
Abstract
Osteoclasts are bone-resorbing cells of monocytic origin. An imbalance between bone formation and resorption can lead to osteoporosis or osteopetrosis. Osteoclastogenesis is triggered by RANKL- and IP3-induced Ca2+ influx followed by activation of NFATc1, a master transcription factor for osteoclastogenic gene regulation. During differentiation, osteoclasts undergo cytoskeletal remodeling to migrate and attach to the bone surface. Simultaneously, they fuse with each other to form multinucleated cells. These processes require PI3-kinase-dependent cytoskeletal protein activation to initiate cytoskeletal remodeling, resulting in the formation of circumferential podosomes and fusion-competent protrusions. In multinucleated osteoclasts, circumferential podosomes mature into stabilized actin rings, which enables the formation of a ruffled border where intensive membrane trafficking is executed. Membrane lipids, especially phosphoinositides, are key signaling molecules that regulate osteoclast morphology and act as second messengers and docking sites for multiple important effectors. We examine the critical roles of phosphoinositides in the signaling cascades that regulate osteoclast functions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Osteoclasts are a unique cell type highly specialized for resorbing bone matrix. Hyperactivation of osteoclasts can result in bone-degenerative disorders such as osteoporosis and osteolytic bone metastasis, while lack or hypoactivation causes osteopetrosis. Active osteoclasts are polykaryons formed by cell–cell fusion of highly motile progenitors of the monocyte–macrophage lineage. For bone resorption, osteoclasts attach firmly to the bone surface by forming stable actin rings. Through the area enclosed by actin rings, osteoclasts secrete digestive acids and proteases and transport degraded matrix components by endocytosis/transcytosis into the cell and to the apical surface. In this way, osteoclasts facilitate bone remodeling and the recycling of bone nutrients, particularly calcium and phosphates.
Osteoclast precursors on the bone surface are stimulated by macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL) produced by osteoblast lineage cells, resulting in the activation of the immediate early transcription factors NF-κB and c-Fos. These transcription factors are essential for the activation of signaling cascades that drive osteoclastogenesis [1]. Activation of RANK is an early event in osteoclastogenesis, leading to phospholipase C (PLC) activation, membrane hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to form diacylglycerol and inositol-1,4,5-trisphosphate (IP3), IP3-mediated Ca2+ release and the activation of the Ca2+-dependent phosphatase calcineurin. Activated calcineurin dephosphorylates and thereby activates nuclear factor of activated T cells cytoplasmic 1 (NFATc1) [2–4], a transcription factor that activates the expression of multiple osteoclastogenic genes, including the membrane fusion promoter (fusogen) DC-STAMP, the actin ring component β3 integrin and the bone degrading hydrolases tartrate-resistant acid phosphatase (TRAP) and cathepsin K [5].
In addition, osteoclast differentiation depends on dramatic changes in cytoskeletal dynamics. Activated osteoclast precursors develop columnar actin puncta, known as podosomes, at the ventral surface [6, 7]. In the early phase of osteoclast differentiation, these actin puncta organize into dynamic rings, and as the cells fuse, these circumferential podosomes eventually mature into stabilized structures known as actin rings that adhere to the bone and isolate the contact site between the osteoclast ruffled membrane and the bone surface from the extracellular fluid [6]. Efficient bone resorption is then achieved through the secretion of protons and hydrolases, including TRAP and protease cathepsin K, at the ruffled border formed inside these belts [8], and concomitant incorporation of the degraded materials by endocytosis.
Cell–cell fusion and vesicle-ruffled border fusion both require intricate orchestration of the plasma membrane and vesicular membranes, involving signaling cascades mediated by membrane lipids. Membrane lipids, including phosphoinositides (PIs), contribute to a wide range of basic biological processes, such as polarity formation, chemotaxis, intercellular trafficking, and cytokinesis [9]. PIs are essential not only as membrane constituents in Eukaryotes and as precursors of second messengers like IP3 but also serve as specialized membrane docking sites for effectors of various signaling cascades [9]. Accumulating evidence suggests that PIs and PI-interacting proteins such as Rho, Arf, and Rab small GTPases function as modulators of osteoclast differentiation [10, 11]. In this review, we focus on recent advances in understanding the regulation of osteoclastogenesis by membrane-derived lipid mediators.
Upregulation of intracellular Ca2+ concentration by IP3 activates NFATc1, a master transcription factor for osteoclastogenesis
Sustained activation of transcription factor NFATc1 is a crucial step in osteoclast differentiation and maturation. Forced expression of NFATc1 in bone marrow macrophages induces osteoclast differentiation, while NFATc1-deficient embryonic stem cells fail to differentiate into osteoclasts following stimulation with RANKL [3, 12]. The canonical mechanism of NFATc1 activation is through dephosphorylation by calcineurin, a Ca2+/calmodulin-dependent phosphatase, and subsequent nuclear translocation [13]. During osteoclastogenesis, intracellular Ca2+ levels oscillate in response to RANKL stimulation, which is thought to cause long-term activation of NFATc1. Since RANKL-induced Ca2+ oscillations are abolished in IP3 receptor (IP3R) knockout cells, Ca2+ release from the endoplasmic reticulum (ER) through IP3R channels is required to generate or sustain these Ca2+ oscillations [14]. Both the IP3R ligand IP3 and the protein kinase C activator diacylglycerol (DAG) are produced from the membrane phospholipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] by PLC. Therefore, metabolism of membrane phospholipids by PLC activation is critical for RANKL-induced Ca2+ signaling and subsequent NFATc1 activation during osteoclastogenesis.
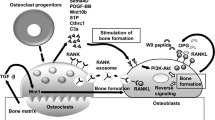
In concert with RANK signaling, immunoglobulin-like receptors such as osteoclast-associated receptor (OSCAR) and the triggering receptor expressed in myeloid cells 2 (TREM-2) transduce Nfatc1 induction signals [15, 16]. Both receptors are associated with adaptor proteins such as DNAX-activation protein (DAP) 12 or the Fc receptor common γ subunit (FcRg) that possess the immunoreceptor tyrosine-based activation motif (ITAM) [17]. After ITAM tyrosine phosphorylation, a complex containing Bruton’s tyrosine kinase (Btk), tyrosine kinase expressed in hepatocellular carcinoma (Tec), the adaptor molecules B cell linker protein (BLNK) and Src homology 2 domain-containing leukocyte protein of 76 kD (SLP76) is formed that facilitates cooperation between RANK and ITAM signaling [18]. This combined signal leads to sustained PLCγ2 phosphorylation, suggesting that integration of RANK and ITAM signaling is required for the efficient activation of PLCγ2 and subsequent Ca2+ oscillations (Fig. 1). Furthermore, following elevation of intracellular Ca2+ but prior to calcium oscillations, Nfatc1 transcription is enhanced by Ca2+/calmodulin-dependent kinase IV (CaMK IV). In turn, CaMK IV phosphorylates the cAMP response element-binding protein (CREB), inducing Fos expression [19].

