
Abstract.
DNA and histone methylation are linked and subjected to mitotic inheritance in mammals. Yet how methylation is propagated and maintained between successive cell divisions is not fully understood. A series of enzyme families that can add methylation marks to cytosine nucleobases, and lysine and arginine amino acid residues has been discovered. Apart from methyltransferases, there are also histone modification enzymes and accessory proteins, which can facilitate and/or target epigenetic marks. Several lysine and arginine demethylases have been discovered recently, and the presence of an active DNA demethylase is speculated in mammalian cells. A mammalian methyl DNA binding protein MBD2 and de novo DNA methyltransferase DNMT3A and DNMT3B are shown experimentally to possess DNA demethylase activity. Thus, complex mammalian epigenetic mechanisms appear to be dynamic yet reversible along with a well-choreographed set of events that take place during mammalian development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Enzymatic modifications of cytosine bases and histone proteins in the nucleosome core provide heritable epigenetic information in vertebrates that is not encoded in the nucleotide sequence of the cell. Chromatin replication during S phase of the cell cycle offers a window of opportunity for these enzymes and accessory factors to load onto the newly synthesized DNA and robustly propagate all the modification marks, as found in the parental cells. Failure to maintain correct epigenetic information leads to catastrophic consequences for the cell, including incorrect gene expression and apoptosis [1–3]. Importantly, cytosine methylation in mammalian cells is faithfully preserved between successive cell divisions. The preservation of DNA methylation during cell division is catalyzed by DNA (cytosine-5) methyltransferases (DNMTs). The major maintenance DNMT enzyme is DNA (cytosine-5) methyltransferase 1 (DNMT1) and has strong preference for hemimethylated DNA in vitro [4]. Previous studies demonstrate that DNMT1 is stably associated with newly replicated origin in the mammalian cells [5–7]. Therefore, it is plausible that DNMT1 can methylate the newly synthesized daughter strands soon after replication by reading the methylation pattern of the parental strand [8]. Similarly, there is strong evidence supporting the heritability of histone modifications in multicellular organisms. The strongest evidence links histone H3K27 and H3K4 modification catalyzed by the Polycomb group (PcG) and trithorax group (trxG) to mitotic inheritance of lineage-specific gene expression patterns [9]. Although some components of trxG and PcG possess histone methyltransferase activities for H3, other components of trxG and PcG interpret those histone marks. These two protein complexes are shown to be critical regulators of numerous developmental genes. They silence or activate a gene via binding to specific regions of a gene and post-translationally modifying the histones. Some recent works have also demonstrated that PcG-mediated gene silencing may involve non-coding RNA and RNAi machinery [9]. Thus, the model of histone modification inheritance is more complex than DNA methylation due to replication-independent histone deposition on DNA [10]. Nevertheless, there is strong speculation that a large number of histone modifications may follow epigenetic inheritance mechanisms. Recent advances in sequencing technology, higher computational capacity and highly specific antibodies against histone modifications have resulted in greater understanding of epigenetic marks in the context of mammalian genome. The flurry of research activity has resulted in several very excellent publications [11–13]. In this review, we have attempted to give readers a general overview of the epigenetic mechanisms in mammals by discussing histone and DNA modifications along with the involvement of RNA in both developmental and biological context.
DNA methylation and its implications in epigenetic regulation
In the mammalian genome DNA methylation occurs by covalent modification of the fifth carbon (C5) in the cytosine base and the majority of these modifications are present at CpG dinucleotides within the genome. However, in mouse embryonic stem cells, the genomic DNA contains methylated CpA, CpT and CpG sequences [14] instead of exclusive CpG methylation, which is predominately found in somatic cells. Nevertheless, 5-methyl cytosine (Me5C) accounts for about 1% of total DNA bases and therefore is estimated to represent 70–80% of all CpG dinucleotides in the genome [15]. The CpG dinucleotides are distributed unevenly across the human genome, but are concentrated in dense pockets called CpG islands (CGIs). The methylation pattern in any given cell is the outcome of independent but dynamic processes of methylation and demethylation. In the mammalian genome, methylation patterns in differentiated somatic cells are generally stable and inheritable. However, reprogramming (demethylation/remethylation) of methylation pattern takes place during two developmental stages, in germ cells and in preimplantation embryos. In contrast to genome-wide demethylation occurrence in the primordial germ cells, genomes of mature sperms and eggs in mammals are highly methylated as compared to somatic cells [8, 16].
Although the majority of CpGs are methylated in the genome, CpG dinucleotides within CGI promoters are typically unmethylated during development and in normal (non-neoplastic/non-senescent) tissue types. The CGIs are genomic DNA regions with high frequency of CpG dinucleotides. Typically, a CGI is a region with at least 200 bp with a greater than 50% GC and an observed/expected CG ratio greater than 60% [17]. Comprehensive analysis of CGIs in human chromosomes 21 and 22 by Takai and Jones [18] revealed that regions of DNA of greater than 500 bp with a G+C equal to or greater than 55%, and observed CG/expected CG of 0.65 were more likely to be associated with the 5′ regions of genes. With this definition most of the Alu-repetitive elements were excluded. These islands overlap with promoter regions of 50–60% of human genes [19]. However, a subset of promoter CGIs are methylated in a tissue-specific manner during development, showing an exception to the general rule that CGI methylation in normal tissue is limited to X-inactivation and imprinted genes [8, 20]. This observation was supported further by recent findings of genome-wide profiling of DNA methylation demonstrating that non-X-linked promoter CGIs are methylated in normal tissues and escape methylation in germ line cells [21]. Another study has estimated 6–8% of CGIs to be methylated in the genomic DNA of human brain, blood, muscle and spleen [22]. Interestingly, in the same study, CGIs displayed tissue-specific methylation of genes essential for development, suggesting a programmed mechanism of DNA methylation. Another means of DNA methylation propagation is via methylation spreading that begins with genome-wide demethylation that starts shortly after fertilization. Remethylation of most of the genome occurs after the blastocyst stage [23] and continues at a slower pace during the rest of the developmental period. Even though the phenomenon of spreading has not been fully understood, it was proposed as a self-perpetuating interaction between chromatin-modifying proteins and DNA methylation [24]. Indeed, many of the chromatin modification enzymes responsible for gene silencing are found associated with each other in mammalian cells. Some of the examples of DNMT1-associated proteins are HDAC1 [25], histone methyltransferase G9a [26], ATP-dependent chromatin modeling enzyme SNF2H [27], and Polycomb protein EZH2 [28]. Therefore, the above hypothesis that initial DNA or histone methylation will attract repressive complexes, and create a transcriptionally unfavorable chromatin conformation is very plausible. This alteration in chromatin structure, in turn, influences the nearby chromatin and makes it more prone to methylation spreading. This phenomenon is well documented in Arabidopsis, where tandem repeats upstream of endogene SDC element recruit non-CG DNA methylation directed by histone methylation and siRNA, and display spreading of siRNAs and methylation beyond the repetitive DNA [29]. Existing pieces of evidence in mammalian cells show that there are certain cis-acting elements that are dispersed throughout the genome and they can either act as a methylation signal element or methylation boundary during methylation spreading. For example, in the mouse Aprt gene, two upstream B1 repetitive DNA elements were identified to provide de novo methylation signal for spreading [30]. These elements reside in the large stretches of DNA dubbed as methylation centers. Other retrotransposon elements such as B2, Alu (human equivalent of B1), and LINE- 1 (long interspersed nuclear element-1) are also considered to possess de novo signaling activity for methylation spreading [24]. In contrast, the Sp1 binding sites within the Aprt promoter provide the counteracting force against spreading. Indeed, site-directed mutation of one or more Sp1 sites eliminates the binding of transcription factors and allows methylation to spread at the Aprt promoter [31]. However, (ATAA)n repeat sequences in the human GSTP1 gene, Sp1 and CTCF elements in the BRCA1 gene, act as boundary elements for prevention of methylation spreading onto CGIs [32, 33]. Recent experimental work on genome-wide DNA methylation analysis discovered an overrepresentation of putative zinc finger binding sites at the boundaries of methylation-resistant CGIs. This observation suggested that these sites may reinforce transcription factor binding and thereby block methylation spreading and promote transcription [34]. Dynamic equilibrium between methylation spreading and its suspension is likely to be responsible for establishing and maintaining stable DNA methylation patterns in human somatic cells. Furthermore, a combined study of bioinformatic approaches and methylation data from chromosome 21 has demonstrated that DNA sequence, repeat frequencies, and predicted DNA structures correlated with methylation status of CGIs [35].
