
Abstract
Cellular senescence involves a reduction in adult stem cell self-renewal, and epigenetic regulation of gene expression is one of the main underlying mechanisms. Here, we observed that the cellular senescence of human umbilical cord blood-derived multipotent stem cells (hUCB-MSCs) caused by inhibition of histone deacetylase (HDAC) activity leads to down-regulation of high mobility group A2 (HMGA2) and, on the contrary, to up-regulation of p16INK4A, p21CIP1/WAF1 and p27KIP1. We found that let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a were increased during replicative and HDAC inhibitor-mediated senescence of hUCB-MSCs by microRNA microarray and real-time quantitative PCR. Furthermore, the configurations of chromatins beading on these miRNAs were prone to transcriptional activation during HDAC inhibitor-mediated senescence. We confirmed that miR-23a, miR-26a and miR-30a inhibit HMGA2 to accelerate the progress of senescence. These findings suggest that HDACs may play important roles in cellular senescence by regulating the expression of miRNAs that target HMGA2 through histone modification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The epigenetic regulation of gene expression, including chromatin modifications and translational regulation by microRNAs (miRNAs), is an important mechanism controlling stem cell self-renewal and pluripotency [1]. Histone modification by acetylation, deacetylation and methylation controls the chromatin structure by balancing euchromatin and heterochromatin, and regulating the expression of specific genes [2]. In the embryonic stem (ES) cell genome, bivalent modifications marking large regions of trimethylated lysine 27 residues in histone H3 (H3K27me3) that harbor smaller regions of H3K4me3 may maintain the balance of key developmental genes for later activation during ES cell differentiation [3]. During ES cell differentiation and mammalian development, the acetylation of histones H3 and H4 decreases, whereas histone H3K9 methylation increases [4]. In the case of multipotent stem cells (MSCs), treatment with histone deacetylase (HDAC) inhibitors decreases the stemness of the cells, including their multipotency and self-renewal ability [5]. In a previous report, we showed that HDAC activity is important for MSC self-renewal via balancing of polycomb genes (PcGs) and jumonji domain containing 3 (JMJD3) expression, which controls cellular senescence via the regulation of histone deacetylase by p16INK4A [6].
As a group, miRNAs are ~22-nucleotide, non-coding RNAs that regulate gene expression through the degradation or inhibition of translation of target mRNAs. This regulation of gene expression is related to cellular differentiation and proliferation. In particular, the timing of miRNA expression during embryogenesis is critical, and miRNAs have important roles during each developmental stage [7]. Although key roles for miRNAs in controlling stemness have been discovered, the mechanisms regulating miRNA expression are not yet well known. The possibility of epigenetic activation of tumor suppressor miRNAs in human cancer cells has been suggested [8]. More recently, several lines of evidence regarding the epigenetic control of miRNA clusters have been reported. Several DNA regions encoding tumor suppressor miRNAs are inactivated by aberrant hypermethylation in human breast cancer cell lines [9]. Furthermore, after treatment of AGS gastric cancer cells with 5-aza-2′-deoxycytidine (5-Aza-dC), a DNA methyltransferase (DNMT) inhibitor, the expression of a specific miRNA cluster can be restored through DNA demethylation [10].
Epigenetic modifying drugs that inhibit HDAC and DNMT have been intensely studied with regard to their anticancer activity. The biology of miRNAs, which epigenetically regulate cellular homeostasis, has also been well-studied in the field of cancer research. Gene expression studies have shown that hundreds of miRNAs are deregulated in cancer cells, and functional studies have revealed that miRNAs are involved in the processes that drive tumorigenesis [11]. Among all miRNAs, the let-7 family was one of the first groups known to be required for the maintenance of stem cell self-renewal [12]. Progenitor cells show reduced expression of let-7 compared to more differentiated mammary epithelial cells. Moreover, enforced let-7 expression induces a loss of self-renewing stem cells from mixed cultures [13]. In neural stem cells (NSCs) from young mice, the overexpression of let-7b increases Ink4a/Arf expression levels and decreases self-renewal through the inhibition of high mobility group a2 (Hmga2) expression [14]. Although the relationship between let-7 and Hmga2 has previously been reported in mouse NSCs, the mechanism by which let-7 expression is regulated has not yet been established in non-embryonic tissues, leaving room for the possibility of let-7 regulation at the transcriptional level [15]. One report showed that 5% of human miRNAs in T24 bladder cancer cells are up-regulated more than three-fold by treatment with a DNA demethylating agent and an HDAC inhibitor. In particular, miR-127 is significantly induced by decreased DNA methylation levels and increased active histone marks around the promoter region of the miR-127 gene. These results show that some miRNAs are regulated by epigenetic modulation of their promoter regions and can be induced by inhibitors of DNA methylation and HDACs [8]. The reciprocal regulatory mechanisms between these epigenetic regulatory factors, however, have not been well-studied in cancer or stem cell biology.
To elucidate the reciprocal regulatory mechanisms between two key epigenetic factors, HDACs and miRNAs, we investigated how miRNAs that affect cellular senescence by targeting HMGA2 are regulated by HDAC activity.
Materials and methods
Isolation and culture of hMSCs
Human adipose tissue-derived MSCs (hAD-MSCs) [16] and human umbilical cord blood-derived MSCs (hUCB-MSCs) [6, 17] were isolated and cultured as previously described. In brief, hAD-MSCs were established from freshly excised mammary fat tissues obtained from 20- to 30-year-old women during reduction mammoplasty procedures. The hAD-MSCs were maintained in K-NAC medium supplemented with 2 mM N-acetyl-l-cysteine (Sigma-Aldrich, St. Louis, MO) and l-ascorbic acid (0.2 mM, Sigma-Aldrich).
The hUCB-MSCs were obtained from umbilical cord blood immediately after full-term deliveries with the informed consent of the mothers. hUCB-MSCs were used in this experiment, and the hUCB-MSCs were maintained in DMEM (Invitrogen, Carlsbad, CA) containing 10% FBS. All procedures for the preparation and utilization of hAD-MSCs (IRB no. 0611/001-001) and hUCB-MSCs (IRB no. 0603/001-002) for research purposes were approved by the institutional review board of Seoul National University.
Senescence-associated beta-galactosidase staining
Senescence-associated beta-galactosidase (SA β-gal) staining was carried out as described by Narita et al. [18] with some modifications. The MSCs were seeded on six-well plates at a density of 1 × 105/well for late-passage cells and 5 × 104/well for early passage cells. Cells were incubated for 3 days until reaching the appropriate confluence. For siRNA or anti-miRNA treatment, cells were seeded at a density of 2 × 104/ml, and siRNA or anti-miRNA was used to treat the cells at 50–60% confluence. The cells were washed twice with PBS and fixed with 0.5% glutaraldehyde in PBS (pH 7.2) for 5 min at room temperature. Cells were then washed with PBS containing MgCl2 (pH 7.2, 1 mM MgCl2) and stained in X-gal solution overnight at 37°C. The cells were washed twice with PBS, and images were captured with a microscope (IX70, Olympus, Japan).