IP3 instigates the activation and amplification of NFATc1. Depicted above is a schematic illustrating the stages of osteoclast differentiation, including recruitment of progenitors to the bone surface, cell–cell fusion, formation of the actin ring and ruffled border and bone resorption. In the early phase of differentiation (red-boxed), osteoclastogenesis is triggered by RANKL–RANK signaling, which activates PLCγ2 to generate IP3 from PI(4,5)P2 in the plasma membrane. IP3 then stimulates calcium oscillations, which are required for subsequent activation of NFATc1. Knockout of the molecules in red have bone-related phenotypes largely because of impaired osteoclast differentiation (see the text for details). Red dotted lines indicate interactions between PIs and proteins. The dotted arrow indicates the Ca2+ oscillation-independent pathway to NFATc1 activation. This figure is modified from Kuroda et al., World Journal of Orthopedics (in press)
The PLCγ family consists of the widely distributed PLCγ1 and the more restricted PLCγ2, which is primarily expressed by hematopoietic cells [20]. While Plcg1 −/− mice cannot develop normally beyond embryonic day 8.5 [21], Plcg2 −/− mice are viable but exhibit an osteopetrotic phenotype [22], indicating that PLCγ2 is required for osteoclastogenesis (Table 1). At a highly conserved region (HCR) in the RANK C-terminal tail, PLCγ2 forms a stimulus-dependent complex with the TRAF6 and Gab2 adapter proteins [23] (Fig. 1). An HCR deletion mutant of the CD40/RANK chimeric receptor does not alter NF-κB or MAPK activation but abolishes Ca2+ oscillations, indicating that HCR-mediated signaling is indispensable for sustained PLCγ2 activation and that sustained PLCγ2 activation is required to maintain Ca2+ oscillations [23].
Calcium signaling during osteoclastogenesis is also controlled by PI-binding proteins. The regulator of G-protein signaling 10 (RGS10) competitively binds to phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3] and this binding is required for RGS10 membrane localization and the subsequent activation of PLCγ2 and Ca2+ oscillations. The intracellular Ca2+ concentration shifts the balance between RGS10–PI(3,4,5)P3 and RGS10–Ca2+/CaM complexes and this may allow for self-sustaining Ca2+ oscillations through oscillatory regulation of PLCγ2 activation [24] (Fig. 1). Mice lacking RGS10 exhibit severe osteopetrosis due to defects in Ca2+ oscillations and reduced osteoclastogenesis in vivo, underscoring the importance of Ca2+ oscillations for NFATc1 activation and amplification during osteoclast differentiation [24] (Table 1). Inositol polyphosphate 4-phosphatase type IIα (Inpp4bα) can also modulate IP3-triggered Ca2+ signaling and subsequent osteoclastogenesis as suggested by the decreased bone mass observed in Inpp4b −/− mice [25] (Table 1). However, Inpp4bα efficiently hydrolyzes Ins(1,3,4)P3 but not IP3 in vitro [25]. Since Ins(1,3,4)P3 does not open purified IP3 receptors [26], the precise molecular mechanisms by which Inpp4bα ablation enhances Ca2+ signaling remains unknown. Nevertheless, as the human INPP4B was also identified as a susceptibility locus for osteoporosis [25], the balance among these different membrane inositol phospholipids could be a critical regulator of Ca2+ signaling and osteoclastogenesis.
In addition to Ca2+ oscillations, NFATc1 is also activated by an osteoblast-induced, Ca2+-independent pathway. When co-cultured with osteoblasts, cell–cell interactions increase NFATc1 protein levels even in osteoclast precursors derived from IP3R type2 and type3 (IP3R2/3) double knockout mice. Furthermore, osteoblasts promote osteoclast differentiation in the absence of detectable RANKL-induced Ca2+ oscillations [14] (Table 1). Phosphorylation-dependent protein stabilization of NFATc1 by Cot (Cancer Osaka thyroid) serine/threonine kinase, also known as tumor progression locus 2 (Tpl-2), partially explains Ca2+- and calcineurin-independent osteoclastogenesis [27]. Whether membrane phospholipids also contribute to Ca2+ oscillation-independent NFATc1 activation is unknown at present.
PI3-kinases and their lipid products regulate osteoclast function
PI3-kinase is activated downstream of osteoclastogenic stimuli
Once primed for differentiation into osteoclasts by IP3-triggered Ca2+ signaling and activation of NFATc1, a number of additional signaling molecules are activated that regulate osteoclast function. The PI3-kinase is one of the central downstream effectors of the M-CSF receptor c-fms [28, 29], RANK [30], and αv β 3 integrin [31] in osteoclasts. PI3-kinases can be classified into three groups: class I, which consists of regulatory and catalytic subunits such as p85 and p110; class II kinases that do not require adaptor subunits; and class III kinases with a catalytic subunit p110 that shares homology with the yeast PI3-kinase Vps34p. Activation of class I PI3-kinase downstream of RANK leads to the production of PI(3,4,5)P3 from PI(4,5)P2. Class I and II PI3-kinases produce PI(3,4)P2 from PI(4)P; alternatively, PI(3,4)P2 can be generated by dephosphorylation of PI(3,4,5)P3 by PI(3,4,5)P3 5-phosphatases such as Src homology 2-containing inositol-5-phosphatase 1 (SHIP1) [32] (Fig. 2). In general, PI(3,4,5)P3 and PI(3,4)P2 serve as stimulants of cell proliferation, survival, and directional migration by anchoring effectors like Akt [32]. PI3-kinase activity can be quenched by generation of PI(4,5)P2 from PI(3,4,5)P3 through hydrolysis by a tumor suppressor gene product, phosphatase and tensin homolog deleted from chromosome 10 (PTEN) [33] (Fig. 2). Thus, PI3-kinase is bidirectionally regulated by a number of proteins.

PI(3,4,5)P3 regulates osteoclast adhesion, motility, and ruffled border function. In the later phase of differentiation (red-boxed phase in the top schematic), signaling downstream of c-fms, RANK and αvβ3 integrin activates PI3-kinase, triggering the production of PI(3,4,5)P3 in the plasma membrane. PI(3,4,5)P3 then recruits and/or activates cytosolic proteins, which are important for cytoskeletal rearrangement. Knockouts of the molecules in red have bone-related phenotypes because of functional defects in osteoclasts (see the text for details). Blue arrows indicate phosphorylation. Red dotted lines indicate interactions between PIs and proteins. Small GTPases are encircled in green, GEF/GAP in blue, and kinases/phosphatases in yellow
The PI3-kinase is required for activation of Akt and MAPK [34], so it is not surprising that mice lacking the p85α subunit of class I PI3-kinase exhibit impaired osteoclast proliferation and maturation [35] (Table 1) as well as of impaired B cell development and activation [36, 37]. A study using osteoclast-specific p85α/β double knockout mice demonstrates that PI3-kinase-dependent activation of Akt is essential for ruffled border formation and vesicle transport [38] (Table 1). Knockouts of the p110α or p110β catalytic subunit (Pik3ca −/− or Pik3cb −/−, respectively) result in embryonic lethality [39, 40], while p110γ knockouts (Pik3cg −/−) are viable but exhibit defects in T cell proliferation and function as well as reduced neutrophil migration and chemotaxis [41]. Although a study using a specific inhibitor suggests a dominant role for p110α in osteoclast differentiation [42], detailed skeletal analysis of conditional knock out of these class I catalytic subunits and deletion of other classes of PI3-kinase genes will be required to clarify the role of each PI3-kinase class and isoform in osteoclastogenesis. In contrast to mice lacking the p85α PI3-kinase subunit, Inpp5d −/− mice lacking SHIP1 exhibit the reverse phenotype, with more numerous and larger osteoclasts that are hypersensitive to M-CSF and RANKL, as well as less apoptotic and hyper-resorptive, resulting in osteoporosis [43] (Table 1). Another study found that the granulocyte–macrophage progenitors of Inpp5d −/− mice show enhanced proliferative potential [44]. Similarly, PTEN negatively regulates osteoclast differentiation [45] (Table 1). These results strongly suggest that PI(3,4,5)P3 is a primary inducer of osteoclastogenesis (Figs. 1 and 2). It would be of great interest to test if direct addition of liposomes containing PI(3,4,5)P3 to osteoclast precursors accelerates osteoclastogenesis. However, it is possible that the spatiotemporal production of PI(3,4,5)P3 must be tightly controlled to properly drive osteoclastogenesis, given the role of PI(3,4,5)P3 as an anchor and the functional segregation of different membrane compartments of the multinuclear osteoclast.