Aberrant gene expression is one of the key features associated with complex diseases such as cancer, type II diabetes, schizophrenia and autoimmune disease. These diseases are known to be heritable, although they do not follow clear Mendelian inheritance patterns. There are several lines of evidence suggesting that epigenetic abnormalities, together with genetic alterations, are responsible for the deregulation of key regulator genes resulting in these diseases. The epigenetic mechanism provides an alternative explanation for some of the features in complex diseases, including late onset, gender effects, parent-of-origin effects, and fluctuation of symptoms [36]. For example, in cancer cells, normally unmethylated CGIs are often hypermethylated to silence flanking tumor suppressor genes during neoplasia [37, 38]. On the other hand, demethylation (hypomethylation) of normally methylated CGIs can lead to unscheduled activation of genes, as was first shown at MAGE-1 locus, which is normally expressed only in germ line cells but is activated in human tumors [39]. Indeed, the pattern of cancer-associated methylation of CGIs also depends on other factors, such as cell lineage and environmental stimuli. Apart from cancer, a rare genetic disease ICF (immunodeficiency, centromeric instability and facial anomalies) syndrome was correlated with methyltransferase machinery. These ICF patients have mutations in the DNMT3B gene, which leads to hypomethylation of satellite DNA and specific chromosomal decondensation [40]. Thus, DNA methylation and enzymatic apparatus play a significant role during normal embryonic development and diseases.
Histone modification and chromatin function
In eukaryotes, DNA is packaged as chromatin in the nucleus. Chromatin is further organized into two different levels of general structure, silent heterochromatin and active euchromatin. Heterochromatin regions correspond to the bulk of nuclear material and constitute both telomeres and pericentric regions, and these areas tend to be rich in repetitive sequences and low in gene content. The rest of the genome is considered to be euchromatin that contains most of the genes and is considered transcriptionally active. The nucleosomes are the basic unit of chromatin consisting of 147 bp of DNA wrapped around a histone octamer. Two copies of each of the following core histones are present in amononucleosome: H2A, H2B, H3 and H4. All of them have a globular Cterminal domain and an unstructured N-terminal tail [41]. Interestingly, a variety of modifications are associated with these tails [42]. Histone modifications include, methylation of arginine (R), methylation, acetylation, ubiquitination and sumoylation of lysines (K), and phosphorylation of serine (S) and threonine (T) (refer to the next section and Table 1 in this review for detailed description of histone modification enzymes and their cognate substrates) [42, 43]. Even though the significance of most of these modifications is not fully understood, recent advances in the field have determined that lysine acetylation and methylation are key modulator marks for transcriptional activation or repression [42]. Acetylation/deacetylation of lysines correlates with chromatin accessibility and transcription, whereas lysine methylation exerts a different effect depending on the number of the methyl groups and position of lysine residues. For example, modifications that are localized to inactive genes, such as trimethylation of H3K9 and H3K27, are often referred to as heterochromatin modifications. Trimethylation of histone H3 lysine 4 (H3K4) and acetylation of H3 and H4 are associated with active transcription and termed as euchromatin modifications [44, 45]. Plasticity of euchromatin keeps DNA open for biological activity, thus genes can be transcriptionally turned on or off. A combination of high levels of acetylation and trimethylation of H3K4, H3K36, and H3K79 can be detected in transcriptionally active genes, while low levels of these modifications are associated with inactive state. However, it should be noted that acetylation is exclusively associated with active chromatin. Most of the acetylated residues reside in N-terminal tails of histones except for H3K56, which resides in the core domain [42]. Similar to lysine methylation, arginine methylation can be either an active or repressive mark for transcription. Indeed, coactivator-associated arginine methyltransferase CARM1/PRMT4 is physically associated with histone acetyltransferases and acts in a cooperative manner mediating the function of nuclear factor κB (NF-κB) [46]. Another arginine methyltransferase, PRMT5 has been identified to be present in a promoter complex, and it is proposed that PRMT5 may function as a transcription repressor [47]. Similarly, histone lysine methyltransferase G9a also exhibits dual functional specificity acting as both transcriptional suppressor [48] and activator [49]. Apart from gene expression or repression, chromatin modification appears to play a vital role in other cellular processes such as the response to DNA damage. In response to DNA damage, histone modifications may assist in marking the site of damage and may provide a platform for repair to take place. For example, phosphorylation of H2A.X variant in response to DNA double strand breaks (DSBs), provides signals for nonhomologous end joining repair pathway in mammals [50]. Furthermore, methylation of H4K20 and cell cycle checkpoint protein Crb2 are associated with ionizing radiation-induced DNA damage in fission yeast that results in cell cycle arrest at G2/M. Similar DNA damage response is also observed in human cells. For example, mono- and dimethyl H4K20 are involved in recognition and recruitment of p53BP1, a homologue of Crb2, to damaged DNA sites [51].
Recently, a central role of acetyltransferase HBO1 (histone acetyltransferase binding to origin recognition complex) was demonstrated during DNA replication [52]. In the above study, HBO1 was found in a complex with ING family of tumor suppressors that are associated with cell proliferation. Low levels of HBO1 and ING5 correlate with reduction in DNA synthesis and affect progression into S phase [52]. These results suggest that the role of H4 acetylation by HBO1 is an important event for DNA synthesis. Furthermore, HBO1, ING4, ING5, and p53 have been shown to associate in shared protein complexes in cells. The tumor suppressor p53 physically interacts with HBO1 and negatively regulates its HAT activity in vitro and in cells, and thus connects p53-responsive stress signaling and HBO1-dependent chromatin modification pathways [53]. Another histone modification, phosphorylation may play a role in condensation/decondensation of chromatin during replication in mammalian cells. For example, phosphorylation of H3S10 may function to displace the HP1 complex from H3K9 methylated chromatin to facilitate cellular events for decondensation [54].
In summary, DNA methylation and histone modifications are important for the coordinated transcription, replication and repair process. In all those complex cellular events, cross-talk between DNA methylation and histone modifications may help to maintain correct and ordered recruitment of protein factors onto chromatin for coordinated function. Therefore, deregulation of cross-talk(s) can lead to aberrant outcomes of important biological processes in living cells.
Enzymes that participate in chromatin modifications
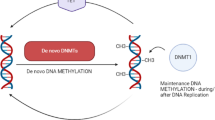
As described before, chromatin modifications in mammals occur at two distinct levels, DNA methylation and histone modifications. Several mammalian DNMTs have been identified, and grouped into two major classes depending on their substrate preference and the resulting function (reviewed in [55, 56]). DNMT3A and DNMT3B are de novo methyltransferases that are responsible for establishing cytosine methylation patterns at unmethylated DNA. Global de novo methylation occurs during early embryogenesis when DNA methylation marks are re-established after genome-wide demethylation for epigenetic reprogramming. Once established, DNA methylation patterns should be stably maintained over cell divisions. This function is fulfilled by the maintenance methyltransferase DNMT1 through its preference for hemimethylated DNA [4, 57], and copying of preexisting methylation patterns onto the newly synthesized DNA strands during DNA replication. Furthermore, different isoforms of DNMT1 (Dnmt1 s and Dnmt1o) participate in maintenance of methylation imprints in preimplantation mouse embryos [58]. Thus, cooperative function between DNMTs provides a way of passing and maintaining epigenetic information between successive cell generations. The targeted deletion of these de novo and maintenance methyltransferases results in various developmental defects [36]. Unlike DNMT1 and DNMT3A/B that contain both regulatory and catalytic domains, DNA methyltransferase DNMT2 has only the catalytic domain exhibiting only weak methyltransferase activity in vitro, and its absence causes no discernable effects in global CpG methylation and developmental phenotype [36]. DNMT3L is a DNMT3-related protein that is expressed only in germ cells and at the stage where de novo methylation occurs [59]. It lacks enzymatic activity but modulates the catalytic activity of DNMT3A and DNMT3B by physically associating with them. Crystal structures for some of these mammalian DNMTs (mouse Dnmts) have been solved, providing additional biochemical and structural insights into the function of the enzymes [60]. To date, the available structures include the PWWP domain of Dnmt3b [61], full-length Dnmt3L with a bound histone H3 N-terminal tail peptide [62], and a complex between the C-terminal domains of Dnmt3a and Dnmt3L [63].
