
Abstract
The loading of antigenic peptides onto major histocompatibility complex class I (MHC I) molecules is an essential step in the adaptive immune response against virally or malignantly transformed cells. The ER-resident peptide-loading complex (PLC) consists of the transporter associated with antigen processing (TAP1 and TAP2), assembled with the auxiliary factors tapasin and MHC I. Here, we demonstrated that the N-terminal extension of each TAP subunit represents an autonomous domain, named TMD0, which is correctly targeted to and inserted into the ER membrane. In the absence of coreTAP, each TMD0 recruits tapasin in a 1:1 stoichiometry. Although the TMD0s lack known ER retention/retrieval signals, they are localized to the ER membrane even in tapasin-deficient cells. We conclude that the TMD0s of TAP form autonomous interaction hubs linking antigen translocation into the ER with peptide loading onto MHC I, hence ensuring a major function in the integrity of the antigen-processing machinery.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The adaptive immune system of jawed vertebrates is responsible for detection and elimination of virus-infected or malignantly transformed cells, thus playing an essential role in survival. Information about the cellular proteome is presented to cytotoxic T cells on the cell surface in the form of complexes of MHC I molecules with antigenic peptides derived from intracellular proteins [1–3]. A large portion of the cellular proteome is degraded by the proteasome. A fraction of these antigenic peptides is transported into the lumen of the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP), a heterodimeric ABC complex composed of TAP1 (ABCB2) and TAP2 (ABCB3). The loading of peptides onto MHC I takes place within the peptide-loading complex (PLC), a multisubunit machinery consisting of TAP1/2, MHC I heavy chain/β2-microglobulin, the chaperone calreticulin, the oxidoreductase ERp57, and tapasin (Tsn). The latter is an essential adapter molecule within the PLC, as it bridges the peptide donor (TAP) with the peptide acceptor (MHC I) [4, 5]. For a large number of MHC I alleles, tapasin is required for loading of high-affinity peptides onto MHC I, a process known as peptide editing [6, 7]. After loading their peptide cargo, MHC I complexes travel via the secretory pathway to the cell surface. Large efforts have been made to understand the assembly, organization, and function of the PLC. For most ER-lumenal parts, structural information is available [8–12]. However, TAP cannot yet be crystallized, and only a homology model of the coreTAP complex is available based on the X-ray structures of bacterial ABC transporter Sav1866 and the NBD1 of TAP1 [13–15].
Each coreTAP subunit consists of six transmembrane helices plus the nucleotide-binding domain. CoreTAP shares significant homology with other ABC transporters and is necessary and sufficient for peptide binding and transport [16]. In contrast, the unique N-terminal domain with four putative transmembrane-spanning segments, called TMD0, shares no homology to any other known protein. Deletion of the first transmembrane-spanning segment of TAP destroys its ability to interact with tapasin [17]. However, there is no structural information available for the TMD0 of TAP1 and TAP2. Despite their critical role in PLC assembly, the TMD0s constitute the least understood part of this macromolecular machinery. TMD0 of TAP2 (hereafter named TMD TAP20 ) is about 30 amino acids shorter than that of TAP1 (TMD TAP10 ). Although their sequences markedly differ, both TMD0s seem to fulfill similar functions in tapasin binding [16], in spite of data indicating a functional asymmetry of greater significance of the rat TMD TAP20 in PLC function [18]. In this study, the functionality and structural integrity of the TMD0s of both TAP subunits were addressed with regard to their subcellular localization, binding, and stoichiometry of tapasin.
Materials and methods
Cloning and constructs
TMD TAP10 and TMD0 TAP2 were cloned into pcDNA3.1(+) (Invitrogen, Darmstadt, Germany) via XhoI/EcoRI and KpnI/NotI, respectively. To generate TMD TAP10 (aa 1–164 of TAP1) with a C-terminal myc-tag and TMD TAP20 (aa 1–127 of TAP2) with a C-terminal HA-tag, the following primer pairs were used: 5′-GTCGACGAATTCATGGCTAGCTCTAGGTG-3′ and 5′-GTCGACCTCGAGTCACAGATCCTCTTCTGAGATGAGTTTTTGTTCGGATCCGCCGGGCACCCAG-3′ for TMD TAP10 , 5′-GTCGACGGTACCAGATCTACCATGCGGCTCCCTGACCTG-3′ and 5′-GTCGACGCGGCCGCTCAAGCGTAGTCTGGGACGTCGTATGGGTAGGATCCCTTCTCCTGGGCTCC-3′ for TMD TAP20 . CoreTAP1 (amino acids 165–748, containing an N-terminal methionine) was amplified with primer pairs 5′-CGATTACTCGAGATGGGTCAGGGCGGCTC-3′and 5′-CGATTAGAATTCCCTTCTGGAGCATCTGC-3′ and cloned into pEGFP-N3 (BD Biosciences, Franklin Lakes, NJ, USA) via XhoI and EcoRI sites. CoreTAP2-mCerulean (amino acids 125-716 plus N-terminal methionine) containing a C-terminal StrepII-tag was amplified with the primer pair 5′-CATGCTTAAGATGGCCCAGGAGAAGGAGCAGGACC-3′and 5′-CCGCTCGAGTCACTTCTCGAATTGTGGGTGAGACCAAGC-3′, then cloned into pcDNA3.1(+) via AflII and XhoI. Tsn-TMD0 constructs were amplified via PCR with the following primers: 5′-AGATCTATGAAGTCCCTGTCTCTGCTCCTCG -3′ as forward primer for both constructs, 5′-ATCGCGGCCGCTCACAGATCCTCTTCTGAGATGAGTTTTTGTTCACCTCCAGGCACCCAAAGACTACC-3′ for Tsn-TMD TAP10 containing a C-terminal myc-tag and 5′-GTCGACGCGGCCGCTCAAGCGTAGTCTGGGACGTCGTATGGGTAGGATCCTTTTTCTTGGGCACCTGGTGGAC-3′ for Tsn-TMD TAP20 with a C-terminal HA-tag. A 34 amino acid long flexible glycine-serine linker was inserted between the C-terminus of tapasin and the N-terminus of the TMD0. Both constructs were cloned into pcDNA3.1(+) via BamHI/NotI. For the TAP1/TAP2 coexpression plasmid, TAP1 was cloned into the MCS2 of pVitro2-neo-mcs (Invivogen, San Diego, CA, USA) via BglII and NheI, TAP2 was cloned into MCS1 via BamHI and SalI.