PI- and raft-dependent c-Src activity is required for osteoclastogenesis
Locally produced PI(3,4,5)P3 and PI(3,4)P2 recruit cytosolic proteins to the plasma membrane. For example, direct interaction between PI(3,4,5)P3 and the Src-homology 2 (SH2) domains of PI3-kinase or the ubiquitous tyrosine kinase c-Src stimulates the formation of a protein complex containing PI3-kinase or c-Src and gelsolin [46]. Membrane targeting of c-Src is also aided by covalent binding of the 14-carbon fatty acid myristate and by the basic amino acid residues at the c-Src N-terminal [47]. In addition, specialized membrane microdomains enriched with cholesterol and sphingolipids (called lipid rafts) are platforms for enrichment of c-Src activity [48]. Membrane-associated c-Src is indispensable for osteoclast functions, as evidenced by the observation that osteoclasts from Src −/− mice manifest impaired formation of actin rings and reduced bone resorption activity, leading to severe osteopetrosis [49, 50] (Table 1). Moreover, this phenotype was not mimicked by deletion of other Src-family kinases [51], indicating the importance of c-Src and its specific binding proteins and substrates in osteoclastogenesis although Hck partially compensates for c-Src as revealed by Hck −/− Src −/−mice [52]. Indeed, c-Src has been shown to regulate cytoskeletal dynamics [53] as well as cell spreading, cell–cell fusion and ruffled border formation [54, 55], seminal early events in osteoclastogenesis vital for subsequent bone resorption. Among these c-Src binding partners are FAK [56, 57], p130Cas [58], WASP [59], Tks5 [60], cortactin [61–63] and GIT2 [64] proteins that mediate adhesion, podosome/fusion-competent protrusion and actin ring formation in osteoclasts (Fig. 2; Table 1). Furthermore, c-Src is also localized at the intercellular vesicular membranes and ruffled border, where it contributes to the secretion of bone-degrading acids and enzymes [65–68].
Cytoskeletal reorganization by small GTPases is controlled by PI(4,5)P2 and PI(3,4,5)P3
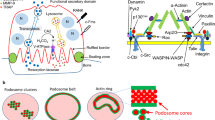
The coalescence and fusion of osteoclasts requires the activation of Rho-family GTPases and molecules that rearrange the actin cytoskeleton [69, 70]. A membrane-type 1 matrix metalloproteinase (MT1-MMP)—p130Cas—Rac signaling pathway was recently shown to be indispensable for this process [71](Fig. 2). Bone marrow cells from Mmp14 −/− mice lacking MT1-MMP are still committed to the osteoclast lineage as they express osteoclast genes like Nfatc1 and the TRAP gene Acp5 but exhibit defects in migration and cell–cell fusion [71] (Table 1). Small GTPases of the Rho and Arf family, such as Rho, Rac, Cdc42 and Arf6, are also recruited and activated/inactivated by PIs on the membrane to regulate osteoclast differentiation and bone resorption [64, 72–74] (Table 1). Indeed, these proteins are central regulators of cytoskeletal remodeling, protrusion formation, and membrane trafficking [75, 76]. Activity is stimulated by guanine nucleotide exchange factors (GEFs) such as the Rac activating Vav family proteins and decreased by GTPase-activating proteins (GAPs), such as Arf6 inhibitors GIT2 or centaurin. These GEFs and GAPs often possess pleckstrin homology (PH) domains through which they directly interact with PI(4,5)P2, PI(3,4)P2 and/or PI(3,4,5)P3 [77, 78]. The GEF Vav3 is crucial for Rac activation and subsequent cytoskeletal rearrangement in osteoclasts as evidenced by osteopetrosis in Vav3 −/− or Vav1 −/− Vav3 −/− mice [79] (Table 1). Osteoclasts from these mice do express osteoclast gene products in response to M-CSF and RANKL, but circumferential podosome formation, cell–cell fusion and bone resorption are impaired [79]. Osteoclasts with reduced expression of the c-Src substrate and Arf6 inhibitor GIT2 induced by RNA interference (RNAi) appear to differentiate normally but lack actin rings [64] (Table 1). While GIT2 lacks the PH domain allowing direct interaction with PIs, it is activated by PI(3,4,5)P3 [80], and this interaction suppresses Arf6 activity. The GTPase Arf6 is required for the formation of membrane protrusions such as invadopodia in cancer cells by promoting endosomal recycling and Rac-mediated cytoskeletal remodeling [76, 81]. Therefore, excessive Arf6 activity in GIT2 knockdown osteoclasts may allow for the formation of circumferential podosomes, which are structures analogous to invadopodia, but obstruct later actin ring formation. Analogous to GIT2 function in osteoclast differentiation, previous studies reported that GIT2 regulates the directional chemotaxis of neutrophils and that the loss of GIT2 in vivo leads to immunodeficiency [82]. Centaurin-α2, another GAP for Arf6 with a PH domain, is also essential for Arf6-dependent cytoskeletal remodeling [78], thereby supporting the importance of the PI(3,4,5)P3-Arf6 pathway in osteoclast maturation (Fig. 2).
The cytoskeletal proteins gelsolin, villin, cofilin, and profilin, which sever or depolymerize actin filaments in vitro, are inactivated by PI(4,5)P2, a PI synthesized by phosphatidylinositol 4-phosphate 5-kinase (PI4P-5 kinase). Conversely, several cytoskeletal proteins that bundle actin filaments or link them to the plasma membrane, including vinculin, talin, ezrin/radixin/moesin (ERM) proteins, WASP/N-WASP and α-actinin, are activated by PI(4,5)P2 and/or PI(3,4,5)P3. Consequently, membrane PIs also control cytoskeletal dynamics and osteoclast functions by regulating the activities of these cytoskeletal proteins. Whole animal knockout of gelsolin (Gsn −/−) [83] or Wasp (Was−/−) [84], or osteoclast-specific knockout of Talin (Tln −/−) [85] increases mouse bone mass or attenuates bone loss following ovariectomy (Table 1). WAS deletion also eliminates the formation of podosome clusters in human primary macrophages [86], thus explaining the impaired migration and invasion of macrophages in patients with Wiskott–Aldrich syndrome (WAS). On the other hand, Was −/− osteoclasts exhibit a greater number of nuclei per cell [84], indicating that cell–cell fusion is enhanced. Both podosomes and fusion-competent protrusions (see below) require cytoskeletal remodeling and membrane deformation. However, according to the knockout phenotype, WASP is exclusively required for podosomes but not for fusion-competent protrusions. In the absence of WASP, additional quantities of actin-regulatory and membrane-deforming molecules interacting with WASP, e.g., Arp2/3 complex and Cdc42, are thought to be used for fusion-competent protrusions, instead of being used for podosomes. This may explain the enhanced cell–cell fusion in Was −/− osteoclasts [84]. It is plausible that the formation of perpendicular actin-rich membrane protrusions like podosomes or horizontal fusion-competent protrusions depends on a balance between PI-regulated complementary cytoskeletal GTPases that either promote actin polymerization or bundling and membrane association. Tropomyosin (Tm) stabilizes actin filaments by functionally antagonizing depolymerization or severing factors such as gelsolin and cofilin [87]. Expression of Tm-2 and Tm-3 is induced in the late phase of osteoclastogenesis and reduced expression or overexpression results in altered spreading, motility, and resorption of osteoclasts [88]. Therefore, a balance of activity among cytoskeletal proteins described above may explain osteoclasts’ resorptive/migratory cycle (polarization/depolarization cycle).