While correct establishment and maintenance of DNA methylation patterns are critical for normal development, DNA demethylation is also equally important for precise execution of developmental programs as evidenced by epigenetic reprogramming events in early embryos and primordial germ cells (PGCs). It is unknown whether DNA demethylation requires demethylase activities or can occur passively through DNA replication in the absence of DNMT1. Although no DNA demethylase activity has been convincingly identified, several mechanisms have been proposed to account for the loss of DNA methylation [64]. For example, DNA deaminases of the Aid/Apobec family have been shown to catalyze deamination of 5-methylcytosine resulting in T:G mismatch, which may lead to DNA demethylation if the mismatch is repaired [65]. Interestingly, a recent study has proposed thatDNMTs themselves have dual roles in CpG methylation and active demethylation of 5-methyl CpGs through deamination during periodic methylation/demethylation cycles of the pS2 gene promoter upon activation by estrogens [66]. Although precise roles of DNMT3A/B in this process are unclear, the study has demonstrated that DNMT3A/B can deaminate both cytosine and 5-methylcytosine in vitro, and that concordant recruitment of DNMT3A/B, DNA glycosylase, and other base excision repair proteins occurs during pS2 promoter demethylation.
Covalent modifications of histones add multiple layers of complexity to chromatin, ranging from small chemical changes such as acetylation and methylation to large peptide addition such as ubiquitylation and sumoylation. Over the past 10 years, several families of histone-modifying enzymes have been identified, as summarized in Table 1. Recent reviews have extensively covered topics of histone modifications, their mechanism of action, and the biological functions derived from individual or combined modifications [42, 67]. Of particular interest, new nomenclature for some families of chromatinmodifying enzymes has recently been proposed since the current nomenclature of the enzymes is rather inconsistent and often creates additional complexity [68]. Most histone modifications are dynamically regulated as evidenced by identification of many enzymes that can remove the modification. One wellstudied example is histone demethylation that is carried out by two classes of enzymes, amine oxidases such as LSD1 and hydroxylases of JmjC family [69]. In contrast, arginine demethylation activity has not been identified yet, although the deimination process converting an arginine to citrulline has been proposed as an alternative mechanism to antagonize arginine methylation [70].
As supported by the number and type of histone-modifying enzymes (Table 1), lysine has emerged as a crucial amino acid residue for histone modifications over the past decade. Interestingly, lysine modifications of non-histone proteins are also mediated by some of the known histone-modifying enzymes, and can be reversed by antagonizing activities just as observed for histone modification. For example, lysine methylation has recently been identified as a novel modification of the p53 tumor suppressor in addition to previously known modifications such as acetylation and ubiquitylation [71]. Histone-modifying enzymes involved in methylation/demethylation of p53 include SYMD2, SET9, and LSD1.
In summary, dynamic modifications of DNA/histones and non-histone proteins by chromatin-modifying enzymes reflect their functional diversity and regulatory complexity.
Other nuclear proteins crucial for epigenetic modifications
Chromatin modifications can directly change chromatin structure by altering the physical properties of individual nucleosomes, primarily by neutralization or addition of charge to target residues. This affects histone-DNA interactions and creates either a more open chromatin architecture or higher-order structures through differential modulation of internucleosomal contacts [67]. In most cases, however, the epigenetic roles of chromatin modifications are augmented by many specialized sets of nuclear proteins that do not participate in chromatin modifications per se but are critical for epigenetic gene regulation. Among many proteins that fall into this category, three types of proteins/complexes are briefly reviewed in this section: chromatin remodeling complexes, effector proteins with various binding modules for different modifications, and insulator proteins.
Chromatin remodeling complexes are energy-driven, multi-protein machinery that allows access to specific DNA regions or histones by altering nucleosomal positions, histone-DNA interactions, and histone octamer positions (Fig. 1A). These chromatin remodellers have a catalytic ATPase to induce changes in local chromatin structure covering one or two nucleosomes. The ATPases in chromatin remodeling complexes are grouped into three subfamilies: the SWI/SNF ATPases, the imitation switch (ISWI) ATPases, and the chromodomain and helicase-like domain (CHD) ATPases. Several recent reviews have summarized the current understanding of diversity and specialization of chromatin remodeling complexes and modulation of remodeller activity by nucleosome modifications [72, 73].

Other nuclear proteins crucial for epigenetic modifications and gene regulation. (A) A simplified example for the role of chromatin remodeling complexes recruited by transcription factors or specific modifications on chromatin [72]. ATP hydrolysis-driven repositioning of nucleosomes exposes an occluded DNA region to allow access of transcriptionalmachinery. (B) Modified histones serve as recognition sites for effector proteins. The illustration shows that trimethylated H3K9 (hexagons) is recognized by HP1 that recruits SUV39H1 and DNMTsto facilitate further H3K9 methylation, HP1 binding, and DNA methylation on adjacent nucleosomes, resulting in repressive chromatin spreading [75, 76]. (C) A model illustrating two major functions of insulators. Insulators (I) placed between an enhancer (E) and promoter blocks enhancer-promoter communication, thereby preventing inappropriate activation of promoters by distant enhancers (left panel). Insulators can also function as a chromatin barrier that limits heterochromatin spreading and prevents repression of neighboring genes (right panel). Condensed heterochromatin is decorated with repressive marks such as H3K9 methylation (hexagons) and DNA methylation (circles).
In many cases, chromatin modifications serve as recognition sites for the recruitment of effector modules that read and implement modification-encoded biological messages [42, 74]. Several distinct binding modules have been identified in various nuclear proteins, coupling a particular histone modification with cognate effector proteins (Table 2). Thus, the composition of modifications on a given histone can either recruit or occlude a set of proteins. Effector proteins may alter chromatin structure by binding two or more nucleosomes as found with HP1 and Polycomb group proteins [74]. Effector proteins can also act as an adaptor to attract additional chromatin-modifying enzymes or remodeling complexes to augment the chromatin alteration initiated by the modification. Such an example can be found in HP1 binding to trimethylated H3K9 [75] and DNMT1 [76]. These initial interactions can recruit SUV39H1 and/or DNMT1 and promote further H3K9 methylation, HP1 binding, and DNA methylation, which may in turn result in further transcriptional gene silencing or chromatin repression [77] (Fig. 1B). Similarly, the PHD domain of BPTF, a component of the NURF chromatin remodeling complex, recognizes trimethylated H3K4 and brings the remodeller with it [78]. Some other effector proteins have enzymatic activities themselves, as exemplified by CHD1 remodeling ATPase, which binds to trimethylated H3K4 and introduces active structure remodeling [42]. Similar effector proteins have been identified for DNA methylation. A series of methyl CpG-binding proteins, such as MBDs and MeCP2, has demonstrated the ability to interpret DNA methylation marks in different biological contexts (reviewed in [56]). Specifically, it has been demonstrated that interpretation of DNA methylation marks by MBDs and MeCP2 has additional assurance via recruitment of HDACs for gene silencing [25, 79, 80]. Recently, another methyl CpG-binding protein UHRF1 has been shown to recruit DNMT1 itself onto chromatin to facilitate the faithful inheritance of genomic methylation patterns [81, 82].