Immunocytochemistry
Immunocytochemical analyses of HMGA2 were performed. Cells cultured with or without 4 mM VPA (valproic acid, Sigma-Aldrich, St. Louis, MO) or 2 mM NB (sodium butyrate, Sigma-Aldrich) for 3, 5 or 7 days were fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 (Sigma-Aldrich). The cells were then incubated with 10% normal goat serum (Zymed Laboratories Inc., San Francisco, CA) and stained with antibodies against HMGA2 (1:200, Abcam, UK), followed by incubation for 1 h with an Alexa 488-labeled secondary antibody (1:1,000; Molecular Probes, Eugene, OR). The nuclei were stained with Hoechst 33258 (1 μg/ml; 10 min), and images were captured with a confocal microscope (Eclipse TE200, Nikon, Japan).
Western blot analysis
Western blot analyses of HDAC1, HDAC2, HMGA2 and p16INK4A were performed as described previously [19]. MSCs cultured with or without HDAC inhibitors for 1, 3, 5 or 7 days were lysed with 50 mM Tris-HCl buffer containing 0.1% Triton X-100 freshly supplemented with a protease/phosphatase inhibitor cocktail. Proteins were then separated using 7.5–15% SDS-PAGE and transferred to nitrocellulose membranes at 350 mA for 5 h. All antibodies were used according to the manufacturer’s instructions, and protein bands were detected using an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, UK).
RT-PCR
Total cellular RNA was extracted from cells with TRIzol reagent™ (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. cDNA was synthesized by adding the purified RNA and oligo-dT primers to Accupower RT premix (Bioneer, Korea), according to the manufacturer’s instructions. PCR was conducted using Accupower PCR premix (Bioneer, Korea). The primer sets sequences used for this study are supplied in Table S2. All PCR products were analyzed by gel electrophoresis on 1.5% agarose gels with ethidium bromide staining, followed by fluorescence digitization using a Bio-Rad GelDoc XR system (Bio-Rad, USA). Semi-quantitative RT-PCR was conducted by quantifying the RT-PCR bands using ImageJ image analysis software (National Institutes of Health, USA). Each gene was normalized against β-actin or RPL13A as a housekeeping gene control. At least three independent analyses were carried out for each gene.
Real-time quantitative PCR
Real-time qPCR was performed using SYBR® Green (Applied Biosystems, Foster City, CA), according to the manufacturer’s protocol. β-actin was used as an internal control. All amplicons were analyzed using Prism 7000 sequence detection system 2.1 software (Applied Biosystems).
siRNA, anti-miRNA and mature miRNA transfection study
Transient transfection assays were performed using commercially available specific siRNAs for inhibition of HDAC1, HDAC2 and HMGA2 along with a non-targeting siRNA (ON Target plus SMARTpool, Dharmacon, USA). Inhibition or overexpression of miRNAs was achieved by commercial antisense miRNAs or mature miRNAs of hsa-miR-23a, hsa-miR-26a and hsa-miR-30a with an appropriate miRNA precursor-negative control (anti-miRNA inhibitor and miRNA precursor-negative control no. 1, Ambion, USA). The siRNA, anti-miRNA and mature miRNA transfections were done according to the manufacturer’s instructions. In brief, cells were seeded at a concentration of 2 × 104/well, and siRNA-containing medium (without the addition of antibiotics) was added when the cells reached 50–60% confluence. Cells were incubated with 50 nM or 100 nM siRNA or 50 nM anti-miRNAs or 50 nM mature miRNAs for 48 or 96 h. To investigate the long-term effects of inhibition, the cells were subcultured for 48–72 h after siRNA, anti-miRNA or mature miRNA transfection. Subcultured cells were stabilized for 24 h and incubated with siRNA, anti-miRNA or mature miRNA for 48–72 h at the same concentration. After inhibition, RNA extraction and subsequent RT-qPCR or SA β-gal staining was performed for genetic or characteristic analyses, respectively.
Chromatin immunoprecipitation (ChIP) assays
The hUCB-MSCs were seeded in 10-cm plates at a density of 0.8–1 × 105 per plate and cultured with or without 4 mM VPA or 2 mM NB for 1 or 3 days. ChIP assays were performed according to the manufacturer’s protocol (ChIP assay kit, Upstate Biotechnology, USA). Chromatin was immunoprecipitated using antibodies, according to the manufacturer’s instructions. Real-time qPCR was performed at a final template dilution of 1:50. The primer sequences used in the ChIP assays in this study are supplied in Table S3.
MTT assay
The effects of hsa-miR-23a inhibition on MSC proliferation were measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, USA) assay as described previously [19]. In brief, cells were plated on 24-well plates at a density of 2 × 104/ml and cultured for 24 h until reaching 50–60% confluence. The cells were then treated with 50 nM miR-23a inhibitor (anti-miR™ miRNA inhibitor, Ambion, USA) and a microRNA inhibitor control for 2 days. At the end of the incubation, 50 μl of MTT stock solution (5 mg/ml) was added, and the plates were incubated for another 4 h at 37°C. Formazan crystals were solubilized with DMSO, and the absorbance was measured with an EL800 microplate reader (BIO-TEK Instruments, USA).
MicroRNA microarray analysis
Young group (passage 5 for hUCB- and hAD-MSCs) and old group (passage 19 for hUCB- and hAD-MSCs; hUCB- and hAD-MSCs treated with 2 mM VPA for 7 days) cells were prepared for miRNA microarray analysis. Total RNA was extracted from the human stem cells using TRI REAGENT (Molecular Research Center, USA), according to the manufacturer’s instructions. Each total RNA sample (2 μg) was labeled and amplified using a ULS aRNA labeling kit (Kreatech diagnostics, The Netherlands). The Cy5-labeled aRNAs were resuspended in 10 μl of hybridization solution (GenoCheck, Korea). We used Agilent human miRNA arrays containing probes and annotation information for all human miRNAs from the Sanger miRBASE public database (Release 12.0, September 2008), with 20 probe replicates per array. We also used the Agilent human whole genome array, which contains probes and annotation information for all human genes sourced from the Genome Reference Consortium Human Build 34 (GRCh34). GeneSpring GX software was used for data normalization, clustering, selection of changed miRNAs and statistical analysis. Gene expression levels were calculated, and relative signal intensities for each gene were generated.