Cell–cell fusion is achieved by fusion-competent protrusions downstream of PI3-kinase
Osteoclasts must overcome a significant energy barrier for the fusion of apposing lipid bilayers given that plasma membranes do not spontaneously fuse. In vitro protein-free experiments indicate that lipid bilayer fusion involves the following steps: establishment of close contact between the bilayers so that they become at least partially dehydrated, formation of highly curved protrusions between bilayers to expose an unstable outer leaflet, resulting in hemifusion, and final formation of a fusion pore, a process that requires the lateral tension concomitant with local or global membrane expansion [89–92]. Osteoclast-specific fusogens such as DC-STAMP [93], OC-STAMP [94], macrophage fusion receptor (MFR) [95], v-ATPase V0 subunit d2 [96], and CD9 in lipid rafts [97] are thought to lower the first intermediate energy barriers by tightly tethering the opposing plasma membranes. Proteins that have membrane-deforming activity or those inducing membrane expansion could then contribute to hemifusion and fusion pore formation. In fact, circumferential podosomes, but not mature actin rings, may supply the lateral tension necessary to drive fusion pore opening [91]. When probed with markers of PI-binding domains, the fusion sites are often enriched with products of PI3-kinases [60]. Inhibition of PI3-kinase activity only during the period of highest fusion frequency results in fusion defects, while the expression of osteoclast genes are unaffected [60]. The phox homology (PX) domain adaptor protein Tks5, known to regulate invadopodia formation in cancer cells [98], was found to act downstream of PI3-kinase and Src in promoting cell–cell fusion. Reduced expression of Tks5 results in the loss of circumferential podosomes and cell–cell fusion [60] (Table 1), which is in accordance with recent findings showing that N-WASP-dependent actin-rich protrusive structures are also key drivers of myoblast fusion [99, 100]. Alternatively, circumferential podosome expansions in osteoclasts often accompany tiny membrane protrusions [101] that might be generated by membrane-deforming proteins such as Bin-Amphiphysin-Rvs161/167 (BAR) domain superfamily proteins [102]. For this reason, we refer to the protrusions observed during osteoclast fusion as fusion-competent protrusions. Studying the shared and distinct fusion mechanisms in multiple biological processes may soon provide a more complete and clear picture of osteoclast fusion.
Efficient bone resorption is achieved through ruffled border formation and intracellular membrane trafficking
Fused osteoclasts reorganize their actin cytoskeletons and eventually form F-actin-rich adhesive structures called actin rings on the ventral membranes contacting the bone surface [6]. The membrane area enclosed by the actin ring, termed the ruffled border, secretes protons and hydrolases that solubilize and digest inorganic and organic bone matrix. The ruffled border was originally found to be the site of lysosomal secretion [103, 104] and defects in its formation were reported in osteoclasts from patients with malignant juvenile osteopetrosis [105]. The a3 subunit of v-ATPases and the Cl−/H+ antiporter CLC-7 localize at both the lysosomes and ruffled border, where they function to acidify secreting lysosomes and the space between the ruffled border and bone surface [106, 107] (Fig. 3). Notably, mutations in the gene encoding the a3 subunit of v-ATPases (OC116) or in the gene encoding CLC-7 (CLCN7) are reported in malignant juvenile osteopetrosis [107, 108]. Similarly, disruption of Atr6i, the gene encoding the a3 subunit of v-ATPases in mice, or mouse Clcn7 causes osteopetrosis [107, 109] (Table 1). Furthermore, the small GTPase Rab7, which regulates vesicle fusion to late endosomes or lysosomes, also localizes to the ruffled border [110] (Fig. 3). Reduced expression of Rab7 impairs actin ring formation, ruffled border formation and bone resorption in vitro [111], while multinucleation (cell–cell fusion) is unaffected (Table 1). Rab3D is another Rab GTPase that is essential for osteoclast function as revealed by Rab3d −/− mice that exhibit an osteopetrotic phenotype [112] (Table 1). Osteoclasts from these mice form disturbed ruffled borders with normal actin rings [112] (Table 1). However, the vesicular trafficking pathway mediated by Rab3D appears different from that mediated by Rab7 as judged by their distinct subcellular localization (Fig. 3). Generally, Rab GTPases function only when the C-terminal cysteine(s) are covalently linked to a farnesyl or geranylgeranyl moiety (called prenylation). The importance of prenylation for Rab GTPase function and osteoclast-mediated bone resorption is demonstrated by gunmetal mice, which have an autosomal recessive mutation in the gene encoding Rab geranylgeranyl transferase (RGGT), resulting in a 70 % reduction in GTPase activity [113] and osteoclasts with normal cytoskeletal architecture but reduced resorptive activity [114] (Table 1).

Vesicular trafficking enables bone resorption. In the resorbing phase (red-boxed phase in the top schematic), Rab GTPases mediate vesicle trafficking, while the fusion of vesicles with the ruffled border to release acids and hydrolases is mediated by Syt VII and Atg5. Dynamin-mediated endocytosis clears degraded materials. Knockouts or mutations of the molecules in red have bone-related phenotypes because of functional defects in osteoclasts (see text for details)
Synaptotagmin VII (Syt VII), a member of the synaptotagmin family that mediates Ca2+-triggered fusion of cytoplasmic/synaptic vesicles to the plasma membrane [115], localizes at the ruffled border, and promotes the secretion of lysosomal contents [116] (Fig. 3). Osteoclasts deficient in Syt VII (Syt7 −/−) fail to localize cathepsin K to the resorptive microenvironment or form ruffled borders; however, the bone density of Syt7 −/− mice is actually reduced partly because of attenuated bone formation by osteoblasts [116] (Table 1). Unlike many other phases of osteoclastogenesis, proteins regulating the formation of the ruffled border are largely unknown. In this regard, autophagic proteins that regulate lysozyme secretion in intestinal Paneth cells [117], insulin secretion in pancreatic β cells [118], or degranulation of mast cells [119] are strong candidate effectors of ruffled border formation and extracellular secretion by osteoclasts. Indeed, osteoclast-specific deletion of autophagy-related (Atg) protein Atg5 leads to increased bone mass and alleviates bone loss caused by ovariectomy [120] (Fig. 3; Table 1). Further, the conjugation of the mammalian Atg8 homolog LC3 with phosphatidylethanolamine (PE) is indispensable for the proper trafficking of cathepsin K to the ruffled border [120]. Both Syt VII and Atg5 are required for ruffled border formation/maturation and vesicle-membrane fusion but not for actin ring formation [116, 120] (Table 1). Protein kinase Cδ is another likely participant in cathepsin K secretion. Mice deficient in Prkcd −/− are osteopetrotic and protected from bone loss induced by ovariectomy [121] (Table 1). Intriguingly, impaired cathepsin K secretion in Prkcd −/− osteoclasts is independent of ruffled border formation and trafficking of lysosomal vesicles [121] (Table 1). Therefore, the DAG-PKCδ pathway may promote cathepsin K secretion through alternate mechanisms.