Finally, insulators are DNA elements that can protect a gene from neighboring transcriptional influences to prevent inappropriate activation or repression of the gene. Insulators have two well-known functions that are represented by enhancer blocking and formation of chromatin barrier, so that they can either prevent distal enhancers from activating a promoter or block heterochromatin spreading that may lead to silencing of neighboring genes (Fig. 1C). Achromatin insulator protein CTCF (CCCTC-binding factor) plays important roles in many aspects of epigenetic regulation including genomic imprinting, X-chromosome inactivation, transcription of non-coding RNAs at repetitive elements, and long-range chromatin interactions (reviewed in [83, 84]). Thus, CTCF-binding sites establish epigenetic boundaries by which correct gene expression is ensured during development, and also contribute to higher-order genome organization within the nucleus.
Chromatin modification and its role in development
Epigenetic mechanisms can affect long-term gene expression, which constitutes the basis for the accurate execution of developmental programs and the maintenance of the cell types over subsequent cell divisions. PcG and trxG genes were first discovered in Drosophila melanogaster as master regulators of homeotic (Hox) gene expression. Polycomb complexes function as repressors of target genes, whose action is balanced by an antagonistic effect of trithorax complexes working on the identical DNA regulatory elements. These elements, PcG or trxG response elements (PREs/TREs), recruit PcG or trxG proteins to form multimeric complexes on PREs/TREs and mediate epigenetic inheritance of silent or active chromatin states through cell divisions, respectively. These PcG and trxG complexes are not required for the initial establishment of homeotic gene expression pattern, but are essential for maintenance of the established state throughout the rest of development (reviewed in [9]). Although PREs/TREs have only been identified and characterized in Drosophila, PcG and trxG genes are also conserved in mammals and play an important role in many developmental processes such as cell lineage specification and stem cell maintenance (reviewed in [85]). For example, recent genome-wide PcG profiling in mouse and human embryonic stem (ES) cells has revealed that most PcG targets in ES cells are regulators of differentiation pathways, suggesting that the PcG proteins keep stem cells in a pluripotent state by suppressing cell fate-specific genes [86, 87]. These PcG target genes can be activated upon differentiation to result in specific cell types with a concomitant loss of PcG proteins, suggesting a possibility that trxG proteins may be involved in this activation by replacing PcG proteins on the target genes.
PcG proteins form two distinct multi-protein complexes, Polycomb repressive complex 1 (PRC1) and PRC2, although mammals have two additional PRC2-related complexes. The components of each complex in different organisms and their recruitment/mechanism of action have been reviewed comprehensively [88, 89]. The catalytically active component of PRC2, EZH2, catalyzes trimethylation of histone H3K27, and this enzymatic activity is required for PRC2-mediated silencing. The H3K27 methylation mark deposited by PRC2 recruits PRC1 via its chromodomain-containing components, which is believed to facilitate condensation of chromatin structure. Other properties of PRC1 also contribute to transcriptional silencing. PRC1-mediated ubiquitylation of histone H2A is critical for Hox gene silencing by an unknown mechanism [90]. In mammalian cells this robust PcGmediated repression appears to be stabilized by DNA methylation since EZH2 can directly recruit DNA methyltransferases to target genes [28]. Furthermore, H3K27 methylation by PcG predisposes the marked genes to de novo methylation leading to aberrant silencing in cancer cells [91]. Although it remains unknown in mammalian cells, there may be additional mechanisms other than histone/DNA modifications in PcG-mediated repression, since studies in Drosophila have implicated other silencing mechanisms such as direct interactions with the transcriptional machinery and transcription of non-coding RNA (ncRNA) (reviewed in [9]). In fact, PcG complexes have been shown to participate in gene silencing during X-chromosome inactivation and genomic imprinting where ncRNAs play a critical role in silencing mechanisms, which is reviewed later in this contribution. As the mechanistic opposite of PcG, trxG proteins also form several multimeric complexes. The trxG-associated MLL1 has been shown to catalyze histone H3K4 trimethylation that is recognized by BPTF, a subunit of NURF chromatin remodeling complex. This targeting of the remodeling complex to histones methylated by trxG is thought to facilitate active chromatin formation by repositioning nucleosomes on the promoter [78].
In addition to activation of genomic programs leading to specific cell types, another equally important epigenetic event during development is that a cell must silence alternative gene expression programs specific to other cell types to secure its fate. The best example of this lineage restriction is found in neurogenesis, during which neural cell fates are acquired in the developing nervous system, and neuron-specific genes are repressed in non-neuronal cells outside the nervous system. This suggests that neuronal chromatin is epigenetically programmed in different cellular contexts. REST (repressor element 1-silencing transcription factor), a repressor of neuronal genes containing a conserved RE1 provides a link between epigenetic mechanisms and neurogenesis by establishing silent chromatin states in cooperation with other corepressors and chromatin modifiers (reviewed in [92]). The corepressor CoREST confers more specialized repression mechanisms, such that the RESTCoREST complex recruits various chromatin modifiers for long-term silencing of neuronal genes in terminally differentiated non-neuronal cells. Chromatin-modifying enzymes and other epigenetic silencing factors involved in this process have been extensively reviewed [92].
In contrast to stable and inheritable silencing of neuronal chromatin in terminally differentiated nonneuronal cells, the situation in ES cells and neuronal progenitors impose another aspect of epigenetic concern on gene expression since these cells should be able to relieve the silent chromatin state upon differentiation to allow a lineage-specific gene expression. A comparative analysis of neuronal gene chromatin in terminally differentiated fibroblasts and pluripotent ES cells has revealed that stem cells and progenitors possess a poised chromatin status for subsequent neuronal differentiation with distinct differences in epigenetic marks and transcriptional features [93]. This study suggests that the core REST complex establishes characteristic chromatin states by recruiting different chromatin modifiers in non-neuronal and ES cells, emphasizing the role of REST and its corepressors in building plasticity of neuronal chromatin.
Taken together, epigenetic mechanisms set a fundamental basis for maintenance of ES cell identity and long-term cellular memory that are crucial for normal development.
Dosage compensation in mammals
In mammals, females have two X chromosomes (XX), while males have only one (XY). This chromosomal difference between the sexes creates a need for dosage compensation systems to adjust the gene dose of X-linked genes. Mammalian dosage compensation is accomplished by silencing of one of the two X chromosomes in females, a process referred to as X chromosome in activation (XCI) (reviewed in [94]). In mouse and human embryos, XCI is initiated in early development. The XCI is regulated by a cis-acting master switch locus, the X-inactivation center (Xic), which includes ncRNA gene Xist (X inactive specific transcript) and its antisense transcription unit Tsix/Xite (Xist spelled backward due to its antisense orientation to Xist). The Xic senses the number of X chromosomes and produces the noncoding Xist RNA from one of the two chromosomes to trigger silencing in cis. Therefore, the initiation of this random inactivation presents important questions on how cells count the number of X chromosomes and choose which one to be inactivated. Recent progress in understanding the mechanisms driving the XCI counting and choice has indicated that multiple regulatory systems may be involved, thus giving rise to multiple models for the initiation of random XCI (reviewed in [95]). Among these interesting findings, trans-interaction of X chromosomes via a novel X-pairing region of Xic has been observed, suggesting that the homologous pairing may enable a cell to detect the number of X chromosomes and coordinate Xist/Tsix expression to determine the future active and inactive X chromosomes (Xa and Xi, respectively) [96]. Another recent study supports an alternative mechanism, a stochastic model where each X chromosome has an independent probability to initiate the XCI within a certain time span. These studies suggest the presence of a novel X-encoded trans-acting XCI activator involved in initiation of XCI, based on observations in tetraploid ES cells [97]. In contrast to random inactivation, in some mammals the parental origin determines which X chromosome is to be inactivated (reviewed in [94]). All tissues of marsupials and the extra-embryonic tissues of mice display imprinted inactivation of the paternal X chromosome. The molecular basis underlying the preferential paternal X inactivation and the nature of imprints are not currently well understood.