Statistical analysis
All experiments were conducted at least in triplicate (n = 3), and results are expressed as the mean ± SD. Statistical analysis was conducted via analysis of variance (ANOVA), followed by Duncan’s multiple range tests or Student’s t-test. p < 0.05 was considered to be significant.
Results
Replicative senescence in hUCB-MSCs down-regulates HDAC1, HDAC2 and HMGA2, and up-regulates CDKN inhibitors
To induce replicative senescence, we repeatedly subcultured hUCB-MSCs in vitro. According to the cumulative population doubling level, human MSCs began to lose their proliferation potential at around the tenth passage (Figure S1). Based on the CPDL data and the morphological characteristics of MSCs, the senescence status of the MSCs was divided into three grades: early (less than 10 passages), intermediate (10–15 passages) and late stage (more than 16 passages). To confirm the senescence of late-stage MSCs, SA β-gal staining was performed. As shown in Fig. 1a, increased activity of β-gal was observed at late stages compared to early stages. In our previous report, HDAC1 and HDAC2, which are representative HDACs in eukaryotes, were decreased in late-stage hUCB-MSCs. It has been reported that trichostatin A, an HDAC inhibitor, represses HMGA2 gene transcription [20] and decreases HMGA2-induced p16INK4A expression during the aging process [14]. Based on these previous reports, we investigated the expression levels of HDAC1, HDAC2, HMGA2 and p16INK4A in hUCB-MSCs at both early and late stages. Both protein (Fig. 1b) and mRNA (Fig. 1c) levels of HDAC1, HDAC2 and HMGA2 were decreased, whereas p16INK4A levels were increased in late-stage senescence of MSCs. The mRNA levels of p21CIP1/WAF1 and p27KIP1 were also increased at late stages (Fig. 1c). Oxidative stress was induced to hUCB-MSC using 300 μM hydrogen peroxide to compare the effects of stress-induced premature senescence on HMGA2 and cyclin-dependent kinase inhibitors such as p16INK4A and p21CIP1/WAF1. Treatment of 300 μM H2O2-mediated oxidative stress increased SA β-gal activity (Figure S3A) and elevation of p21CIP1/WAF1 and down-regulation of HMGA2 mRNA levels were observed (Figure S3B, C and D). However, p16INK4A levels were decreased during oxidative stress-mediated premature senescence, suggesting that not p16INK4A but p21CIP1/WAF1 might involve in this process differently from replicative senescence (Figure S3B and C).

Replicative senescence of hUCB-MSCs. a MSCs undergo replicative senescence upon repeated (more than 15 passages) subculturing in vitro as shown by SA β-gal staining. b–c The expression of HDAC1, HDAC2 and HMGA2 was down-regulated, whereas p16INK4A was up-regulated during repeated subculture-induced senescence of MSCs as shown by immunoblot analysis (b) and real-time qPCR (c). The expression levels of p21CIP1/WAF1 and p27KIP1 were also up-regulated as shown by real-time qPCR (c). * and ** represent statistical significance at the levels of p < 0.05 and p < 0.005, respectively
HDAC inhibitors down-regulate HMGA2 and induce senescence
To determine whether HDAC activity was necessary for the regulation of HMGA2 during cellular senescence in hUCB-MSCs, we used the HDAC inhibitors valproic acid (VPA) and sodium butyrate (NB). Based on our previous report showing that 4 mM VPA or 2 mM NB was sufficient to induce senescence, we treated hUCB-MSCs for 7 days with 4 mM or 8 mM VPA and 2 mM or 4 mM NB, and investigated the expression levels of HMGA2 and other related genes. After treatment with the HDAC inhibitors, real-time qPCR revealed that the expression level of HMGA2 was decreased, whereas p16INK4A and p21CIP1/WAF1 levels were increased in a concentration-dependent manner in both VPA- and NB-treated hUCB-MSCs. The expression of p27KIP1, however, was only increased in cells treated with 8 mM VPA; the other treatment groups did not show elevated expression levels of p27KIP1 (Fig. 2a). We also evaluated the time course of the changes in gene expression following treatment with HDAC inhibitors using real-time qPCR and immunoblot analyses. Both mRNA and protein levels of HMGA2 were significantly increased after 3 days of treatment (Fig. 2b, c). The levels of p16INK4A and p21CIP1/WAF1 were up-regulated after 1 and 3 days, respectively, of treatment with the HDAC inhibitors. In contrast, the expression of p27KIP1 was not increased significantly over the entire period of treatment (Fig. 2b). Given that the expression of HMGA2 was decreased after 3 days of treatment with the HDAC inhibitors, we treated hUCB-MSCs with the HDAC inhibitors for 3, 5 and 7 days and investigated the expression pattern of HMGA2 using immunocytochemistry. The nuclear localization of HMGA2 was confirmed, and the expression of HMGA2 was decreased in a time-dependent manner (Fig. 2d).

HDAC inhibitors down-regulate HMGA2 and induce senescence. a The HDAC inhibitors VPA and NB were used to treat hUCB-MSCs at two concentrations for 7 days. Real-time qPCR analysis was performed to investigate the expression levels of HMGA2, p16INK4A, p21CIP1/WAF1 and p27KIP1 based on the concentration of the HDAC inhibitors. b hUCB-MSCs were treated with 4 mM VPA or 2 mM NB for 1, 3, 5 and 7 days. Real-time qPCR analysis was performed to investigate the expression levels of HMGA2, p16INK4A, p21CIP1/WAF1 and p27KIP1 in a time-dependent manner. c–d After treatment with 4 mM VPA or 2 mM NB for the indicated time periods, immunoblot assay for HMGA2 and p16INK4A (c) and immunocytochemistry for HMGA2 (d) were performed to investigate the protein expression levels
HDAC1 and HDAC2 regulate HMGA2 expression, and HMGA2 inhibition induces cellular senescence
Given that HDAC inhibitors have broad-spectrum biological activity that is not restricted to HDAC inhibition alone, specific inhibition of HDAC1 and HDAC2 was performed using small interfering RNA (siRNA) to complement the results seen with the HDAC inhibitors. First, decreased expression levels of HDAC1 and HDAC2 were confirmed following treatment with siHDAC1 and siHDAC2, respectively, using real-time qPCR, suggesting proper targeting of the siRNAs (Fig. 3a). In addition, both siHDAC1 and siHDAC2 decreased the expression level of HMGA2 (Fig. 3a). To confirm whether the inhibition of HMGA2 indeed led to the senescence of hUCB-MSCs, specific targeting of HMGA2 was carried out using siRNA. After inhibition of HMGA2, a decrease in HMGA2 expression levels was confirmed using real-time qPCR (Fig. 3b). Consistently with our previous report [21], the inhibition of HMGA2 as well as HDAC1 and HDAC2 led to the senescence of hUCB-MSCs as shown by SA β-gal staining, in addition to significantly increased expression of p21CIP1/WAF1 and p16INK4A (Fig. 3b, c).

