle cells: role of miR-25 in regulation of airway smooth muscle phenotype. Am J Respir Cell Mol Biol 42(4):506–513
O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D (2007) MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA 104(5):1604–1609
Taganov KD, Boldin MP, Chang KJ, Baltimore D (2006) NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA 103(33):12481–12486
Mathe E, Nguyen GH, Funamizu N, He P, Moake M, Croce CM, Hussain SP (2012) Inflammation regulates microRNA expression in cooperation with p53 and nitric oxide. Int J Cancer 131(3):760–765
Moschos SA, Williams AE, Perry MM, Birrell MA, Belvisi MG, Lindsay MA (2007) Expression profiling in vivo demonstrates rapid changes in lung microRNA levels following lipopolysaccharide-induced inflammation but not in the anti-inflammatory action of glucocorticoids. BMC Genom 8:240
Lu TX, Munitz A, Rothenberg ME (2009) MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol 182(8):4994–5002
Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, Horn F (2007) Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 110(4):1330–1333
Niu J, Shi Y, Tan G, Yang CH, Fan M, Pfeffer LM, Wu ZH (2012) DNA damage induces NF-kappaB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J Biol Chem 287(26):21783–21795
Moen MD, McKeage K, Plosker GL, Siddiqui MA (2007) Imatinib: a review of its use in chronic myeloid leukaemia. Drugs 67(2):299–320
Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galie N, Gomez-Sanchez MA, Grimminger F, Grunig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA (2013) Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127(10):1128–1138
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10(12):1470–1476
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9(6):654–659
Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Koppel T, Jahantigh MN, Lutgens E, Wang S, Olson EN, Schober A, Weber C (2009) Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal 2(100):ra81
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, Tait JF, Tewari M (2011) Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA 108(12):5003–5008
Wang K, Zhang S, Marzolf B, Troisch P, Brightman A, Hu Z, Hood LE, Galas DJ (2009) Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc Natl Acad Sci USA 106(11):4402–4407
Tijsen AJ, Creemers EE, Moerland PD, de Windt LJ, van der Wal AC, Kok WE, Pinto YM (2010) MiR423-5p as a circulating biomarker for heart failure. Circ Res 106(6):1035–1039
Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, Mayr A, Weger S, Oberhollenzer F, Bonora E, Shah A, Willeit J, Mayr M (2010) Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res 107(6):810–817
Zampetaki A, Mayr M (2012) Analytical challenges and technical limitations in assessing circulating miRNAs. Thromb Haemost 108(4):592–598
Acknowledgments
Research in our laboratories on miRNAs and pulmonary arterial hypertension is funded by the British Heart Foundation and the Medical Research Council.
Disclosure
Professor Andrew H. Baker and Professor Margaret R. MacLean are co-inventors on a patent for inhibition of miR-145 in PAH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Grant, J.S., White, K., MacLean, M.R. et al. MicroRNAs in pulmonary arterial remodeling. Cell. Mol. Life Sci. 70, 4479–4494 (2013). https://doi.org/10.1007/s00018-013-1382-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-013-1382-5