Once the future Xi is chosen, XCI starts with the accumulation of Xist RNA along the Xi. The Xist expression is regulated by the Tsix gene that acts primarily in the nucleus and is transcribed in the antisense direction over the Xist gene [98]. The Xist RNA coating-induced silencing accompanies multiple layers of epigenetic modifications on the Xi, which lock in and stably maintain the inactive state through cell divisions (reviewed in [94, 95]). Chromosomewide studies revealed various X-linked histone modifications, including hypoacetylation of histone H4 [99], trimethylation of H3K9 and H3K27 [100, 101], H4K20 monomethylation [102], H2AK119 monoubiquitylation [103], as well as substitution of core histone H2A with the histone variant macroH2A [104]. In addition to the histone modification profile, the Xa and Xi allele-specific DNA methylation patterns have also been established [105]. Analysis of Dnmt1 −/− embryos has shown that methylation is required for stable maintenance of gene silencing on the Xi [106]. As discussed above, a wide range of chromatin modifiers are known to be involved in XCI, including PcG complexes, histone deacetylases, and DNMT (reviewed in [94]). Although the exact combination of histone modifications on the Xi may vary during development and in different lineages and cell types, the order of chromatin modifications leading to X inactivation was postulated based on the observations during female mouse ES cell differentiation (reviewed in [94, 95]). First, Xist RNA transcription and accumulation on the Xi in cis trigger silencing through as yet unknown mechanisms. Then, recruitment of PRC1 and PRC2 mediates H2AK119 monoubiquitylation and H3K27 trimethylation, respectively. At this early stage of XCI, the inactivation process is reversible and dependent on the presence of Xist RNA. As cell differentiation proceeds, the Xi undergoes deposition of histone macroH2A and histone H4 hypoacetylation, followed by promoter-specific DNA methylation on the Xi. At this phase, the XCI is irreversible and Xist RNA is not required for maintenance of the inactive state. Apart from chromatin modifications on Xi, the Xi also shows the shift to late replication during random inactivation [107] and Xist RNA defines a repressive nuclear compartment early on in the XCI process [108]. Thus, the epigenetic marks and temporal/spatial segregation mechanisms contribute to the initiation and maintenance of XCI. Despite significant progress in understanding of molecular mechanisms of XCI, there are still many unanswered questions. For example, the counting and choice process of random inactivation awaits further elucidation of its molecular basis. Similarly, the mechanisms by which Xist RNA triggers recruitment of chromatin-modifying complexes remain unknown. Furthermore, it is still elusive how cis-acting elements and trans-acting factors coordinate and spread silencing across the chromosome.
Genomic imprinting
Diploid organisms such as mammals carry two copies of autosomal genes, one from each parent. In most cases, both parental alleles have equal potential to be expressed in cells. However, a subset of autosomal genes are subject to genomic imprinting by which the expression is limited to one of the two parental alleles depending on the parent-of-origin of the gene. Genomic imprinting is an epigenetic mechanism conserved in placental mammals, and failure to establish correct imprinting has been shown to cause defects in embryonic and neonatal growth and can result in neurological disorders such as Prader-Willi syndrome [109]. To date about ∼80 imprinted genes have been identified in mice, the majority of which are clustered in the genome while there are some solo imprinted genes [110]. Each imprinted gene cluster often encompasses several protein-coding genes over 100–3000 kb DNA, and at least one non-coding RNA (ncRNA) gene [111]. For example, the maternally imprinted Igf2r cluster spans ∼500 kb, and contains three maternally expressed protein-coding genes and the 108 kb Air ncRNA gene that is essential for imprinted gene expression [112, 113]. Expression of imprinted genes in each cluster is generally controlled by a single major cis-acting element, the imprinting control region (ICR) [114]. ICRs are CpG-rich DNA sequences that are methylated in only one of the two parental gametes, and thus carry the parental information. This DNA methylation imprint is acquired during gametogenesis. Prior to sex determination, the parental imprints are erased in germ cells formed in the embryonic gonad. As the embryo develops into a male or female, gametic imprints are placed on paternally imprinted genes during sperm production and on maternally imprinted genes during egg formation, respectively. After fertilization, this methylation imprint is maintained on the same parental chromosome through cell divisions. Establishment and maintenance of the imprints require a series of epigenetic machinery. Gametic imprints are established in germ cells by the de novo methyltransferase Dnmt3a [115]. Another member of Dnmt3 family, Dnmt3L, has been shown to be essential for maternal imprinting in female germ cells, whereas its disruption in male germ cells results in meiotic catastrophe caused by retrotransposon reactivation [116, 117]. These imprinted marks are stably propagated through successive cell divisions by maintenance methyltransferase Dnmt1 and its oocyte-specific isoform Dnmt1o [118, 119]. Furthermore, these gametic imprints can be erased in germ lines during genome-wide reprogramming by an unknown demethylation mechanism(s). Although DNA methylation is the most important mechanism for imprinting, it does not appear to be the only mechanism. Histone modification by a mouse PcG protein Eed has been demonstrated to affect a few paternally repressed genes; however, it has a relatively minor effect compared to that of DNA methylation and may only contribute to maintenance of imprints [120]. Similarly, the absence of histone methyltransferase G9a has been shown to exert pronounced effects on paternal repression of placenta-specific imprinted genes [121].
As mentioned above, each imprinted gene cluster contains at least one ncRNA gene that plays a crucial role in silencing of the multiple protein-coding genes in the cluster by cis-acting mechanisms. Current understanding of imprinted gene expression/silencing has been gained from the studies on six well-characterized clusters, of which four (Igf2r/Air, IC2/Kcnq1, Gnas, and PWS/AS) are maternally imprinted, and two (Igf2/H19 and Dlk1) are paternally imprinted by DNA methylation acquired in the male gamete (as reviewed in [111]). Despite the differences in gene organization and ICR functions in different clusters, a few common features of imprinted gene expression/silencing can be derived. The unmethylated ICRs are implicated in all six clusters as positive regulators of ncRNA expression. In maternally imprinted clusters (Igf2r/Air and IC2/Kcnq1), the unmethylated ICR works as a promoter for a paternally expressed ncRNA that is an antisense orientation to at least one of the genes in the cluster. While deletion of the methylated maternal ICR has no effect on maternally inherited alleles, deletion of the unmethylated paternal ICR reverses the parental-specific expression pattern such that ncRNA expression is lost and biallelic gene expression is obtained by abrogation of paternal silencing. Truncation of ncRNAs at these loci also has similar effects on paternal gene expression, relieving silencing of the paternally inherited alleles (reviewed in [122]). ICRs in paternally imprinted clusters appear to utilize different mechanisms to control the imprinted gene expression. For example, the H19 ncRNA at the Igf2/H19 locus is expressed from the unmethylated maternal chromosome but the ICR does not act as a promoter. Rather, it serves as a boundary element for CTCF (CCCTCbinding factor) that is a chromatin insulator protein [83]. The CTCF protein binds the unmethylated maternal ICR blocking the interaction of downstream enhancers with Igf2 and Ins promoters , while it does not affect the interaction between the enhancers and H19 ncRNA promoter. On the paternal chromosome, the DNA methylation imprint prevents CTCF binding, thus allowing the enhancers to drive the expression of Igf2 and Ins genes [123]. Interestingly, the CTCF protein has been shown to have an additional function at the Igf2/H19 locus, protecting the maternal allele from methylation post-fertilization [124].
Although there is an obvious involvement of ncRNAs in imprinted gene silencing, it is unclear how they can repress even non-overlapped genes that are several hundred kilobase pairs apart from either side of the imprinted ncRNA gene. The major question on this issue is to determine whether imprinted ncRNAs silence genes through the transcript itself or through the action of transcription. Several models have recently been reviewed to address this question [122]. Given the similarities in silencing mechanisms between genomic imprinting and X-chromosome inactivation many useful insights into imprinting mechanisms may be obtained by examining whether what is known about X-chromosome inactivation can be applied to genomic imprinting. Another important question that remains unanswered is how the gametic methylation machinery distinguishes parental-specific alleles and establishes DNA methylation marks at different regions at different loci.