While digesting the bone matrix, osteoclasts must properly dispose of large amounts of calcium, phosphate, and digested collagen that would otherwise rise to cytotoxic levels. Osteoclasts transport these products and transcytose the vesicles containing these materials to the apical region of the plasma membrane, called functional secretory domain, into the extracellular space [122, 123] (Fig. 3). The ruffled border is thus the site of extensive endocytic activity and expresses known endocytic proteins like clathrin, AP-2 and dynamin [124, 125]. Exogenous small tracer molecules rapidly enter the osteoclast and are found at the ruffled border within minutes [126]. Therefore, at least some endocytosis from the ruffled border is thought to be receptor-independent and non-specific, resembling macropinocytosis. Dynamin, a PH domain-containing GTPase essential for podosome formation and endocytosis, is pivotal for the coat-dependent specific uptake [127, 128] (Fig. 3). Overexpression of dynamin stimulates osteoclast migration and resorption and this stimulation depends on the presence of dynamin GTPase activity [129] (Table 1). Dynasore, a specific inhibitor of dynamin [130], may be a useful agent for treating osteoporosis if selectively delivered to osteoclasts.
It has been suggested that raft-dependent membrane trafficking from the ventral or apical membrane is necessary to maintain a functional ruffled border [110, 131], but it is still unclear as to how osteoclasts organize and segregate the functionally distinct membrane regions or as to how regions like the ruffled border and apical membrane coordinate membrane recycling so that exocytosis and endocytosis are optimized.
Conclusions
In this review, we presented evidence demonstrating that membrane lipids, particularly PIs, are crucial for osteoclast differentiation and bone resorption. As osteoclastogenesis is largely dependent on IP3-mediated Ca2+ oscillations, signaling pathways that lead to IP3 production are of special importance. Indeed, knockout of various molecules in Ca2+- or IP3-dependent pathways result in osteopetrotic phenotypes because of impaired osteoclast differentiation and bone resorption (Fig. 1; Table 1). Once committed, remodeling of both the actin cytoskeleton and the plasma membrane drives the morphological changes associated with osteoclastogenesis. During morphological transformation and maturation, there are three major barriers to overcome according to knockout or knockdown phenotypes: (1) cell–cell fusion, (2) actin ring formation, and (3) ruffled border formation. (1) To overcome the energy barrier of membrane fusion, fusogens on the plasma membrane and efficient migration and formation of fusion-competent protrusions are required. Production of PI(3,4,5)P3 and/or PI(3,4)P2 on the membrane triggers migration and protrusion formation in pre-fusion osteoclasts (Fig. 2; Table 1). Without cell–cell fusion, mononuclear osteoclasts can still proceed to the next barrier. (2) Actin ring formation requires actin regulatory molecules that act to stabilize dense actin bundles (Fig. 2 and Table 1). Without actin rings, osteoclasts cannot proceed to the next barrier. The actin ring acts to segregate hydrolytic enzymes, acids, and toxic digestive products from the extracellular environment. Actin ring formation is also dependent on inositol phospholipids that regulate a variety of small GTPases associated with the membrane and cytoskeleton. (3) The ruffled border of osteoclasts allows these cells to efficiently release degradative enzymes and resorb digestion products for recycling by membrane trafficking and vesicle–membrane fusion (Fig. 3; Table 1).
References
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM (1999) Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402:304–309
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J (1999) RANK is essential for osteoclast and lymph node development. Genes Dev 13:2412–2424
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3:889–901
Matsuo K, Galson DL, Zhao C, Peng L, Laplace C, Wang KZ, Bachler MA, Amano H, Aburatani H, Ishikawa H, Wagner EF (2004) Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem 279:26475–26480
Negishi-Koga T, Takayanagi H (2009) Ca2 + -NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev 231:241–256
Jurdic P, Saltel F, Chabadel A, Destaing O (2006) Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol 85:195–202
Saltel F, Chabadel A, Bonnelye E, Jurdic P (2008) Actin cytoskeletal organisation in osteoclasts: a model to decipher transmigration and matrix degradation. Eur J Cell Biol 87(8–9):459–468
Zhao H, Ross F (2007) Mechanisms of osteoclastic secretion. Ann N Y Acad Sci 1116:238–244
Takenawa T, Itoh T (2001) Phosphoinositides, key molecules for regulation of actin cytoskeletal organization and membrane traffic from the plasma membrane. Biochim Biophys Acta 1533:190–206
Chellaiah MA (2006) Regulation of podosomes by integrin alphavbeta3 and Rho GTPase-facilitated phosphoinositide signaling. Eur J Cell Biol 85:311–317
Ory S, Brazier H, Pawlak G, Blangy A (2008) Rho GTPases in osteoclasts: orchestrators of podosome arrangement. Eur J Cell Biol 87:469–477
Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H (2005) Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med 202:1261–1269
Hogan PG, Chen L, Nardone J, Rao A (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17:2205–2232
Kuroda Y, Hisatsune C, Nakamura T, Matsuo K, Mikoshiba K (2008) Osteoblasts induce Ca2 + oscillation-independent NFATc1 activation during osteoclastogenesis. Proc Natl Acad Sci USA 105:8643–8648
Kim N, Takami M, Rho J, Josien R, Choi Y (2002) A novel member of the leukocyte receptor complex regulates osteoclast differentiation. J Exp Med 195:201–209
Cella M, Buonsanti C, Strader C, Kondo T, Salmaggi A, Colonna M (2003) Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J Exp Med 198:645–651
Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, Taniguchi T, Takayanagi H, Takai T (2004) Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 428:758–763
Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, Takai T, Kodama T, Morio T, Geha RS, Kitamura D, Kurosaki T, Ellmeier W, Takayanagi H (2008) Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 132:794–806
Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, Kodama T, Chatila TA, Bito H, Takayanagi H (2006) Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med 12:1410–1416
Wilde JI, Watson SP (2001) Regulation of phospholipase C gamma isoforms in haematopoietic cells: why one, not the other? Cell Signal 13:691–701