Cell lines and transfection
HeLa cells were cultured in DMEM (PAA Laboratories, Pasching, Austria) supplemented with 10 % fetal calf serum (FCS; Biochrom, Berlin, Germany). M553, a human tapasin-deficient melanoma cell line [19, 20], was maintained in RPMI 1640 with 10 % FCS. Transfection of M553 was performed using XtremeGene HP (Roche, Grenzach-Wyhlen, Germany) according to the manufacturer’s instructions. HeLa cells were transfected with 18 mM branched polyethyleneimine (PEI) with a DNA-to-PEI ratio of one to three. After 24–48 h, cells were harvested and used for the indicated experiments.
Antibodies
For immunofluorescence experiments, the following antibodies were used: mouse anti-myc 4A6 (Millipore, Billerica, MA, USA), mouse anti-HA HA-7 (Abcam, Cambridge, UK), mouse anti-TAP1 148.3 [21], and mouse anti-StrepII (IBA BioTAGnology, Göttingen, Germany). As organelle makers, rabbit anti-Calreticulin (polyclonal IgG fraction; Sigma-Aldrich, Steinheim, Germany), anti-ERGIC-53 (Sigma-Aldrich), and anti-GM130 (Golgi; EP892Y; Abcam) were used. Detection of the primary antibodies was done with donkey anti-mouse-Alexa488 (Invitrogen), goat anti-rabbit-Cy3 (Dianova, Hamburg, Germany), and, for simultaneous detection of GFP and StrepII together with organelle marker, goat anti-mouse-Alexa633 (Invitrogen). For immunoblotting, anti-myc 4A6, anti-HA HA-7, anti-SRP54 (BD Bioscience), anti-MHC I hc HC10 [22] and mAb 7F6, raised against amino acids 21GPAVIECWFVEDASGKG35 of human tapasin, were used.
Membrane preparation and carbonate extraction
Membranes were prepared from 5 × 106 transiently transfected HeLa cells. The cell pellet was mixed with 50 mM ice-cold Tris buffer (pH 7.3) containing 250 mM sucrose and protease inhibitor mix (Serva, Heidelberg, Germany). Cells were pulped with a glass homogenizer. Post-nuclear supernatants were collected by centrifugation for 10 min at 700g and membranes were sedimented by centrifugation at 100,000g for 30 min at 4 °C. Membranes were resuspended in 0.1 M Na2CO3 buffer pH 11.5 and incubated on ice for 15 min, followed by centrifugation at 100,000g for 20 min at 4 °C. The supernatant was collected. The remaining pellet was resuspended in carbonate buffer pH 11.5, adjusted to 1.6 M sucrose and overlaid with 1.25 M and 0.25 M sucrose in the same buffer. Centrifugation was performed for 90 min at 100,000g and the floating membranes were collected. Proteins were precipitated by chloroform–methanol and subsequently analyzed by SDS-PAGE (15 %).
Immunofluorescence and image processing
Transiently transfected HeLa cells grown on cover slips were fixed 30 min with 4 % formaldehyde in PBS at room temperature, quenched with 50 mM glycine for 10 min, and permeabilized with 0.1 % Triton X-100 for 20 min. After blocking with 5 % bovine serum albumin for 30 min, cells were stained with the primary antibodies followed by secondary antibodies for 1 h at room temperature. Nuclei were visualized by DAPI staining (Dianova). Preparations were mounted in 10 % (w/v) Mowiol (Calbiochem/Merck, Nottingham, UK). Samples were analyzed with a confocal laser-scanning microscope (LSM 510; Zeiss, Germany) equipped with a Plan-Apochromat 63x/1.4 Oil DIC objective. Deconvolution of the images was performed using non-blind 2D deconvolution of AutoQuantX2 (MediaCybernetics, Bethesda, MD, USA). Non-transfected cells were removed from the picture prior to determination of Pearson’s coefficients, and a threshold was manually set to remove background. For colocalization analysis, the JACoP plug-in [23] for ImageJ was used to calculate Pearson’s coefficients out of 8–10 individual cells per construct and organelle marker. For coreTAP1 and coreTAP2 co-expression, Pearson’s coefficients were calculated for coreTAP1-GFP and the organelle marker.