Inheritance of silent loci and genome defense
Completion of the human and mouse genome sequence revealed that transposable elements (TEs) play a major role in shaping the mammalian genome, in particular, in its evolution [125, 126]. These elements account for 45% and 37% of human and mouse genome, respectively. Families of repetitive elements include long terminal repeats (LTR)-retrotransposon, long interspersed nuclear elements (LINE), short interspersed nuclear elements (SINE), and DNA transposons.
Retrotransposons transpose with the help of reverse transcriptase and they can be divided into two subfamilies depending on the presence or absence of direct repeats at the end of the element called LTR. LINE elements do not contain LTRs and account for 17% of the total human genome. A small percentage of these autonomous non-LTR retrotransposons in the human genome remain active [127]. Intracisternal Aparticles (IAPs), MaLR and Etn elements are active LTR retrotransposons present in the mouse genome [128]. In contrast, SINE elements are non-autonomous, non-LTR retrotransposons. The Alu repeats are most common SINE families in human genome and account for 10% of the whole genome mass [129]. B1 and B2 are major SINE elements in mouse genome [130]. DNA transposons do not require reverse transcriptase for integration event into the genome. Instead, a self-encoded protein called transposase can recognize terminal inverted repeats (TIR) of the DNA transposons for genome integration. To date, no evidence has been available for the presence of active DNA transposon, although many copies of inactive fossil DNA transposons are present [125].
There are many ways that transposable elements can interfere with the structure and regulation of gene expression in the genome. They include insertion, deletion or an inversion of genomic sequences. Recombination between non-allelic repeats can lead to rearrangements/translocations, and strong constitutive promoters of retrotransposons can express chimeric mRNA [128, 131]. Transposable elements also can serve as promoters, enhancers, silencers, and alternative splicing site and thereby modulate the expression of related genes [132]. In contrast to the huge number and different modes of gene disruption associated with these transposons, the damage that transposons cause to their host is generally minor. For instance, only 1 in 600 germ line mutations in human can be attributed to transposon insertions [133]. In fact, the damage caused by transposons is largely limited by active repression of these endogenous parasitic elements. Most transposon copies reside in heterochromatin, which by definition contains regions of silent DNA so that they possess little harm to the host genome.
Mammalian (and other vertebrates) genome structure is protected against these parasitic transposable elements. DNA cytosine methylation and modification of histone tails (methylation at H3K9 and deacetylation) are associated with the host-defense system [134, 135]. Drosophila suffers from abundant transposon-mediated mutations and lacks DNA methylation, which adds supportive evidence to the above scenario [136]. In mouse, the transcription of IAP is normally repressed but is greatly induced in embryos lacking DNMT1, demonstrating that methylation is responsible for the repressed state of these elements [137]. Human endogenous retroviruses (HERVs) resemble simple retroviruses in structure. The demethylation of HERVs has been examined in a limited number of cancers (germ cell tumors and cancers of the ovary, testicles and bladder). In these cases, HERV hypomethylation increases with malignancy [138]. In vitro transcription assays using site-specific mutagenesis and methylation demonstrate that methylation of critical CpG dinucleotides within the LINE promoter is enough to ensure repression of transcription. In a number of cancers, hypomethylation of LINE elements is evident, compared to their normal counterparts or unaffected adjacent tissues [139, 140]. LINE hypomethylation can occur early in cancer initiation, notably in colon and prostate cancers. In most other cancers studied (leukemias, urothelial, ovarian and breast cancers), LINE demethylation increases with the degree of malignancy. Therefore, depending on the cancer type, LINE hypomethylation may be useful as an early detector of cancer or a prognostic indicator [141].
Modification of histones also plays a role in suppressing TE transcription. Chromatins associated with TEs are enriched for methylation of histone H3K9, which is a signal for transcriptional suppression. Mutation in Suv39, a H3K9 methyltransferase, leads to reactivation of TE transcription in mouse ES cells [135]. In A. thaliana, DDM1 is required for TE silencing. Mutation of ddm1 results in a loss of DNA and H3K9 methylation, also leading to active TE [142]. Lsh1, mouse homolog of ddm1 is required for TE suppression, and elevated TE transcripts were observed in mutant Lsh1 −/− embryos [143].
Another layer of regulation of TE comes with RNA interference (RNAi) that is mediated through micro-RNAs (miRNAs). These ∼22-nucleotide-long small RNA molecules can negatively control their target gene expression. To date there are more than 460 miRNAs documented [144]. Although RNAi-mediated DNA methylation and TE silencing are well understood for A. thaliana [145], the mechanism by which RNAi mediates chromatin modification is not established in mammals. It is known that the DNA methyltransferase DNMT3A binds to artificially introduced siRNA and directs DNA methylation, which is consistent with a requirement of this enzyme in the downstream event in RNAi [146]. However, complete understanding of RNAi-regulated epigenetic mechanism in mammals still awaits further investigations.
Future prospects
Research in the last two decades demonstrated an emerging pattern of cross-talk between different epigenetic pathways. Some of these pathways were similar and conserved between both yeast and mammalian cells. For example, a cross-talk between RNAi pathways and histone modification reading protein Chp1 of yeast is similar to the Xist RNA of the mammalian cells that plays a role in deposition of DNA and histone methylation marks for X chromosome inactivation, although yeast cells are devoid of DNA methylation. One of the nagging but difficult questions in epigenetic mechanisms is the timing of the events. It is plausible to imagine that chromatin replication during S phase of the cell cycle may offer a greater flexibility for such information to pass from one generation to next. This hypothesis is supported by the presence of several complexes of epigenetic factors such as DNMT1-G9a-PCNA [26], CAF1-MBD1-SETDB1 [147], DNMT1-HDACs [25, 148] and the Polycomb protein EZH2-DNMT1 complex that directs H3K27 methylation [28] during mammalian chromatin replication. However, these observations do not answer all the questions. Indeed, mislocalization of DNMT1 from the replication fork only had a small effect on the overall genomic methylation by reducing the methylation efficiency [149]. Perhaps there are post-replicative chromatin modifications that occur after the initial wave of replicative chromatin modification during cell division. Currently, it is not known what roles modified histones play after the semi-conservative chromatin replication. With the recent discovery of several histone demethylases that can erase epigenetic marks, epigenetic modifications appear to be much more reversible rather than fixed. This brings us to another challenging area of how epigenetic marks are erased or rewritten during development and diseases. These phenomena are also not understood during ES cell development, especially how a multi-potent stem cell can give rise to several different cell type, each being genetically identical but with unique epigenetic signatures and different cellular phenotypes. Such distinctive epigenetic phenotypes are hallmarks of adult monozygotic human twins [150]. Finally, we need a better understanding of the molecular phenomenon of epigenetics in mammalian development and diseases. With modern technological innovations, such as high-throughput DNA sequencing, whole genome bisulfite sequencing and chromatin immunoprecipitation-sequencing, we can explore chromatin modifications in a more efficient manner. What we know today is just a small percentage of the exciting field of epigenetics.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the orginal author(s) and source are credited.
References
Lande-Diner, L., Zhang, J., Ben-Porath, I., Amariglio, N., Keshet, I., Hecht, M., Azuara, V., Fisher, A. G., Rechavi, G. and Cedar, H. (2007) Role ofDNAmethylation in stable gene repression. J. Biol. Chem. 282, 12194–12200.
Chen, T., Hevi, S., Gay, F., Tsujimoto, N., He, T., Zhang, B., Ueda, Y. and Li, E. (2007) Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 39, 391–396.
Jackson-Grusby, L., Beard, C., Possemato, R., Tudor, M., Fambrough, D., Csankovszki, G., Dausman, J., Lee, P., Wilson, C., Lander, E. and Jaenisch, R. (2001) Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 27, 31–39.
Pradhan, S.,Bacolla, A., Wells, R.D. and Roberts, R. J. (1999) Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 274, 33002–33010.
Takebayashi, S., Tamura, T., Matsuoka, C. and Okano, M. (2007) Major and essential role for the DNA methylation mark in mouse embryogenesis and stable association of DNMT1 with newly replicated regions. Mol. Cell. Biol. 27, 8243–8258.