Ji QS, Winnier GE, Niswender KD, Horstman D, Wisdom R, Magnuson MA, Carpenter G (1997) Essential role of the tyrosine kinase substrate phospholipase C-gamma1 in mammalian growth and development. Proc Natl Acad Sci USA 94:2999–3003
Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R (2006) PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Investig 116:2869–2879
Taguchi Y, Gohda J, Koga T, Takayanagi H, Inoue J (2009) A unique domain in RANK is required for Gab2 and PLCgamma2 binding to establish osteoclastogenic signals. Genes Cells 14:1331–1345
Yang S, Li YP (2007) RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2 +]i oscillation regulation. Genes Dev 21:1803–1816
Ferron M, Boudiffa M, Arsenault M, Rached M, Pata M, Giroux S, Elfassihi L, Kisseleva M, Majerus PW, Rousseau F, Vacher J (2011) Inositol polyphosphate 4-phosphatase B as a regulator of bone mass in mice and humans. Cell Metab 14:466–477
Ferris CD, Huganir RL, Supattapone S, Snyder SH (1989) Purified inositol 1,4,5-trisphosphate receptor mediates calcium flux in reconstituted lipid vesicles. Nature 342:87–89
Kuroda Y, Hisatsune C, Mizutani A, Ogawa N, Matsuo K, Mikoshiba K (2012) Cot kinase promotes Ca2 + oscillation/calcineurin-independent osteoclastogenesis by stabilizing NFATc1 protein. Mol Cell Biol 32:2954–2963
Varticovski L, Druker B, Morrison D, Cantley L, Roberts T (1989) The colony stimulating factor-1 receptor associates with and activates phosphatidylinositol-3 kinase. Nature 342:699–702
Palacio S, Felix R (2001) The role of phosphoinositide 3-kinase in spreading osteoclasts induced by colony-stimulating factor-1. Euro J Endocrinol/Euro Federation Endocr Soc 144:431–440
Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, Choi Y (1999) TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell 4:1041–1049
Lakkakorpi PT, Wesolowski G, Zimolo Z, Rodan GA, Rodan SB (1997) Phosphatidylinositol 3-kinase association with the osteoclast cytoskeleton, and its involvement in osteoclast attachment and spreading. Exp Cell Res 237:296–306
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296:1655–1657
Vazquez F, Devreotes P (2006) Regulation of PTEN function as a PIP3 gatekeeper through membrane interaction. Cell Cycle 5:1523–1527
King WG, Mattaliano MD, Chan TO, Tsichlis PN, Brugge JS (1997) Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol Cell Biol 17:4406–4418
Munugalavadla V, Vemula S, Sims EC, Krishnan S, Chen S, Yan J, Li H, Niziolek PJ, Takemoto C, Robling AG, Yang FC, Kapur R (2008) The p85alpha subunit of class IA phosphatidylinositol 3-kinase regulates the expression of multiple genes involved in osteoclast maturation and migration. Mol Cell Biol 28:7182–7198
Suzuki H, Terauchi Y, Fujiwara M, Aizawa S, Yazaki Y, Kadowaki T, Koyasu S (1999) Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science 283:390–392
Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, Cantley LC (1999) Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science 283:393–397
Shinohara M, Nakamura M, Masuda H, Hirose J, Kadono Y, Iwasawa M, Nagase Y, Ueki K, Kadowaki T, Sasaki T, Kato S, Nakamura H, Tanaka S, Takayanagi H (2012) Class IA phosphatidylinositol 3-kinase regulates osteoclastic bone resorption through Akt-mediated vesicle transport. J Bone Miner Res 27(12):2464–2475
Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem 274:10963–10968
Bi L, Okabe I, Bernard DJ, Nussbaum RL (2002) Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mammalian genome. Off J Int Mamm Genome Soc 13:169–172
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM (2000) Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287:1040–1046
Grey A, Chaussade C, Empson V, Lin JM, Watson M, O’Sullivan S, Rewcastle G, Naot D, Cornish J, Shepherd P (2010) Evidence for a role for the p110-alpha isoform of PI3K in skeletal function. Biochem Biophys Res Commun 391:564–569
Takeshita S, Namba N, Zhao JJ, Jiang Y, Genant HK, Silva MJ, Brodt MD, Helgason CD, Kalesnikoff J, Rauh MJ, Humphries RK, Krystal G, Teitelbaum SL, Ross FP (2002) SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med 8:943–949
Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, Borowski A, Jirik F, Krystal G, Humphries RK (1998) Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev 12:1610–1620
Sugatani T, Alvarez U, Hruska KA (2003) PTEN regulates RANKL- and osteopontin-stimulated signal transduction during osteoclast differentiation and cell motility. J Biol Chem 278:5001–5008
Chellaiah MA, Biswas RS, Yuen D, Alvarez UM, Hruska KA (2001) Phosphatidylinositol 3,4,5-trisphosphate directs association of Src homology 2-containing signaling proteins with gelsolin. J Biol Chem 276:47434–47444
Sigal CT, Zhou W, Buser CA, McLaughlin S, Resh MD (1994) Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids. Proc Natl Acad Sci USA 91:12253–12257
Matsubara T, Ikeda F, Hata K, Nakanishi M, Okada M, Yasuda H, Nishimura R, Yoneda T (2010) Cbp recruitment of Csk into lipid rafts is critical to c-Src kinase activity and bone resorption in osteoclasts. J Bone Miner Res 25:1068–1076
Lakkakorpi PT, Nakamura I, Young M, Lipfert L, Rodan GA, Duong LT (2001) Abnormal localisation and hyperclustering of (alpha) (V)(beta)(3) integrins and associated proteins in Src-deficient or tyrphostin A9-treated osteoclasts. J Cell Sci 114:149–160
Soriano P, Montgomery C, Geske R, Bradley A (1991) Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64:693–702
Lowell CA, Soriano P (1996) Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev 10:1845–1857
Lowell CA, Niwa M, Soriano P, Varmus HE (1996) Deficiency of the Hck and Src tyrosine kinases results in extreme levels of extramedullary hematopoiesis. Blood 87:1780–1792
Destaing O, Sanjay A, Itzstein C, Horne WC, Toomre D, De Camilli P, Baron R (2008) The tyrosine kinase activity of c-Src regulates actin dynamics and organization of podosomes in osteoclasts. Mol Biol Cell 19:394–404
Winograd-Katz SE, Brunner MC, Mirlas N, Geiger B (2011) Analysis of the signaling pathways regulating Src-dependent remodeling of the actin cytoskeleton. Eur J Cell Biol 90(2–3):143–156
Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR (1992) Requirement of pp 60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Investig 90:1622–1627
Duong LT, Lakkakorpi PT, Nakamura I, Machwate M, Nagy RM, Rodan GA (1998) PYK2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of alpha (v)beta3 integrin, and phosphorylated by src kinase. J Clin Investig 102:881–892
Lakkakorpi PT, Bett AJ, Lipfert L, Rodan GA, le Duong T (2003) PYK2 autophosphorylation, but not kinase activity, is necessary for adhesion-induced association with c-Src, osteoclast spreading, and bone resorption. J Biol Chem 278:11502–11512