Coimmunoprecipitation
For coimmunoprecipitation, sheep anti-mouse Dynabeads (Invitrogen) were loaded with mouse anti-myc 4A6 (TMD TAP10 and Tsn-TMD TAP10 ) or mouse anti-HA HA-7 (TMD TAP20 and Tsn-TMD TAP20 ) for 2 h at 4 °C. Control precipitations were performed using a non-specific isotype-matched mouse antibody. After coating, the beads were washed three times with 1 ml IP buffer containing 20 mM Tris/HCl, pH 7.4, 0.1 % BSA, 150 mM NaCl, and 5 mM MgCl2. For each sample, 2.5 × 106 HeLa cells were plated in a 14.5-cm dish the day before the transfection. Then, 24 h (Tsn-TMD0s) or 48 h (TMD0s) after transfection, the cells were harvested and solubilized in 1 ml buffer containing 20 mM Tris/HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 % digitonin, and 1 % protease inhibitor mix (Serva) for 1 h at 4 °C. Solubilized cells were centrifuged for 30 min at 100,000g at 4 °C. After pre-clearing, the supernatant was incubated with pre-coated magnetic beads for 1 h at 4 °C. After washing, beads were isolated by a magnet and bound protein was eluted with 30 μl SDS sample buffer for 10 min at 65 °C. Eluates were analyzed by SDS-PAGE and subsequent immunoblotting.
Protease K digestion
Membranes were prepared as described above. After centrifugation at 100,000g, membranes were resuspended in ice-cold PBS containing 5 mM CaCl2. Proteinase K (0.6 units, Sigma Aldrich) were added in the absence or presence of 0.1 % Triton-X 100, and incubated on ice for 30 min. Digestion was stopped by the addition of PMSF (5 mM final), followed by incubation at 95 °C for 10 min in SDS sample buffer. Samples were separated by SDS-PAGE (10 %) and subsequent immunoblotting with the indicated antibodies.
Results
Expression and membrane insertion of the isolated TMD0s
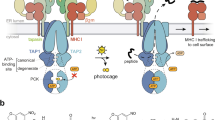
The coreTAP subunits can be expressed without affecting membrane targeting, insertion, folding, dimerization, and transport activity [16]. However, it remains open whether the unique and extra TMD0 represents an autonomous domain with the ability to fold independently of coreTAP and other components of the peptide-loading complex. We thus divided human TAP1 and TAP2 into TMD0 and coreTAP to further study their functions (Fig. 1a). The boundary for dissection of the domains lies within the second cytoplasmic loop, resulting in TMD TAP10 (aa 1–164) and TMD TAP20 (aa 1–127). For immunodetection, the TMD0s of TAP1 and TAP2 were tagged with a C-terminal myc- and HA-epitope, respectively (Fig. 1b). Strikingly, both TMD0s were expressed at high levels in transiently transfected HeLa cells. TMD TAP10 as well as TMD TAP20 were found to be in the membrane fraction (20 and 16 kDa, respectively) together with the integral type I membrane protein tapasin and resistant to alkaline extraction, whereas the peripheral membrane protein, the signal recognition particle SRP54, is in the supernatant (Fig. 1b). These results demonstrate that the TMD0 of each TAP subunit is targeted and inserted as integral membrane proteins.

Unique N-terminal domains (TMD0s) of TAP1 and TAP2 represent an autonomous membrane protein fold. a Structural model of the TAP complex [3, 11, 50]. CoreTAP is shown as a homology model based on the structure of Sav1866 [13, 15]. The extra N-terminal domain TMD0 of each TAP subunit, illustrated as a four-helix bundle, recruits the tapasin-ERp57 conjugate via transmembrane domain interaction. b TMD TAP10 and TMD TAP20 are integral membrane domains. Membranes were prepared from transiently transfected HeLa cells and alkaline extraction was performed. I an aliquot (1/10) of the membranes before alkaline extraction to confirm expression, M and S membrane fraction and supernatant after treatment with carbonate buffer pH 11.5, respectively. Immunoblots were developed against TMD TAP1/20 , tapasin as integral type I membrane protein, and SRP54 as peripheral membrane protein
Subcellular localization of the TAP domains
The ER is the major compartment for MHC I peptide loading, although TAP activity was also found in post-ER compartments [24]. We therefore investigated the subcellular localization of the N-terminal domains. HeLa cells were transiently transfected with each TMD0 or wt TAP and analyzed by immunofluorescence microscopy (Fig. 2). As expected, wt TAP was found to be mostly in the ER. Both TMD TAP10 and TMD TAP20 co-localized to a large extent with the ER. Minor fractions of both TMD0s were found in the ERGIC. Interestingly, the overlap with the ERGIC was more pronounced for TMD TAP20 than for TMD TAP10 . The colocalization with the Golgi marker GM130 was only weak, demonstrating that isolated TMD0s are efficiently retained in the early secretory pathway.