Leonhardt, H., Page, A. W., Weier, H. U. and Bestor, T. H. (1992) A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71, 865–873.
Chuang, L. S., Ian, H. I., Koh, T.W., Ng, H. H., Xu,G. and Li, B. F. (1997) Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277, 1996–2000.
Bird, A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21.
Schuettengruber, B., Chourrout, D., Vervoort, M., Leblanc, B. and Cavalli, G. (2007) Genomeregulation by Polycomb and trithorax proteins. Cell 128, 735–745.
Kusch, T. and Workman, J. L. (2007) Histone variants and complexes involved in their exchange. Subcell. Biochem. 41, 91–109.
Bernstein, B. E., Mikkelsen, T. S., Xie, X., Kamal, M., Huebert, D. J., Cuff, J., Fry, B., Meissner, A., Wernig, M., Plath, K., Jaenisch, R., Wagschal, A., Feil, R., Schreiber, S. L. and Lander, E. S. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326.
Meissner,A., Mikkelsen, T. S.,Gu, H., Wernig, M., Hanna, J., Sivachenko, A., Zhang, X., Bernstein, B. E., Nusbaum, C., Jaffe, D. B., Gnirke, A., Jaenisch, R. and Lander, E. S. (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770.
Pan, G., Tian, S., Nie, J., Yang, C., Ruotti, V., Wei, H., Jonsdottir, G. A., Stewart, R. and Thomson, J. A. (2007) Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1, 299–312.
Ramsahoye,B.H., Biniszkiewicz, D., Lyko, F., Clark, V., Bird, A. P. and Jaenisch, R. (2000) Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 97, 5237–5242.
Ehrlich, M., Gama-Sosa, M. A., Huang, L. H., Midgett, R. M., Kuo, K. C., McCune, R. A. and Gehrke, C. (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 10, 2709–2721.
Reik, W., Dean, W. and Walter, J. (2001) Epigenetic reprogramming in mammalian development. Science 293, 1089–1093.
Gardiner-Garden, M. and Frommer, M. (1987)CpG islands in vertebrate genomes. J. Mol. Biol. 196, 261–282.
Takai, D. and Jones, P. A. (2002) Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 99, 3740–3745.
Wang,Y. and Leung, F.C. (2004)Anevaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics 20, 1170–1177.
Strichman-Almashanu, L. Z., Lee, R. S., Onyango, P. O., Perlman, E., Flam, F., Frieman, M. B. and Feinberg, A. P. (2002) A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 12, 543–554.
Shen, L., Kondo,Y., Guo,Y.,Zhang, J., Zhang, L., Ahmed, S., Shu, J., Chen, X., Waterland, R. A. and Issa, J. P. (2007) Genome-wide profiling ofDNAmethylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 3, 2023–2036.
Illingworth,R., Kerr,A., Desousa,D., Jorgensen, H., Ellis, P., Stalker, J., Jackson, D., Clee, C., Plumb, R., Rogers, J., Humphray, S., Cox, T., Langford, C. and Bird, A. (2008) A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 6, e22.
Santos, F., Hendrich, B., Reik, W. and Dean, W. (2002) Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 241, 172–182.
24 Turker, M. S. (2002) Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 21, 5388–5393.
Fuks, F., Burgers, W. A., Brehm, A., Hughes-Davies, L. and Kouzarides, T. (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 24, 88–91.
Esteve, P. O., Chin, H. G., Smallwood, A., Feehery, G. R., Gangisetty, O., Karpf, A. R., Carey, M. F. and Pradhan, S. (2006) Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 20, 3089–3103.
Robertson, A. K., Geiman, T. M., Sankpal, U. T., Hager, G. L. and Robertson, K.D. (2004) Effects of chromatin structure on the enzymatic and DNA binding functions of DNA methyltransferases DNMT1 and Dnmt3a in vitro. Biochem.Biophys. Res. Commun. 322, 110–118.
Vire, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., Morey, L., Van Eynde, A., Bernard, D., Vanderwinden, J. M., Bollen, M., Esteller, M., Di Croce, L., de Launoit, Y. and Fuks, F. (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874.
Henderson, I. R. and Jacobsen, S. E. (2008) Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev. 22, 1597–1606.
Yates, P. A., Burman, R.W., Mummaneni, P., Krussel, S. and Turker, M. S. (1999) Tandem B1 elements located in a mouse methylation center provide a target for de novo DNA methylation. J. Biol. Chem. 274, 36357–36361.
Mummaneni, P., Yates, P., Simpson, J., Rose, J. and Turker, M. S. (1998) The primary function of a redundant Sp1 binding site in the mouse aprt gene promoter is to block epigenetic gene inactivation. Nucleic Acids Res. 26, 5163–5169.
Millar,D. S., Paul, C. L.,Molloy, P. L. and Clark, S. J. (2000) A distinct sequence (ATAAA)n separates methylated and unmethylated domains at the 5′-end of the GSTP1 CpG island. J. Biol. Chem. 275, 24893–24899.
Butcher, D. T., Mancini-DiNardo, D. N., Archer, T. K. and Rodenhiser, D. I. (2004) DNA binding sites for putative methylation boundaries in the unmethylated region of the BRCA1 promoter. Int. J. Cancer 111, 669–678.
Fan, S., Fang, F., Zhang, X. and Zhang, M. Q. (2007) Putative zinc finger protein binding sites are over-represented in the boundaries of methylation-resistant CpG islands in the human genome. PLoS ONE 2, e1184.
Bock, C., Paulsen, M., Tierling, S., Mikeska, T., Lengauer, T. and Walter, J. (2006) CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet. 2, e26.
Li, E. and Bird, A. (2007) DNA methylation In mammals. In: Epigenetics, pp. 341–356, Allis, C., Jenuwein, T., Reinberg, D. and Caparros, M. (eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
Feinberg, A. P. and Vogelstein, B. (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301, 89–92.
Issa, J. P. and Baylin, S. B. (1996) Epigenetics and human disease. Nat. Med. 2, 281–282.
De Smet, C., De Backer, O., Faraoni, I., Lurquin, C., Brasseur, F. and Boon, T. (1996) The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc. Natl. Acad. Sci. USA 93, 7149–7153.
Gisselsson, D., Shao, C., Tuck-Muller, C. M., Sogorovic, S., Palsson, E., Smeets, D. and Ehrlich, M. (2005) Interphase chromosomal abnormalities and mitotic missegregation of hypomethylated sequences in ICF syndrome cells. Chromosoma 114, 118–126.
Luger, K., Mader,A.W., Richmond, R. K., Sargent,D. F. and Richmond, T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260.
Kouzarides, T. (2007) Chromatin modifications and their function. Cell 128, 693–705.
Santos-Rosa, H. and Caldas, C. (2005) Chromatin modifier enzymes, the histone code and cancer. Eur. J. Cancer 41, 2381–2402.
Fischle, W., Wang, Y., Jacobs, S. A., Kim, Y., Allis, C. D. and Khorasanizadeh, S. (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17, 1870–1881.
Pray-Grant, M.G., Daniel, J. A., Schieltz, D., Yates, J. R. 3rd, and Grant, P. A. (2005) Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature 433, 434–438.
Covic, M., Hassa, P. O., Saccani, S., Buerki, C., Meier, N. I., Lombardi, C., Imhof, R., Bedford, M. T., Natoli, G. and Hottiger, M.O. (2005) Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J. 24, 85–96.
Fabbrizio, E., El Messaoudi, S., Polanowska, J., Paul, C., Cook, J. R., Lee, J. H., Negre, V., Rousset, M., Pestka, S., Le Cam, A. and Sardet, C. (2002) Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3, 641–645.
Tachibana, M., Sugimoto, K., Nozaki, M., Ueda, J., Ohta, T., Ohki, M., Fukuda, M., Takeda, N., Niida, H., Kato, H. and Shinkai, Y. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791.
Yuan, X., Feng, W., Imhof, A., Grummt, I. and Zhou, Y. (2007) Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol. Cell 27, 585–595.