Nakamura I, Jimi E, Duong LT, Sasaki T, Takahashi N, Rodan GA, Suda T (1998) Tyrosine phosphorylation of p130Cas is involved in actin organization in osteoclasts. J Biol Chem 273:11144–11149
Chellaiah MA, Kuppuswamy D, Lasky L, Linder S (2007) Phosphorylation of a Wiskott–Aldrich syndrome protein-associated signal complex is critical in osteoclast bone resorption. J Biol Chem 282:10104–10116
Oikawa T, Oyama M, Kozuka-Hata H, Uehara S, Udagawa N, Saya H, Matsuo K (2012) Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell–cell fusion. J Cell Biol 197:553–568
Tehrani S, Faccio R, Chandrasekar I, Ross FP, Cooper JA (2006) Cortactin has an essential and specific role in osteoclast actin assembly. Mol Biol Cell 17:2882–2895
Luxenburg C, Parsons JT, Addadi L, Geiger B (2006) Involvement of the Src-cortactin pathway in podosome formation and turnover during polarization of cultured osteoclasts. J Cell Sci 119:4878–4888
Matsubara T, Myoui A, Ikeda F, Hata K, Yoshikawa H, Nishimura R, Yoneda T (2006) Critical role of cortactin in actin ring formation and osteoclastic bone resorption. J Bone Miner Metab 24:368–372
Heckel T, Czupalla C, Expirto Santo AI, Anitei M, Arantzazu Sanchez-Fernandez M, Mosch K, Krause E, Hoflack B (2009) Src-dependent repression of ARF6 is required to maintain podosome-rich sealing zones in bone-digesting osteoclasts. Proc Natl Acad Sci USA 106:1451–1456
Horne WC, Neff L, Chatterjee D, Lomri A, Levy JB, Baron R (1992) Osteoclasts express high levels of pp 60c-src in association with intracellular membranes. J Cell Biol 119:1003–1013
Tanaka S, Takahashi N, Udagawa N, Sasaki T, Fukui Y, Kurokawa T, Suda T (1992) Osteoclasts express high levels of p60c-src, preferentially on ruffled border membranes. FEBS Lett 313:85–89
Edwards JC, Cohen C, Xu W, Schlesinger PH (2006) c-Src control of chloride channel support for osteoclast HCl transport and bone resorption. J Biol Chem 281:28011–28022
Furuyama N, Fujisawa Y (2000) Regulation of collagenolytic protease secretion through c-Src in osteoclasts. Biochem Biophys Res Commun 272:116–124
Brazier H, Pawlak G, Vives V, Blangy A (2009) The Rho GTPase Wrch1 regulates osteoclast precursor adhesion and migration. Int J Biochem Cell Biol 41:1391–1401
Leung R, Wang Y, Cuddy K, Sun C, Magalhaes J, Grynpas M, Glogauer M (2010) Filamin A regulates monocyte migration through Rho small GTPases during osteoclastogenesis. J Bone Miner Res 25:1077–1091
Gonzalo P, Guadamillas MC, Hernandez-Riquer MV, Pollan A, Grande-Garcia A, Bartolome RA, Vasanji A, Ambrogio C, Chiarle R, Teixido J, Risteli J, Apte SS, del Pozo MA, Arroyo AG (2010) MT1-MMP is required for myeloid cell fusion via regulation of Rac1 signaling. Dev Cell 18:77–89
Chellaiah MA (2005) Regulation of actin ring formation by rho GTPases in osteoclasts. J Biol Chem 280:32930–32943
Ito Y, Teitelbaum SL, Zou W, Zheng Y, Johnson JF, Chappel J, Ross FP, Zhao H (2010) Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J Clin Investig 120:1981–1993
Croke M, Ross FP, Korhonen M, Williams DA, Zou W, Teitelbaum SL (2011) Rac deletion in osteoclasts causes severe osteopetrosis. J Cell Sci 124:3811–3821
Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420:629–635
D’Souza-Schorey C, Chavrier P (2006) ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7:347–358
Han J, Luby-Phelps K, Das B, Shu X, Xia Y, Mosteller RD, Krishna UM, Falck JR, White MA, Broek D (1998) Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science 279:558–560
Venkateswarlu K, Brandom KG, Yun H (2007) PI-3-kinase-dependent membrane recruitment of centaurin-alpha2 is essential for its effect on ARF6-mediated actin cytoskeleton reorganisation. J Cell Sci 120:792–801
Faccio R, Teitelbaum SL, Fujikawa K, Chappel J, Zallone A, Tybulewicz VL, Ross FP, Swat W (2005) Vav3 regulates osteoclast function and bone mass. Nat Med 11:284–290
Vitale N, Patton WA, Moss J, Vaughan M, Lefkowitz RJ, Premont RT (2000) GIT proteins, A novel family of phosphatidylinositol 3,4, 5-trisphosphate-stimulated GTPase-activating proteins for ARF6. J Biol Chem 275:13901–13906
Sabe H (2003) Requirement for Arf6 in cell adhesion, migration, and cancer cell invasion. J Biochem 134:485–489
Mazaki Y, Hashimoto S, Tsujimura T, Morishige M, Hashimoto A, Aritake K, Yamada A, Nam JM, Kiyonari H, Nakao K, Sabe H (2006) Neutrophil direction sensing and superoxide production linked by the GTPase-activating protein GIT2. Nat Immunol 7:724–731
Chellaiah M, Kizer N, Silva M, Alvarez U, Kwiatkowski D, Hruska KA (2000) Gelsolin deficiency blocks podosome assembly and produces increased bone mass and strength. J Cell Biol 148:665–678
Calle Y, Jones GE, Jagger C, Fuller K, Blundell MP, Chow J, Chambers T, Thrasher AJ (2004) WASp deficiency in mice results in failure to form osteoclast sealing zones and defects in bone resorption. Blood 103:3552–3561
Teitelbaum SL (2011) The osteoclast and its unique cytoskeleton. Ann N Y Acad Sci 1240:14–17
Linder S, Nelson D, Weiss M, Aepfelbacher M (1999) Wiskott–Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci USA 96:9648–9653
DesMarais V, Ghosh M, Eddy R, Condeelis J (2005) Cofilin takes the lead. J Cell Sci 118:19–26
Kotadiya P, McMichael BK, Lee BS (2008) High molecular weight tropomyosins regulate osteoclast cytoskeletal morphology. Bone 43:951–960
Yang L, Huang HW (2003) A rhombohedral phase of lipid containing a membrane fusion intermediate structure. Biophys J 84:1808–1817
Kozlovsky Y, Kozlov MM (2002) Stalk model of membrane fusion: solution of energy crisis. Biophys J 82:882–895
Shillcock JC, Lipowsky R (2005) Tension-induced fusion of bilayer membranes and vesicles. Nat Mater 4:225–228
Kozlov MM, McMahon HT, Chernomordik LV (2010) Protein-driven membrane stresses in fusion and fission. Trends Biochem Sci 35:699–706
Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, Morita K, Ninomiya K, Suzuki T, Miyamoto K, Oike Y, Takeya M, Toyama Y, Suda T (2005) DC-STAMP is essential for cell–cell fusion in osteoclasts and foreign body giant cells. J Exp Med 202:345–351
Yang M, Birnbaum MJ, MacKay CA, Mason-Savas A, Thompson B, Odgren PR (2008) Osteoclast stimulatory transmembrane protein (OC-STAMP), a novel protein induced by RANKL that promotes osteoclast differentiation. J Cell Physiol 215:497–505
Saginario C, Sterling H, Beckers C, Kobayashi R, Solimena M, Ullu E, Vignery A (1998) MFR, a putative receptor mediating the fusion of macrophages. Mol Cell Biol 18:6213–6223
Lee SH, Rho J, Jeong D, Sul JY, Kim T, Kim N, Kang JS, Miyamoto T, Suda T, Lee SK, Pignolo RJ, Koczon-Jaremko B, Lorenzo J, Choi Y (2006) v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med 12:1403–1409