Subcellular localization of TMD TAP1/20 . Indirect immunofluorescence on transiently transfected HeLa cells (TMD0s or wt TAP1/2 as a control) was performed using TMD TAP10 , TMD TAP20 and TAP1 specific (myc, HA and 148.3, respectively, green) as well as organelle specific antibodies [ER anti-calreticulin; ER-Golgi intermediate compartment (ERGIC): ERGIC-53; Golgi GM-130; red]. a TMD TAP10 , b TMD TAP20 , c wt TAP. Images were taken using confocal laser scanning microscopy. Pearson coefficients were calculated from 8–10 individual images. Scale bar 10 μm
We next investigated the localization of coreTAP. Previous studies demonstrated the peptide transport activity of the coreTAP complex into ER-derived microsomes from Sf9 cells using a glycosylation dependent assay [16]. Due to differences in the post-translational modifications between the insect cell and mammalian cell system, this evidence for ER retention of the coreTAP complex might not necessarily be transferrable to human cells. To clarify this point, coreTAP1 and coreTAP2 were fused C-terminally to eGFP and mCerulean, respectively. Confocal laser scanning microscopy directly visualized coreTAP1-eGFP, whereas coreTAP2 was indirectly detected via its C-terminal Strep-tag, due to its low expression level and the relatively weak fluorescence of the mCerulean. Similar to the TMD0s, coreTAP1 and coreTAP2 localized to the ER when expressed individually in HeLa cells (Fig. 3a, b). We reasoned that this ER localization might be caused by the formation of heterodimers with endogenous TAP. To examine if heterodimers of both coreTAP subunits were also restricted to the ER, we coexpressed coreTAP1 and coreTAP2 (Fig. 3c). The Pearson’s coefficients for the tested compartments were similar to those of overexpressed wt TAP (Fig. 2c), indicating that the coreTAP shows the same subcellular distribution as wt TAP.

Subcellular localization of coreTAP. HeLa cells were transiently transfected with coreTAP1-eGFP (a), coreTAP2-mCerulean (b), or a combination of both coreTAP constructs (c), and analyzed by confocal laser scanning microscopy. CoreTAP2-mCerulean was indirectly visualized via its C-terminal StrepII-tag with a StrepII antibody. Organelles were stained with antibodies against marker proteins [ER anti-calreticulin; ER-Golgi intermediate compartment (ERGIC): anti-ERGIC-53; Golgi: anti-GM130]. Pearson coefficients were calculated from 8 to 10 individual images. Scale bar 10 μm
TMD0s of TAP1 and TAP2 form autonomous platforms for tapasin recruitment
It has been previously demonstrated that the coreTAP complex does not interact with tapasin [16, 17, 25]. Thus, the tapasin-binding site could be formed at the interface of TMD0 and coreTAP, or by the TMD0 alone. To distinguish between these possibilities, coimmunoprecipitation experiments were performed with the TMD0s of both TAP subunits and endogenous tapasin in HeLa cells. TMD TAP10 and TMD TAP20 were precipitated using anti-myc or anti-HA antibody, respectively (Fig. 4). Importantly, each TMD0 interacts with tapasin, confirming that the TMD0s are correctly folded and form an independent interaction domain for tapasin.

Recruitment of tapasin by TMD TAP1/20 . TMD TAP10 or TMD TAP20 were transiently expressed in HeLa cells and subjected to coimmunoprecipitation using myc or HA antibodies. As mock control, an isotype antibody was used. For both TMD0s, co-precipitation of tapasin was observed. Asterisks: immunoglobulin heavy chain. The solubilizate (S) represents 1/20 aliquot of the precipitate
TMD0s of TAP1 and TAP2 localize to the ER independently of tapasin
TMD TAP10 and TMD TAP20 lack any known ER retention signal but interact with tapasin, which harbors a di-lysine ER retrieval signal [26]. We therefore asked whether binding to tapasin retains the TMD0 in the ER. To address this, we expressed wt TAP or the TMD0s in the tapasin-deficient cell line M553 [19, 20] and analyzed their subcellular distribution by immunostaining (Fig. 5). Similar to HeLa cells, wt TAP co-localized with the ER marker and only a minimal fraction was found in the ERGIC and Golgi. The same distribution was observed for TMD TAP10 and TMD TAP20 , demonstrating that tapasin interaction is expendable for correct localization. Again, the fraction of TMD TAP20 that was found in the ERGIC was enhanced compared to that of TMD TAP10 .