HDAC1 and HDAC2 regulate HMGA2 expression, and HMGA2 inhibition induces cellular senescence. a Specific inhibition of HDAC1 and HDAC2 using siRNA was performed as described in “Materials and methods”. The expression levels of HDAC1 and HDAC2 were decreased as shown by RT-PCR analysis. Graphs represent the semi-quantification of RT-PCR results from at least three independent experiments. b Specific inhibition of HDAC1 and HDAC2 decreased the expression of HMGA2. c Specific down-regulation of HDAC1, HDAC2 and HMGA2 was sufficient to cause cellular senescence in MSCs as shown by SA β-gal staining. The expression levels of HMGA2 and p16INK4A, p21CIP1/WAF1 and p27KIP1 were determined by real-time qPCR analysis
Expression of miRNAs targeting HMGA2 is up-regulated in senescent hUCB-MSCs
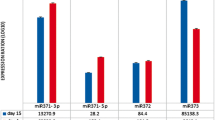
Based on the aforementioned results, the inhibition of HDACs, specifically HDAC1 and HDAC2, appears to decrease HMGA2 expression. Among several reported mechanisms, the let-7 miRNA family includes factors known to regulate HMGA2 expression by targeting the 3′ UTR of HMGA2 mRNA [14, 22–24]. Keeping this in mind, we hypothesized that miRNAs up-regulated during the senescence process, including the let-7 family, might participate in decreasing the expression of HMGA2. We found that all isotypes of the let7 family were up-regulated following treatment with the HDAC inhibitors (Figure S4). To investigate miRNAs other than the let-7 family that are regulated during senescence following treatment with HDAC inhibitors, miRNA microarray analysis comparing early stage cells with senescent cells was performed. We found 37 miRNAs that were significantly up-regulated (p < 0.05) in the senescent hUCB-MSCs compared to the early stage group. Using web-based miRNA database programs such as Miranda, Targetscan and Pictar, which use databases of miRNAs to mine target mRNAs, miRNAs targeting the HMGA2 mRNA were investigated (Table S1 and Fig. 4b). Among 381 miRNAs indicated to target the HMGA2 mRNA, 25 were up-regulated in senescent hUCB-MSCs compared to the early stage group (Fig. 4a, b). Among the miRNAs indicated by at least two database programs to target HMGA2 mRNA, let-7a1, let-7d and let-7f1, which constitute a let-7 cluster, and miR-23a, miR-26a and miR-30a were selected for further experiments, and their expression levels were analyzed by real-time qPCR. As shown in Fig. 4c, the miRNAs selected by these criteria were up-regulated by HDAC inhibitor-mediated induction of senescence. Nevertheless, there were differences in the efficiency and specific inhibition of both HDAC1 and HDAC2 by their respective siRNAs, leading to the up-regulation of these miRNAs and suggesting that HDAC activity may act as a specific regulatory signal for certain miRNAs such as let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a (Fig. 4d). To compare the effects of HDAC inhibitors with oxidative stress on HMGA2-targeing miRNAs expression, we assessed the expression level of let 7a and miR-23a. Unlike HDAC inhibitor, oxidative stress down-regulated the expression of let-7a and miR-23a, suggesting that the mechanisms by which HMGA2 decrease might be different in between these two causes of cellular senescence in hUCB-MSCs (Figure S3E).

Precursor and mature miRNAs that target HMGA2 are up-regulated in HDAC inhibitor-mediated senescence of hUCB-MSCs. a The miRNAs targeting HMGA2 were searched using a screening program for the miRNA database. b HMGA2-targeting miRNAs among those that were up-regulated in the senescence group in the microarray analysis are represented by a Venn diagram. c After treatment of hUCB-MSCs with 4 mM VPA or 2 mM NB for 3 days, the expression of individual microRNAs was analyzed using real-time qPCR. d Following specific inhibition of HDAC1 and HDAC2 using siRNAs, the expression of individual microRNAs was analyzed using real-time qPCR. e After treatment of hUCB-MSCs with HDAC inhibitors at the indicated concentrations and durations, the expression levels of precursor forms of miRNAs were analyzed by RT-PCR. f Semi-quantification of the PCR products was performed by ImageJ analysis. The graph shows the relative gene expression of cells treated with the HDAC inhibitors compared with control cells. * and ** represent statistical significance at the levels of p < 0.05 and p < 0.01, respectively
Precursor forms of miRNAs increase after inhibition of HDACs
Regulation of miRNA expression can be achieved at the transcriptional or post-transcriptional level. Although miRNA regulation at the post-transcriptional level is becoming increasingly understood [7, 25], the factors that may affect the transcriptional regulation of miRNAs are not well characterized. Based on the observation that HMGA2-targeting miRNAs increase following HDAC inhibitor treatment as shown in Fig. 4c, d, we investigated the expression levels of the precursor forms of let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a using RT-PCR analysis to determine whether the up-regulation of miRNA expression occurred at the transcriptional level. The precursor forms of these miRNAs were significantly up-regulated following treatment with the inhibitors for HDAC 1 and/or 3 days Based on these results, we hypothesized that miRNA expression might be regulated at the transcriptional level following treatment with HDAC inhibitors (Fig. 4e, f). Given that miRNA biogenesis begins with transcription by RNA polymerase II, followed by processing by RNase III, Drosha and Dicer, we investigated the expression levels of these factors during HDAC inhibitor-mediated cellular senescence in hUCB-MSCs. The expression levels of Drosha and Dicer were unchanged or slightly decreased during hUCB-MSC senescence induced by treatment with HDAC inhibitors, suggesting that the expression of let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a, which target HMGA2, might rather be transcriptionally up-regulated than be enhanced by post-transcriptional miRNA processing. In contrast, the regulation of Lin28, which is reported to down-regulate let-7 at the post-transcriptional level, inconsistently fluctuated following treatment with HDAC inhibitors, suggesting that Lin28 may not be involved in the up-regulation of let-7 during HDAC inhibitor-induced senescence in hUCB-MSCs (Figure S5).