Ishii M, Iwai K, Koike M, Ohshima S, Kudo-Tanaka E, Ishii T, Mima T, Katada Y, Miyatake K, Uchiyama Y, Saeki Y (2006) RANKL-induced expression of tetraspanin CD9 in lipid raft membrane microdomain is essential for cell fusion during osteoclastogenesis. J Bone Miner Res 21:965–976
Seals DF, Azucena EF Jr, Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, Courtneidge SA (2005) The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 7:155–165
Sens KL, Zhang S, Jin P, Duan R, Zhang G, Luo F, Parachini L, Chen EH (2010) An invasive podosome-like structure promotes fusion pore formation during myoblast fusion. J Cell Biol 191:1013–1027
Gruenbaum-Cohen Y, Harel I, Umansky KB, Tzahor E, Snapper SB, Shilo BZ, Schejter ED (2012) The actin regulator N-WASp is required for muscle-cell fusion in mice. Proc Natl Acad Sci USA 109:11211–11216
Takito J, Nakamura M, Yoda M, Tohmonda T, Uchikawa S, Horiuchi K, Toyama Y, Chiba K (2012) The transient appearance of zipper-like actin superstructures during the fusion of osteoclasts. J Cell Sci 125:662–672
Oikawa T, Matsuo K (2012) Possible role of IRTKS in Tks5-driven osteoclast fusion. Commun Integ Biol 5:508–512
Baron R, Neff L, Louvard D, Courtoy PJ (1985) Cell-mediated extracellular acidification and bone resorption: evidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J Cell Biol 101:2210–2222
Baron R, Neff L, Brown W, Courtoy PJ, Louvard D, Farquhar MG (1988) Polarized secretion of lysosomal enzymes: co-distribution of cation-independent mannose-6-phosphate receptors and lysosomal enzymes along the osteoclast exocytic pathway. J Cell Biol 106:1863–1872
Helfrich MH, Gerritsen EJ (2001) Formation of non-resorbing osteoclasts from peripheral blood mononuclear cells of patients with malignant juvenile osteopetrosis. Br J Haematol 112:64–68
Toyomura T, Murata Y, Yamamoto A, Oka T, Sun-Wada GH, Wada Y, Futai M (2003) From lysosomes to the plasma membrane: localization of vacuolar-type H + -ATPase with the a3 isoform during osteoclast differentiation. J Biol Chem 278:22023–22030
Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ (2001) Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104:205–215
Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ, Kubisch C (2000) Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet 9:2059–2063
Li YP, Chen W, Liang Y, Li E, Stashenko P (1999) Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet 23:447–451
Palokangas H, Mulari M, Vaananen HK (1997) Endocytic pathway from the basal plasma membrane to the ruffled border membrane in bone-resorbing osteoclasts. J Cell Sci 110(Pt 15):1767–1780
Zhao H, Laitala-Leinonen T, Parikka V, Vaananen HK (2001) Downregulation of small GTPase Rab7 impairs osteoclast polarization and bone resorption. J Biol Chem 276:39295–39302
Pavlos NJ, Xu J, Riedel D, Yeoh JS, Teitelbaum SL, Papadimitriou JM, Jahn R, Ross FP, Zheng MH (2005) Rab3D regulates a novel vesicular trafficking pathway that is required for osteoclastic bone resorption. Mol Cell Biol 25:5253–5269
Detter JC, Zhang Q, Mules EH, Novak EK, Mishra VS, Li W, McMurtrie EB, Tchernev VT, Wallace MR, Seabra MC, Swank RT, Kingsmore SF (2000) Rab geranylgeranyl transferase alpha mutation in the gunmetal mouse reduces Rab prenylation and platelet synthesis. Proc Natl Acad Sci USA 97:4144–4149
Taylor A, Mules EH, Seabra MC, Helfrich MH, Rogers MJ, Coxon FP (2011) Impaired prenylation of Rab GTPases in the gunmetal mouse causes defects in bone cell function. Small GTPases 2:131–142
Pang ZP, Sudhof TC (2010) Cell biology of Ca2 + -triggered exocytosis. Curr Opin Cell Biol 22:496–505
Zhao H, Ito Y, Chappel J, Andrews NW, Teitelbaum SL, Ross FP (2008) Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Dev Cell 14:914–925
Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HW 4th (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456:259–263
Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, Kawamori R, Fujitani Y, Watada H (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8:325–332
Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A, Ichimura Y, Ezaki J, Nishida K, Komazawa-Sakon S, Niyonsaba F, Ishii T, Yanagawa T, Kominami E, Ogawa H, Okumura K, Nakano H (2011) Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol 127(1267–76):e6
DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW (2011) Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell 21:966–974
Cremasco V, Decker CE, Stumpo D, Blackshear PJ, Nakayama KI, Nakayama K, Lupu TS, Graham DB, Novack DV, Faccio R (2012) PKCdelta deficiency perturbs bone homeostasis by selective uncoupling of cathepsin K secretion and ruffled border formation in osteoclasts. J Bone Miner Res 27(12):2452–2463
Salo J, Lehenkari P, Mulari M, Metsikko K, Vaananen HK (1997) Removal of osteoclast bone resorption products by transcytosis. Science 276:270–273
Nesbitt SA, Horton MA (1997) Trafficking of matrix collagens through bone-resorbing osteoclasts. Science 276:266–269
Mulari MT, Zhao H, Lakkakorpi PT, Vaananen HK (2003) Osteoclast ruffled border has distinct subdomains for secretion and degraded matrix uptake. Traffic 4:113–125
Stenbeck G, Horton MA (2004) Endocytic trafficking in actively resorbing osteoclasts. J Cell Sci 117:827–836
Stenbeck G, Horton MA (2000) A new specialized cell-matrix interaction in actively resorbing osteoclasts. J Cell Sci 113(Pt 9):1577–1587
Ochoa GC, Slepnev VI, Neff L, Ringstad N, Takei K, Daniell L, Kim W, Cao H, McNiven M, Baron R, De Camilli P (2000) A functional link between dynamin and the actin cytoskeleton at podosomes. J Cell Biol 150:377–389
Ferguson SM, De Camilli P (2012) Dynamin, a membrane-remodeling GTPase. Nat Rev Mol Cell Biol 13:75–88
Bruzzaniti A, Neff L, Sanjay A, Horne WC, De Camilli P, Baron R (2005) Dynamin forms a Src kinase-sensitive complex with Cbl and regulates podosomes and osteoclast activity. Mol Biol Cell 16:3301–3313
Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–850
Mulari MT, Nars M, Laitala-Leinonen T, Kaisto T, Metsikko K, Sun Y, Vaananen HK (2008) Recombinant VSV G proteins reveal a novel raft-dependent endocytic pathway in resorbing osteoclasts. Exp Cell Res 314:1641–1651
Acknowledgments
This work was supported by the Promotion of Environmental Improvement for Independence of Young Researchers, Kanrinmaru Project, of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, a Grant-in-Aid for Young Scientists (A) (23689020) from MEXT, a Grant-in-Aid for Challenging Exploratory Research from MEXT (23659162) and the Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Oikawa, T., Kuroda, Y. & Matsuo, K. Regulation of osteoclasts by membrane-derived lipid mediators. Cell. Mol. Life Sci. 70, 3341–3353 (2013). https://doi.org/10.1007/s00018-012-1238-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1238-4