Subcellular localization of TMD TAP10 and TMD TAP20 in tapasin deficient cells. TMD TAP10 (a), TMD TAP20 (b), and wt TAP1/2 (c) were transiently expressed in the tapasin deficient cell line M553, stained with the antibodies as indicated in Fig. 2, and analyzed via indirect immunofluorescence. Organelles were stained with antibodies against marker proteins [ER anti-calreticulin; ER-Golgi intermediate compartment (ERGIC): anti-ERGIC-53; Golgi: anti-GM130]. Pearson coefficients were calculated from 8 to 10 individual images. Scale bar 10 μm
Each TMD0 provides one single binding site for tapasin
How many tapasin molecules are present within a fully assembled PLC is still a matter of debate. The tapasin-to-TAP ratios reported varied from 1:1 and 2:1 to 4:1 [5, 27, 28]. To address this question, we tethered tapasin to the TMD0 of each TAP subunit via a flexible glycine–serine linker of 34 aa, leading to Tsn-TMD TAP10 and Tsn-TMD TAP20 . As a result, the stoichiometry is fixed and one tapasin binding site is already occupied by the fused tapasin (Fig. 6a). Similar to the isolated TMD0s, the fusion constructs are located to the ER in HeLa cells, after transient expression (data not shown). Our recent experiments targeting the topology of TAP2 revealed that the N-terminus of TAP2 faces the cytosol. For further clarification, the membrane topology of the fusion constructs was confirmed by Proteinase K treatment (Fig. 6b). The myc- or HA-tag at C-terminus of Tsn-TMD0 was accessible from the cytosol, where the ER-lumenal calreticulin is degraded only after Triton-X 100 permeabilization of the membrane. In addition, the linker is accessible for Proteinase K, cleaving the tapasin from TMD0. These results demonstrate the correct membrane insertion of tapasin and that the linker and C-terminus of TMD0s are exposed to the cytosol. By coimmunoprecipitation, we could demonstrate that no additional, endogenous tapasin was recruited to Tsn-TMD TAP10 or Tsn-TMD TAP20 (Fig. 6c), revealing that there is only one single binding site for tapasin on each TMD0. Although Tsn-TMD TAP10 recruited slightly less of MHC I than Tsn-TMD TAP20 , both Tsn-TMD0 fusion proteins were able to bind MHC I, showing that the fused tapasin is folded and functional.

Each TMD0 harbors one single tapasin-binding site. a Model of the Tsn-TMD0 TAP1/2 fusion constructs interacting with MHC I molecules. b Membrane topology of the Tsn-TMD TAP1/20 fusion constructs. Crude membranes were prepared from HeLa cells transiently transfected with Tsn-TMD TAP10 or Tsn-TMD TAP20 . Membranes (Membr.) were treated with Proteinase K (0.6 units) in the absence or presence of Triton-X 100 (T-X 100) for 30 min on ice followed by immunoblotting. c Tsn-TMD TAP10 or Tsn-TMD TAP20 were transiently expressed in HeLa cells and subjected to coimmunoprecipitation with anti-myc, anti-HA, or an isotype control antibody. Asterisks: immunoglobulin heavy chain. The solubilizate (S) represents 1/20 aliquot of the precipitate
Discussion
In this study, we demonstrated that the isolated TMD0s of TAP1 and TAP2 are independently targeted to the ER membrane and form autonomous interaction hubs for tapasin recruitment. They are not required for the ER localization and transport function of the coreTAP complex [16]. A similar functional partition into a core ABC transport complex and extra N-terminal domains was shown for other ABC transporters. Some of these N-terminal extensions have an impact on the subcellular trafficking or function of the ABC transporter, while others seem to serve different purposes, extending the fascinating properties of ABC transporters towards receptor and channel functions. The lysosomal polypeptide transporter ABCB9 (TAP-like), which forms a homodimeric complex, possesses a TMD0 that can be expressed separately. Although this TMD0 is dispensable for mere transport activity, it is required for the lysosomal localization of the transporter. The core transport complex lacking the TMD0s is targeted to the plasma membrane but redirected into lysosomes upon separate expression of TMD0 [29]. However, an intrinsic affinity of TMD0 for the core translocation complex, as seen for ABCB9, was not observed for TAP (data not shown).
Mitochondrial ABC transporters, also members of the ABCB family, have N-terminal extensions containing very long mitochondrial pre-sequences [30–32]. In contrast to non-mitochondrial ABC transporters, these N-terminal extensions are not membrane spanning per se. Nevertheless, they are essential for correct targeting, since deletion of the leader sequence in ABCB10 and its yeast homologue MDL1 resulted in mistargeting of the transporter to the ER membrane [31, 32]. Vice versa, fusion of the N-terminal extension of ABCB7 (residue 1–135) to the dihydrofolate reductase leads to mitochondrial targeting of the fusion protein [30].