HDAC inhibitors regulate epigenetic histone marks in the miRNA genome
Based on the aforementioned results, factors that are well-established as regulators of miRNA biogenesis at the post-transcriptional level appear unlikely to be related to the up-regulation of let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a during HDAC inhibitor-mediated senescence. In contrast, considering the up-regulation of individual miRNA precursor forms, miRNA biogenesis might be modulated at the transcriptional level during the progress of cellular senescence following the inhibition of HDAC activity. To determine whether the transcription of miRNAs was affected by HDAC activity, we investigated the histone modifications in the chromatin at the site of each miRNA coding region using chromatin immunoprecipitation (ChIP) analysis. We found that acetylation of histone H3 and H4 and trimethylation of histone H3 at the lysine 4 residue (H3K4), all of which activate transcription, were increased in the proximity of the coding regions of let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a (Fig. 5b, c). Furthermore, histone H3K9Me3 and H3K27Me3, which are known to repress transcription, were decreased in the vicinity of these miRNAs. The histone modification patterns in miRNA coding regions in the VPA-treated group were similar to those in the NB-treated group (Fig. 5b, c). Given that the majority of miRNAs are transcribed from genomic DNA by RNA polymerase II, we investigated the level of enrichment of RNA polymerase II at each miRNA coding region with ChIP to support the hypothesis of miRNA transcriptional activation via histone modifications. ChIP analysis and subsequent real-time qPCR analysis showed an increase in RNA polymerase II bound to DNA sequences in the vicinity of the miRNA coding regions (Fig. 5d). To confirm whether the transcriptional regulation of these miRNAs by the inhibition of HDACs is specific to unprocessed primary forms of these miRNAs which target HMGA2, we looked into other mRNA (OCT4) and miRNA (miR-329) than HMGA2-targeting miRNAs. First, we confirmed that the expression levels of OCT4, a well-known marker for undifferentiated embryonic stem cells and pluripotency and miR-329, one of the highly expressed miRNAs in mouse pluripotent stem cells, which is conserved in human [26]. We found that OCT4 and miR-329 expression levels were decreased after treatment of 4 mM VPA or 2 mM NB for more than 1 day (Figure S6A). ChIP analysis showed that binding of histone H3K27me3 increased at the OCT4 promoter region after treatment of HDAC inhibitors (Figure S6B-C). Interestingly, the genomic region encoding miR-329 showed prominently increased enrichment of H3K9me3 and H3K27me3 but decreased binding of H3K4me3 after treatment of HDAC inhibitors in hUCB-MSCs (Figure S6B-C). Both the OCT4 promoter and miR-329 coding region showed decreased binding of RNA polymerase II; on the contrary, the enrichment of EZH2, a methyltransferase of histone H3K27, was increased after treatment of HDAC inhibitors. In addition, increased enrichment of MeCP2 at the promoter region of OCT4 suggested that DNA methylation may contribute to repression of OCT4 transcription by treatment of HDAC inhibitor (Figure S6D). Taken together, these data suggest that HDAC activity may regulate transcription of HMGA2-targeting miRNAs by modifying histone patterns bound to their genomic contexts.

miRNA expression is regulated by histone modification. a–d After treatment with HDAC inhibitors at the indicated concentrations for 1 day, a ChIP assay and real-time qPCR analysis were performed. The binding of five types of acetylated or methylated histone forms (b–c) and RNA polymerase II (d) was analyzed at the DNA sites encoding the indicated miRNAs (a). The graph shows the relative enrichment of target protein at the miRNA coding sites of cells treated with the HDAC inhibitors compared with control cells. * and ** represent statistical significance at the levels of p < 0.05 and p < 0.01, respectively
miR-23a, miR-26a and miR-30a regulate HMGA2 expression and senescence of hUCB-MSCs
It is well-known that the let-7 miRNA targets the HMGA2 mRNA. There have been no reports, however, about whether miR-23a, miR-26a and miR-30a virtually target the HMGA2 mRNA in the context of cellular senescence or otherwise. Therefore, we investigated whether miR-23a, miR-26a and miR-30a target HMGA2 mRNA by using a specific miRNA inhibitors and mature miRNAs. Following miRNA inhibitor-mediated inhibition of miR-23a, miR-26a and miR-30a expression, we observed down-regulation of the miRNAs and subsequent up-regulation of HMGA2 mRNA expression by real-time qPCR. On the contrary, overexpression of the miRNAs induced down-regulation of HMGA2 mRNA expression, suggesting that the miRNAs might target HMGA2 (Fig. 6a, b). To investigate the effects of the miRNAs on the expression levels of further downstream factors of senescence such as p16INK4A and p21CIP1/WAF1, real-time qPCR was performed after transient inhibition or over-expression of miRNAs using anti- or mature miRNA oligonucleotides. Expression levels of miR-23a and miR-30a were interrelated with p16INK4A and p21CIP1/WAF1 expression levels (Fig. 6c, d). Anti-miRNA-mediated inhibition of miR-23a and miR-30a decreased expression of p16INK4A and p21CIP1/WAF1. On the other hand, mature-miR-23a and miR-30a increased the expression level of the p16INK4A and p21CIP1/WAF1 (Fig. 6c, d). Over-expression of miR-26a also increased p16INK4A; however, inhibition of miR-26a did not decrease the expression of p16INK4A. Regulation of p21CIP1/WAF1 expression by miR-26a was not consistent with that regulated by miR-23a and miR-30a (Fig. 6c, d). Inhibition or over-expression of miR-23a and 30a affected cellular senescence as evidenced by SA β-gal staining (Fig. 6e). Anti-miR-23a and anti-miR-30a prevented cells from an increase of β-galactosidase activity. Mature-miRNAs of them enhanced staining status of SA β-gal. After regulation of miR-26a, however, SA β-gal staining status did not show prominent changes suggesting that the role miR-26a may not be specific to HMGA2 (Fig. 6e). Taken together, these results confirm that miR-23a, miR-26a and miR-30a regulate HMGA2 expression in cellular senescence.

miR-23a, miR-26a and miR-30a miRNA inhibition or overexpression regulate HMGA2 expression and senescence. Anti-miRNAs or mature miRNAs were transfected as described in Materials and Methods. a–b Following inhibition or overexpression of miR-23a, miR-26a and miR-30a using anti- or mature-miRNA, real-time qPCR was performed to evaluate the expression levels of each miRNA (a) and HMGA2 (b). c–d After modulation of miR-23a, miR-26a and miR-30a expression, the expression levels of p16INK4A (c) and p21CIP1/WAF1(d) were investigated by real-time PCR. e After inhibition or overexpression of miR-23a, miR-26a and miR-30a, cellular senescence was investigated by SA β-gal staining
Discussion
In this study, we identified an HDAC-HMGA2-p16INK4A regulatory pathway during the senescence process of hUCB-MSCs. We used this pathway as a model of regulation by miRNA expression. Furthermore, we confirmed that HDAC regulates miRNAs targeting the HMGA2 mRNA via histone modifications during the senescence process induced by HDAC inhibitors.