Most full-length ABC transporters of the subfamily C also display an extra N-terminal domain (named MSD or TMD0), which typically comprises five transmembrane-spanning segments (reviewed in [33]). MRP1 and MRP2, for example, harbor TMD0s with strategic functions in targeting of the transporters to the plasma membrane [34, 35]. As for TAP, the TMD0 of MRP1 is dispensable for transport activity per se [36]. The sulfonylurea receptor SUR1 (ABCC8) is the regulatory subunit of the KATP channel KIR6.2. Mutations in KIR6.2 are associated with congenital hyperinsulinism [37, 38] and neonatal diabetes [39, 40]. TMD SUR10 is the interaction and regulation domain to KIR6.2 [41, 42]. The TMD SUR10 , expressed in the absence of the remaining core ABC transporter, retains its ability to associate with and gate KIR6.2 [43]. In conclusion, some ABC transporters present an interesting division of work using several independent membrane-spanning domains: one forming the gated translocation pathway and one with functions in regulation, trafficking, or binding of interaction partners. However, the TMD0s of TAP1 and TAP2 constitute prime examples of ER-resident ABC proteins.
A number of mechanisms are known that retain or retrieve proteins in or to the ER. Soluble proteins are retrieved from post-ER compartments by the KDEL motif that binds to the KDEL receptor. For type I membrane proteins, C-terminal di-lysine motifs (KKxx or KxKxx) interact with COPI vesicles for retrieval to the ER [44]. Type II membrane proteins are retained by a double-arginine motif at the N-terminus [45]. However, neither coreTAP nor any of the TMD0s contain these characteristic signals. Nevertheless, direct evidence for ER localization of both TMD0s is delivered in this work by immunofluorescence analysis. TMD TAP10 and TMD TAP20 were predominantly found in the ER, with a minor fraction entering the post-ER compartment, ERGIC. This distribution resembles that of wt TAP and is in agreement with a previous study that found a minor fraction of TAP active in the ERGIC [24]. Of note, the escape of TMD TAP20 into the ERGIC is enhanced compared to TMD TAP10 . These data imply that the TMD0s of both TAP subunits are retained in the ER, but the unidentified retention or retrieval factor acts more strongly in TMD TAP10 than in TMD TAP20 . This retention/retrieval signal must be directly located within each TMD0, because ER localization is not mediated by the interaction with the KKxx motif-containing partner tapasin. The localization of wt TAP was formerly suggested to be independent of tapasin [24]. Here, we demonstrate that this is also the case for the isolated membrane interaction hubs TMD0 of TAP1 and TAP2. In addition, we provide direct evidence that coreTAP1 and coreTAP2 localize to the ER membrane. Escape to post-ER compartments was weak and comparable to that of full-length TAP, demonstrating that the mechanism for TAP ER retention is preserved in coreTAP.
The observation that the independently expressed TMD0s of both TAP subunits are able to recruit tapasin gives direct evidence that the TMD0s are folded and functional independent of coreTAP. Furthermore, coimmunoprecipitation experiments with Tsn-TMD0 fusions demonstrate that each TMD0 only bears one single binding site for tapasin. This suggests a tapasin-to-TAP subunit ratio of 1:1, which is in perfect agreement with the latest study on this subject [28]. However, we cannot formally rule out the possibility that a second tapasin-binding site is formed by combination of the very C-terminal parts of TMD TAP10 and TMD TAP20 as well as the N-terminus of coreTAP1 and coreTAP2. Tsn-TMD TAP10 was reproducibly found to recruit less MHC I than Tsn-TMD TAP20 (Fig. 6). This might reflect a greater importance for TMD TAP20 in MHC I loading. This is a reasonable speculation, since for chicken and all other avian MHC I loci sequenced so far, only TAP2 harbors a TMD0, while TAP1 has no TMD0 and is therefore equivalent to coreTAP1 [46, 47]. The exact interaction sites of TMD TAP10 and TMD TAP20 for tapasin remain unknown. For tapasin, a number of candidate residues have been proposed, including residues F397, F401, G405, K408, and W412 of mouse tapasin [48], and K408 of human tapasin [49]. For TAP, it was previously shown that removal of the first transmembrane helix destroys the tapasin-TAP interaction [17]. Whether this is due to an altered overall folding of the truncated N-terminal domain or missing essential residues will require future approaches. The structural analysis of the TMD is one approach to resolve this important interaction hub.
In conclusion, we show for the first time that the TMD0s of both TAP subunits form autonomous interaction scaffolds for the assembly of the MHC I peptide-loading complex in the ER membrane. Each TMD0 connects ERp57, calreticulin, and peptide-receptive MHC I via a single tapasin molecule to the peptide supplier TAP. According to this, the PLC can be subdivided into three functional modules: (1) peptide binding and transport by the coreTAP complex, (2) peptide loading and editing by the Tsn-ERp57/MHC I subcomplex, and (3) the conjunction of these functions, accomplished by the interaction hubs TMD TAP10 and TMD TAP20 .
Abbreviations
- Crt:
-
Calreticulin
- ER:
-
Endoplasmic reticulum
- ERGIC:
-
ER-Golgi intermediate compartment
- MHC I:
-
Major histocompatibility complex class I
- PLC:
-
Peptide-loading complex
- TAP:
-
Transporter associated with antigen processing
- TMD:
-
Transmembrane domain
- Tsn:
-
Tapasin
- wt:
-
Wild-type
References
Neefjes J, Jongsma ML, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11:823–836
Cresswell P (2005) Antigen processing and presentation. Immunol Rev 207:5–7
Parcej D, Tampé R (2010) ABC proteins in antigen translocation and viral inhibition. Nat Chem Biol 6:572–580
Sadasivan B, Lehner PJ, Ortmann B, Spies T, Cresswell P (1996) Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity 5:103–114
Ortmann B, Copeman J, Lehner PJ, Sadasivan B, Herberg JA, Grandea AG, Riddell SR, Tampé R, Spies T, Trowsdale J, Cresswell P (1997) A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP complexes. Science 277:1306–1309
Williams AP, Peh CA, Purcell AW, McCluskey J, Elliott T (2002) Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity 16:509–520
Howarth M, Williams A, Tolstrup AB, Elliott T (2004) Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc Natl Acad Sci USA 101:11737–11742
Madden DR (1995) The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol 13:587–622
Kozlov G, Maattanen P, Schrag JD, Pollock S, Cygler M, Nagar B, Thomas DY, Gehring K (2006) Crystal structure of the bb’ domains of the protein disulfide isomerase ERp57. Structure 14:1331–1339
Nørgaard Toft K, Larsen N, Steen Jørgensen F, Højrup P, Houen G, Vestergaard B (2008) Small angle X-ray scattering study of calreticulin reveals conformational plasticity. Biochim Biophys Acta 1784:1265–1270
Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM (2009) Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity 30:21–32
Chouquet A, Païdassi H, Ling WL, Frachet P, Houen G, Arlaud GJ, Gaboriaud C (2011) X-ray structure of the human calreticulin globular domain reveals a peptide-binding area and suggests a multi-molecular mechanism. PLoS ONE 6:e17886
Dawson RJ, Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443:180–185
Gaudet R, Wiley DC (2001) Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. EMBO J 20:4964–4972
Oancea G, O’Mara ML, Bennett WF, Tieleman DP, Abele R, Tampé R (2009) Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc Natl Acad Sci USA 106:5551–5556
Koch J, Guntrum R, Heintke S, Kyritsis C, Tampé R (2004) Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP). J Biol Chem 279:10142–10147
Koch J, Guntrum R, Tampé R (2006) The first N-terminal transmembrane helix of each subunit of the antigenic peptide transporter TAP is essential for independent tapasin binding. FEBS Lett 580:4091–4096
Leonhardt RM, Keusekotten K, Bekpen C, Knittler MR (2005) Critical role for the tapasin-docking site of TAP2 in the functional integrity of the MHC class I-peptide-loading complex. J Immunol 175:5104–5114
Cormier JN, Panelli MC, Hackett JA, Bettinotti MP, Mixon A, Wunderlich J, Parker LL, Restifo NP, Ferrone S, Marincola FM (1999) Natural variation of the expression of HLA and endogenous antigen modulates CTL recognition in an in vitro melanoma model. Int J Cancer 80:781–790
Marincola FM, Shamamian P, Alexander RB, Gnarra JR, Turetskaya RL, Nedospasov SA, Simonis TB, Taubenberger JK, Yannelli J, Mixon A et al (1994) Loss of HLA haplotype and B locus down-regulation in melanoma cell lines. J Immunol 153:1225–1237
Meyer TH, van Endert PM, Uebel S, Ehring B, Tampé R (1994) Functional expression and purification of the ABC transporter complex associated with antigen processing (TAP) in insect cells. FEBS Lett 351:443–447
Stam NJ, Vroom TM, Peters PJ, Pastoors EB, Ploegh HL (1990) HLA-A- and HLA-B-specific monoclonal antibodies reactive with free heavy chains in western blots, in formalin-fixed, paraffin-embedded tissue sections and in cryo-immuno-electron microscopy. Int Immunol 2:113–125
Bolte S, Cordelières FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microscopy 224:213–232
Ghanem E, Fritzsche S, Al-Balushi M, Hashem J, Ghuneim L, Thomer L, Kalbacher H, van Endert P, Wiertz E, Tampé R, Springer S (2010) The transporter associated with antigen processing (TAP) is active in a post-ER compartment. J Cell Sci 123:4271–4279
Procko E, Raghuraman G, Wiley DC, Raghavan M, Gaudet R (2005) Identification of domain boundaries within the N-termini of TAP1 and TAP2 and their importance in tapasin binding and tapasin-mediated increase in peptide loading of MHC class I. Immunol Cell Biol 83:475–482
Paulsson KM, Jevon M, Wang JW, Li S, Wang P (2006) The double lysine motif of tapasin is a retrieval signal for retention of unstable MHC class I molecules in the endoplasmic reticulum. J Immunol 176:7482–7488
Li S, Sjögren HO, Hellman U, Pettersson RF, Wang P (1997) Cloning and functional characterization of a subunit of the transporter associated with antigen processing. Proc Natl Acad Sci USA 94:8708–8713