HMGA proteins are abundant in pluripotent ES cells and most malignant human tumors, but are not detectable in normal somatic cells. They act as activators and repressors of gene expression and most likely facilitate DNA structural changes during the formation of specialized nucleoprotein structures at selected promoter regions [27, 28]. HMGA2 is closely related to the maintenance of self-renewal and pluripotency in stem cells, and the role of HMGA2 in the self-renewal of mouse NSCs has been reported [14]. In mouse NSCs, HMGA2 contributes to the maintenance of self-renewal by reducing the expression of p16INK4A and p19ARF; furthermore, the regulation of HMGA2 by let-7b has also been suggested [14]. In the present study, we used hUCB-MSCs to confirm the down-regulation of HMGA2 and the up-regulation of p16INK4A in both replicative and HDAC inhibitor-mediated senescence (Fig. 1, 2 and Figure S2). We were able to reproduce the results of Nishino et al. by showing that specific inhibition of HMGA2 induced the cellular senescence of hUCB-MSCs, followed by increased p16INK4A and p21CIP1/WAF1 expression (Fig. 3). In a previous report, Nishino [14] showed that Hmga2 is related to the control of stem cell aging, and the regulation of p16INK4A and p19ARF expression was revealed as a potential mechanisms for induction of cellular scenescence. For both our data and previously published results, however, the way in which Hmga2 represses the Ink4a/Arf locus has not been established. It should be determined whether HMGA2 cooperates with established repressors of the locus such as polycomb group proteins, and the HMGA2-driven epigenetic events that silence the locus should be identified [24]. Thus far, two key regulatory mechanisms of HMGA2 expression have been reported. One involves the let-7 family of miRNAs, which target the HMGA2 mRNA. The 3′-UTR of human HMGA2 contains seven putative let-7 complementary sites [29]. The inhibitory effect of the let-7 family on HMGA2 has been confirmed in various cancer cell lines and mouse NSCs [14, 22, 23, 29]. The other mechanism involves the transcriptional regulation of HMGA2 via Sp1 and Sp3 during HDAC inhibitor treatment [20]. Considering that some miRNAs play important roles in maintaining the self-renewal and pluripotency of stem cells, we focused on the relationship between miRNAs and HMGA2 during the senescence process. Using miRNA microarray and database analyses, we identified a number of hypothetical HMGA2-targeting miRNAs that were up-regulated during cellular senescence in hUCB-MSCs (Table S1, Fig. 4, 5, 6). Among these miRNAs, we confirmed that miR-23a, miR-26a and miR-30a virtually regulate HMGA2 expression (Fig. 6). In a previous report, Gao et al. [30] suggested that mitochondrial glutaminase is one of the targets of miR-23a; however, other functions of miR-23a have not yet been unveiled. Therefore, our novel finding that miR-23a targets HMGA2 in the context of the cellular senescence of hUCB-MSCs is important for uncovering the biological functions of miR-23a.
To be functional, miRNAs must mature via a unique biogenesis machinery. Drosha, DGCR8 and Dicer are the primary factors that execute the biogenesis of miRNAs, and their expression levels and functional changes can affect the maturation process of miRNAs. Chiosea et al. [31, 32] reported that the expression of Dicer, one of the key miRNA processing enzymes, is up-regulated in prostate cancer cells, suggesting a mechanism by which global miRNA over-expression may occur in various cancer cell lines. In that context, we found that the expression of Drosha and Dicer was decreased slightly, unlike the specific augmentation observed for the group of HMGA2-targeting miRNAs, including let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a. These results suggest that there are mechanisms of miRNA expression regulation different from those involving the maturation enzymes. In contrast, the finding of reduced levels of miRNA processing enzymes during hUCB-MSC senescence may explain how other groups of miRNAs are down-regulated.
Recent evidence suggests that the processing of several primary miRNA transcripts (pri-miRNAs) is blocked post-transcriptionally in embryonic stem cells, embryonal carcinoma cells and primary tumors. Viswanathan et al. showed that Lin28, a developmentally regulated RNA binding protein, selectively blocks the processing of pri-let-7 miRNAs in embryonic cells. They identified Lin28 as a negative regulator of miRNA biogenesis and suggested that Lin28 may play a central role in blocking miRNA-mediated differentiation in stem cells and in certain cancers [33]. In addition, Heo et al. [7] observed that Lin28 proteins act mainly in the cytoplasm by inducing the uridylation of precursor let-7 (pre-let-7) at its 3′ end. We also evaluated the expression of LIN28 during cellular senescence to confirm whether it affected the expression of let-7 family members. We found no consistent changes in LIN28 expression, indicating that LIN28 does not participate in the regulation of let-7 family members during the cellular senescence of hUCB-MSCs (Figure S5).
Several lines of evidence support the epigenetic regulation of miRNA transcription [8]. Lehmann et al. [9] reported that hypermethylation of the miRNA genome is strongly correlated with the methylation of known tumor suppressor genes, and that treatment of various breast cancer cell lines with a DNA demethylating agent, 5-aza-2′-deoxycytidine, reduces gene methylation of miRNA-9-1 and concomitantly reactivates its expression. In addition, another report found that expression of a large miRNA cluster (C19MC) was activated in human cancer cells through the demethylation of a CpG-rich region [10]. Although epigenetic regulation of miRNA transcription has been reported, the focus has been limited to DNA methylation of CpG-rich regions. There is a report suggesting that histone acetylation plays an important role in chromatin remodeling and is required for gene activation [34]. Although several studies have suggested this possibility, there has been no direct evidence showing the regulation of miRNAs by histone modifications in a mammalian system [35, 36]. Here, we found that HDAC regulates the transcription of a group of miRNAs through the modification of histones. These results offer direct evidence that activating and repressive epigenetic marks on histones regulate miRNA transcription, extending our understanding of the cross-talk between systems of epigenetic control. It still needs to be determined whether the inhibition of HDAC, DNA methylation and miRNAs cross-regulate one another, especially given the reports that HDAC inhibitors affect the activity of DNMT [37, 38].
Transcription of the miRNA genome is mainly driven by RNA polymerase II and is believed to share the characteristics and regulatory mechanisms of class II genes. Class II genes usually have three promoter regions: (1) a roughly 100-bp long core promoter domain, (2) the area immediately upstream of this region and (3) distal domains that are thousands of base pairs (bps) in length [39, 40]. Typically, RNA polymerase II binds to specific sequence elements within the core promoter [40]. Using ChIP analysis with random primers, we found that RNA polymerase II binds to genomic coordinates ranging from −500 bp to +500 bp of the coding regions of miRNAs such as let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a. These regions may include promoter regions or the miRNA coding region itself if these transcriptional units exist for miRNA regulation. The increase in RNA polymerase II binding to these regions directly supports the transcriptional activation of miRNAs such as let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a during the process of senescence. This finding, along with the active histone marks in the vicinity of miRNA coding regions, may represent a mechanism by which the transcription of miRNAs is increased.
Taken together, our novel findings show that HDACs may play key roles in the regulation of certain miRNAs, including let-7a1, let-7d, let-7f1, miR-23a, miR-26a and miR-30a, in the context of the cellular senescence of hUCB-MSCs. These miRNAs target HMGA2 mRNA, which in turn regulates cellular senescence, including p16INK4A (Fig. 7). Given the previously reported interaction between DNA methylation and miRNAs, these results suggest cross-talk between epigenetic regulatory mechanisms, including histone modification, DNA methylation and miRNAs. Further studies are necessary to elucidate the detailed mechanisms underlying these processes.

Schematic diagram describing how transcriptional regulation of miRNAs, which target mRNA of HMGA2, occurs in HDAC inhibitor-mediated senescent MSCs. Alteration of histone codes in the proximity of miRNAs by treatment of HDAC inhibitor in hUCB-MSCs may lead the miRNAs to be prone to transcriptional activation. Increase of HMGA2 targeting miRNAs represses HMGA2 expression levels and causes p16INK4A and p21CIP1/WAF1, which eventually cause cellular senescence and diminution of self-renewal in hUCB-MSCs
References
Boheler KR (2009) Stem cell pluripotency: a cellular trait that depends on transcription factors, chromatin state and a checkpoint deficient cell cycle. J Cell Physiol 221:10–17
Shukla V, Vaissiere T, Herceg Z (2008) Histone acetylation and chromatin signature in stem cell identity and cancer. Mutat Res 637:1–15
Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K, Odom DT, Otte AP, Volkert TL, Bartel DP, Melton DA, Gifford DK, Jaenisch R, Young RA (2006) Control of developmental regulators by polycomb in human embryonic stem cells. Cell 125:301–313
Meshorer E, Misteli T (2006) Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol 7:540–546
Lee S, Park JR, Seo MS, Roh KH, Park SB, Hwang JW, Sun B, Seo K, Lee YS, Kang SK, Jung JW, Kang KS (2009) Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Prolif 42:711–720
Jung JW, Lee S, Seo MS, Park SB, Kurtz A, Kang SK, Kang KS (2010) Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cell Mol Life Sci 67:1165–1176
Heo I, Joo C, Cho J, Ha M, Han J, Kim VN (2008) Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell 32:276–284
Saito Y, Jones PA (2006) Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle 5:2220–2222
Lehmann U, Hasemeier B, Christgen M, Muller M, Romermann D, Langer F, Kreipe H (2008) Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol 214:17–24
Tsai KW, Kao HW, Chen HC, Chen SJ, Lin WC (2009) Epigenetic control of the expression of a primate-specific microRNA cluster in human cancer cells. Epigenetics 4:587–592
Negrini M, Nicoloso MS, Calin GA (2009) MicroRNAs and cancer–new paradigms in molecular oncology. Curr Opin Cell Biol 21:470–479
Bussing I, Slack FJ, Grosshans H (2008) let-7 microRNAs in development, stem cells and cancer. Trends Mol Med 14:400–409
Ibarra I, Erlich Y, Muthuswamy SK, Sachidanandam R, Hannon GJ (2007) A role for microRNAs in maintenance of mouse mammary epithelial progenitor cells. Genes Dev 21:3238–3243
Nishino J, Kim I, Chada K, Morrison SJ (2008) Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf expression. Cell 135:227–239
Hammond SM, Sharpless NE (2008) HMGA2, microRNAs, and stem cell aging. Cell 135:1013–1016
Park JR, Jung JW, Lee YS, Kang KS (2008) The roles of Wnt antagonists Dkk1 and sFRP4 during adipogenesis of human adipose tissue-derived mesenchymal stem cells. Cell Prolif 41:859–874
Seo KW, Lee SR, Bhandari DR, Roh KH, Park SB, So AY, Jung JW, Seo MS, Kang SK, Lee YS, Kang KS (2009) OCT4A contributes to the stemness and multi-potency of human umbilical cord blood-derived multipotent stem cells (hUCB-MSCs). Biochem Biophys Res Commun 384:120–125
Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW (2006) A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 126:503–514
Jung JW, Cho SD, Ahn NS, Yang SR, Park JS, Jo EH, Hwang JW, Jung JY, Kim SH, Kang KS, Lee YS (2005) Ras/MAP kinase pathways are involved in Ras specific apoptosis induced by sodium butyrate. Cancer Lett 225:199–206
Ferguson M, Henry PA, Currie RA (2003) Histone deacetylase inhibition is associated with transcriptional repression of the Hmga2 gene. Nucleic Acids Res 31:3123–3133
Jung JW, Lee S, Seo MS, Park SB, Kurtz A, Kang SK, Kang KS (2010) Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cell Mol Life Sci 67:1165–1176
Lee YS, Dutta A (2007) The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 21:1025–1030
Park SM, Shell S, Radjabi AR, Schickel R, Feig C, Boyerinas B, Dinulescu DM, Lengyel E, Peter ME (2007) Let-7 prevents early cancer progression by suppressing expression of the embryonic gene HMGA2. Cell Cycle 6:2585–2590
Tzatsos A, Bardeesy N (2008) Ink4a/Arf regulation by let-7b and Hmga2: a genetic pathway governing stem cell aging. Cell Stem Cell 3:469–470
Xu B, Huang Y (2009) Histone H2a mRNA interacts with Lin28 and contains a Lin28-dependent posttranscriptional regulatory element. Nucleic Acids Res 37:4256–4263
Liu L, Luo GZ, Yang W, Zhao X, Zheng Q, Lv Z, Li W, Wu HJ, Wang L, Wang XJ, Zhou Q (2010) Activation of the imprinted Dlk1-Dio3 region correlates with pluripotency levels of mouse stem cells. J Biol Chem 285:19483–19490