Rufer E, Leonhardt RM, Knittler MR (2007) Molecular architecture of the TAP-associated MHC class I peptide-loading complex. J Immunol 179:5717–5727
Demirel O, Bangert I, Tampé R, Abele R (2010) Tuning the cellular trafficking of the lysosomal peptide transporter TAPL by its N-terminal domain. Traffic 11:383–393
Csere P, Lill R, Kispal G (1998) Identification of a human mitochondrial ABC transporter, the functional orthologue of yeast Atm1p. FEBS Lett 441:266–270
Graf SA, Haigh SE, Corson ED, Shirihai OS (2004) Targeting, import, and dimerization of a mammalian mitochondrial ATP binding cassette (ABC) transporter, ABCB10 (ABC-me). J Biol Chem 279:42954–42963
Gompf S, Zutz A, Hofacker M, Haase W, van der Does C, Tampé R (2007) Switching of the homooligomeric ATP-binding cassette transport complex MDL1 from post-translational mitochondrial import to endoplasmic reticulum insertion. FEBS J 274:5298–5310
Tusnády GE, Sarkadi B, Simon I, Váradi A (2006) Membrane topology of human ABC proteins. FEBS Lett 580:1017–1022
Westlake CJ, Cole SPC, Deeley RG (2005) Role of the NH2-terminal membrane spanning domain of multidrug resistance protein 1/ABCC1 in protein processing and trafficking. Mol Biol Cell 16:2483–2492
Bandler PE, Westlake CJ, Grant CE, Cole SPC, Deeley RG (2008) Identification of regions required for apical membrane localization of human multidrug resistance protein 2. Mol Pharmacol 74:9–19
Bakos E, Evers R, Szakács G, Tusnády GE, Welker E, Szabó K, Haas M, van Deemter L, Borst P, Váradi A, Sarkadi B (1998) Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J Biol Chem 273:32167–32175
Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF, Bryan J (1995) Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268:426–429
James C, Kapoor RR, Ismail D, Hussain K (2009) The genetic basis of congenital hyperinsulinism. J Med Genet 46:289–299
Bryan J, Munoz A, Zhang X, Dufer M, Drews G, Krippeit-Drews P, Aguilar-Bryan L (2007) ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch 453:703–718
Aittoniemi J, Fotinou C, Craig TJ, de Wet H, Proks P, Ashcroft FM (2009) Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos Trans R Soc Lond B 364:257–267
Chan KW, Zhang H, Logothetis DE (2003) N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J 22:3833–3843
Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JMCL, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JPH, Sumnik Z, van Rhijn A, Wales JKH, Clark P, Gorman S, Aisenberg J, Ellard S, Njølstad PR, Ashcroft FM, Hattersley AT (2004) Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. New Engl J Med 350:1838–1849
Babenko AP, Aguilar-Bryan L, Bryan J (1998) A view of sur/KIR6.X KATP channels. Annu Rev Physiol 60:667–687
Cosson P, Letourneur F (1994) Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science 263:1629–1631
Schutze MP, Peterson PA, Jackson MR (1994) An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J 13:1696–1705
Moon DA, Veniamin SM, Parks-Dely JA, Magor KE (2005) The MHC of the duck (Anas platyrhynchos) contains five differentially expressed class I genes. J Immunol 175:6702–6712
Walker BA, Hunt LG, Sowa AK, Skjødt K, Göbel TW, Lehner PJ, Kaufman J (2011) The dominantly expressed class I molecule of the chicken MHC is explained by coevolution with the polymorphic peptide transporter (TAP) genes. Proc Natl Acad Sci USA 108:8396–8401
Papadopoulos M, Momburg F (2007) Multiple residues in the transmembrane helix and connecting peptide of mouse tapasin stabilize the transporter associated with the antigen-processing TAP2 subunit. J Biol Chem 282:9401–9410
Petersen JL, Hickman-Miller HD, McIlhaney MM, Vargas SE, Purcell AW, Hildebrand WH, Solheim JC (2005) A charged amino acid residue in the transmembrane/cytoplasmic region of tapasin influences MHC class I assembly and maturation. J Immunol 174:962–969
Abele R, Tampé R (2009) Peptide trafficking and translocation across membranes in cellular signaling and self-defense strategies. Curr Opin Cell Biol 21:508–515
Acknowledgments
We thank Drs. David Parcej and Andreas Hinz for providing the coreTAP2 constructs. The German Research Foundation (SFB 807 Transport and Communication across Biological Membranes and TA157/7 to R.T.), the European Drug Initiative on Channels and Transporters (EDICT to R.T.) funded by the European Commission Seventh Framework Program, and the Japan Society for the Promotion of Science (JSPS grant 20228001 to K.U.) supported this work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hulpke, S., Tomioka, M., Kremmer, E. et al. Direct evidence that the N-terminal extensions of the TAP complex act as autonomous interaction scaffolds for the assembly of the MHC I peptide-loading complex. Cell. Mol. Life Sci. 69, 3317–3327 (2012). https://doi.org/10.1007/s00018-012-1005-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1005-6