Li O, Vasudevan D, Davey CA, Droge P (2006) High-level expression of DNA architectural factor HMGA2 and its association with nucleosomes in human embryonic stem cells. Genesis 44:523–529
Eda A, Tamura Y, Yoshida M, Hohjoh H (2009) Systematic gene regulation involving miRNAs during neuronal differentiation of mouse P19 embryonic carcinoma cell. Biochem Biophys Res Commun 388:648–653
Shell S, Park SM, Radjabi AR, Schickel R, Kistner EO, Jewell DA, Feig C, Lengyel E, Peter ME (2007) Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci USA 104:11400–11405
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765
Chiosea S, Jelezcova E, Chandran U, Acquafondata M, McHale T, Sobol RW, Dhir R (2006) Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am J Pathol 169:1812–1820
Chiosea S, Jelezcova E, Chandran U, Luo J, Mantha G, Sobol RW, Dacic S (2007) Overexpression of dicer in precursor lesions of lung adenocarcinoma. Cancer Res 67:2345–2350
Viswanathan SR, Daley GQ, Gregory RI (2008) Selective blockade of microRNA processing by Lin28. Science 320:97–100
Kim W, Benhamed M, Servet C, Latrasse D, Zhang W, Delarue M, Zhou DX (2009) Histone acetyltransferase GCN5 interferes with the miRNA pathway in Arabidopsis. Cell Res 19:899–909
Bandres E, Agirre X, Bitarte N, Ramirez N, Zarate R, Roman-Gomez J, Prosper F, Garcia-Foncillas J (2009) Epigenetic regulation of microRNA expression in colorectal cancer. Int J Cancer 125:2737–2743
Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC (2006) Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res 66:1277–1281
Walton TJ, Li G, Seth R, McArdle SE, Bishop MC, Rees RC (2008) DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor beta and induce apoptosis in prostate cancer cell-lines. Prostate 68:210–222
You JS, Kang JK, Lee EK, Lee JC, Lee SH, Jeon YJ, Koh DH, Ahn SH, Seo DW, Lee HY, Cho EJ, Han JW (2008) Histone deacetylase inhibitor apicidin downregulates DNA methyltransferase 1 expression and induces repressive histone modifications via recruitment of corepressor complex to promoter region in human cervix cancer cells. Oncogene 27:1376–1386
Wang X, Xuan Z, Zhao X, Li Y, Zhang MQ (2009) High-resolution human core-promoter prediction with CoreBoost_HM. Genome Res 19:266–275
Liang R, Bates DJ, Wang E (2009) Epigenetic control of MicroRNA expression and aging. Curr Genomics 10:184–193
Acknowledgment
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST, 2010-0020265).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Lee and J-W. Jung contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2010_457_MOESM1_ESM.tif
Figure S1. Cumulative population doubling level of hUCB-MSCs was calculated after repeated subculture. In each passage cells were cultured for 4 days and then subcultured for calculation of CPDL. Prominent decrease of inclination in the graph is observed after 10th passage (TIFF 228 kb)
18_2010_457_MOESM2_ESM.tif
Figure S2. Changes in HDAC1, HDAC2, HMGA2 and p16INK4A expression levels were assessed using RT-PCR in the course of HDAC inhibitor-mediated and/or replicative senescence of hUCB-MSC. (a) HDAC1, HDAC2, HMGA2 and p16INK4A expression levels were compared in between early (p6) and late stage (p19) hUCB-MSCs. (b) Dose-dependent regulation of HMGA2 and p16INK4A expression after 5- or 7-day treatment of VPA or NB. (c) A time-course analysis of HMGA2 and p16INK4A expression levels followed by treatment of 4mM VPA or 2mM NB 2mM (TIFF 458 kb)
18_2010_457_MOESM3_ESM.tif
Figure S3. In order to induce oxidative stress-mediated senescence, hUCB-MSCs were treated with 300uM H2O2 for 1-day, 3-, 5- or 7-days. (a) After treatment, MSCs underwent senescence as shown by SA β-gal staining. p16INK4A and p21CIP1/WAF1 expression levels were assessed by RT-PCR(b) and real-time qPCR(c). (d~e) HMGA2 expression(d) and LET7-a and hsa-miR-23a expression(e) were down regulated after 300uM H2O2 treatment as shown by real-time qPCR (TIFF 0.99 mb)
18_2010_457_MOESM4_ESM.tif
Figure S4. let-7 family of miRNAs are up-regulated after treatment of HDAC inhibitors. let-7 isotypes were analyzed by RT-PCR followed by agarose gel electrophoresis. Semi-quantification of PCR product was performed by Image J analysis. Graph shows relative gene expression levels of HDAC inhibitor treated cells compared with control cells. * and ** represent statistical significance at the levels of p<0.05 and p<0.01, respectively (TIFF 284 kb)
18_2010_457_MOESM5_ESM.tif
Figure S5. Factors related with biogenesis of miRNA. HDAC inhibitors were treated to hUCB-MSCS and RT-PCR analyses followed by agarose gel electrophoresis were performed for the expression levels of Dicer, Drosha and LIN28. Semi-quantification of PCR product was performed by Image J analysis. Graph shows relative gene expression levels of HDAC inhibitor treated cells compared with control cells. * and ** represent statistical significance at the levels of p<0.05 and p<0.01, respectively (TIFF 272 kb)
18_2010_457_MOESM6_ESM.tif
Figure S6. Histone modification patterens at OCT4 promoter and the genomic context of miR-329-1 after treatment of HDAC inhibitors. (a~d) After treatment with HDAC inhibitors at the indicated concentrations for 1 d or 3 d, real-time qPCR analysis and ChIP assay were performed. (a) Expression level of OCT4 and miR-329-1 was investigated by real-time qPCR. Five types of acetylated or methylated histone forms (c) were analyzed at OCT4 promoter and in the vincinity of DNA encoding miR-329-1. The graph shows relative enrichment of target proteins at OCT4 promoter and in the vincinity of DNA encoding miR-329-1 of cells treated with the HDAC inhibitors compared with control cells. (d) Relative enrichment of RNA polymerase II, MeCP2 and EZH2 were also investigated by ChIP analysis. * and ** represent statistical significance at the levels of p<0.05 and p<0.01, respectively (TIFF 310 kb)
18_2010_457_MOESM7_ESM.tif
Table S1. miRNAs targeting HMGA2 are up-regulated in the senescent hMSCs. We performed microRNA microarray analysis as described in the Materials and Methods section. Significantly up-regulated miRNAs (p<0.05) are listed in Table S1. The miRNAs targeting HMGA2 were identified using screening programs for the miRNA database (Miranda, Pictar and Targetscan) and are indicated by the ‘o’ mark (TIFF 1.21 mb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lee, S., Jung, JW., Park, SB. et al. Histone deacetylase regulates high mobility group A2-targeting microRNAs in human cord blood-derived multipotent stem cell aging. Cell. Mol. Life Sci. 68, 325–336 (2011). https://doi.org/10.1007/s00018-010-0457-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0457-9