Chowdhury, D., Keogh, M. C., Ishii, H., Peterson, C. L., Buratowski, S. and Lieberman, J. (2005) gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 20, 801–809.
Botuyan, M. V., Lee, J., Ward, I. M., Kim, J. E., Thompson, J. R., Chen, J. and Mer, G. (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373.
Doyon, Y., Cayrou, C., Ullah, M., Landry, A. J., Cote, V., Selleck,W., Lane, W. S., Tan, S., Yang, X. J. and Cote, J. (2006) ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 21, 51–64.
Iizuka, M., Sarmento, O. F., Sekiya, T., Scrable, H., Allis, C.D. and Smith, M. M. (2008) Hbo1 Links p53-dependent stress signaling to DNA replication licensing. Mol. Cell. Biol. 28, 140–153.
Fischle, W., Tseng, B. S., Dormann, H. L., Ueberheide, B.M., Garcia, B. A., Shabanowitz, J., Hunt, D. F., Funabiki, H. and Allis, C. D. (2005) Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122.
Goll, M. G. and Bestor, T. H. (2005) Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514.
Klose, R. J. and Bird, A. P. (2006) Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97.
Bestor, T. H. (1992) Activation of mammalian DNA methyltransferase by cleavage of a Zn binding regulatory domain. EMBO J. 11, 2611–2617.
Hirasawa, R., Chiba, H., Kaneda, M., Tajima, S., Li, E., Jaenisch, R. and Sasaki, H. (2008) Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 22, 1607–1616.
Schaefer, C. B., Ooi, S. K., Bestor, T. H. and Bourc’his, D. (2007) Epigenetic decisions inmammalian germ cells. Science 316, 398–399.
Cheng, X. and Blumenthal, R. M. (2008) Mammalian DNA methyltransferases: A structural perspective. Structure 16, 341–350.
Qiu, C., Sawada, K., Zhang, X. and Cheng, X. (2002) The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 9, 217–224.
Ooi, S. K., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z., Erdjument-Bromage, H., Tempst, P., Lin, S. P., Allis, C. D., Cheng, X. and Bestor, T. H. (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717.
Jia, D., Jurkowska, R.Z., Zhang, X., Jeltsch, A. and Cheng, X. (2007) Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251.
Cedar, H. and Verdine, G. L. (1999) Gene expression. The amazing demethylase. Nature 397, 568–569.
Morgan, H. D., Dean, W., Coker, H. A., Reik, W. and Petersen-Mahrt, S. K. (2004) Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J. Biol. Chem. 279, 52353–52360.
Metivier, R., Gallais, R., Tiffoche, C., Le Peron, C., Jurkowska, R. Z., Carmouche, R. P., Ibberson, D., Barath, P., Demay, F., Reid, G., Benes, V., Jeltsch, A., Gannon, F. and Salbert, G. (2008) Cyclical DNA methylation of a transcriptionally active promoter. Nature 452, 45–50.
Bhaumik, S. R., Smith, E. and Shilatifard, A. (2007) Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 14, 1008–1016.
Allis, C. D., Berger, S. L., Cote, J., Dent, S., Jenuwien, T., Kouzarides, T., Pillus, L., Reinberg, D., Shi, Y., Shiekhattar, R., Shilatifard, A., Workman, J. and Zhang, Y. (2007) New nomenclature for chromatin-modifying enzymes. Cell 131, 633–636.
Shi, Y. and Whetstine, J. R. (2007) Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 25, 1–14.
Cuthbert, G. L., Daujat, S., Snowden, A. W., Erdjument-Bromage, H., Hagiwara, T., Yamada, M., Schneider, R., Gregory, P. D., Tempst, P., Bannister, A. J. and Kouzarides, T. (2004) Histone deimination antagonizes arginine methylation. Cell 118, 545–553.
Huang, J. and Berger, S. L. (2008) The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. 18, 152–158.
Saha, A., Wittmeyer, J. and Cairns, B. R. (2006) Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 7, 437–447.
Kwon, C. S. and Wagner, D. (2007) Unwinding chromatin for development and growth: A few genes at a time. Trends Genet. 23, 403–412.
Ruthenburg, A. J., Li, H., Patel, D. J. and Allis, C. D. (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell. Biol. 8, 983–994.
Jacobs, S. A. and Khorasanizadeh, S. (2002) Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 295, 2080–2083.
Smallwood, A., Esteve, P. O., Pradhan, S. and Carey, M. (2007) Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 21, 1169–1178.
Fuks, F., Hurd, P. J., Deplus, R. and Kouzarides, T. (2003) The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 31, 2305–2312.
Wysocka, J., Swigut, T., Xiao, H., Milne, T. A., Kwon, S. Y., Landry, J., Kauer, M., Tackett, A. J., Chait, B. T., Badenhorst, P., Wu, C. and Allis, C. D. (2006) A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442, 86–90.
Drewell, R. A., Goddard, C. J., Thomas, J. O. and Surani, M. A. (2002) Methylation-dependent silencing at the H19 imprinting control region by MeCP2. Nucleic Acids Res. 30, 1139–1144.
Kondo, E., Gu, Z., Horii, A. and Fukushige, S. (2005) The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol. Cell. Biol. 25, 4388–4396.
Bostick, M., Kim, J. K., Esteve, P. O., Clark, A., Pradhan, S. and Jacobsen, S. E. (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317, 1760–1764.
Sharif, J., Muto, M., Takebayashi, S., Suetake, I., Iwamatsu, A., Endo, T. A., Shinga, J., Mizutani-Koseki, Y., Toyoda, T., Okamura, K., Tajima, S., Mitsuya, K., Okano, M. and Koseki, H. (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912.
Filippova, G. N. (2008) Genetics and epigenetics of the multifunctional protein CTCF. Curr. Top. Dev. Biol. 80, 337–360.
Wallace, J. A. and Felsenfeld, G. (2007) We gather together: Insulators and genome organization. Curr. Opin. Genet. Dev. 17, 400–407.
Ringrose, L. and Paro, R. (2007) Polycomb/trithorax response elements and epigenetic memory of cell identity. Development 134, 223–232.
Boyer, L. A., Plath, K., Zeitlinger, J., Brambrink, T., Medeiros, L. A., Lee, T. I., Levine, S. S., Wernig, M., Tajonar, A., Ray, M. K., Bell, G.W., Otte, A. P., Vidal, M., Gifford, D. K., Young, R.A. and Jaenisch, R. (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353.
Lee, T. I., Jenner, R. G., Boyer, L. A., Guenther, M. G., Levine, S. S., Kumar, R. M., Chevalier, B., Johnstone, S. E., Cole, M. F., Isono, K., Koseki, H., Fuchikami, T., Abe, K., Murray, H. L., Zucker, J. P., Yuan, B., Bell, G. W., Herbolsheimer, E., Hannett, N. M., Sun, K., Odom, D. T., Otte, A. P., Volkert, T. L., Bartel, D. P., Melton, D. A., Gifford, D. K., Jaenisch, R. and Young, R. A. (2006) Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125, 301–313.
Schwartz, Y. B. and Pirrotta, V. (2007) Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 8, 9–22.
Kohler, C. and Villar, C. B. (2008) Programming of gene expression by Polycomb group proteins. Trends Cell Biol. 18, 236–243.
Cao, R., Tsukada, Y. and Zhang, Y. (2005) Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell 20, 845–854.
Schlesinger, Y., Straussman, R., Keshet, I., Farkash, S., Hecht, M., Zimmerman, J., Eden, E., Yakhini, Z., Ben-Shushan, E., Reubinoff, B. E., Bergman, Y., Simon, I. and Cedar, H. (2007) Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 39, 232–236.
Ballas, N. and Mandel, G. (2005) The many faces of REST oversee epigenetic programming of neuronal genes. Curr. Opin. Neurobiol. 15, 500–506.
Ballas, N., Grunseich, C., Lu, D.D., Speh, J.C. and Mandel, G. (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657.
Heard, E. and Disteche, C.M. (2006) Dosage compensation in mammals: Fine-tuning the expression of the X chromosome. Genes Dev. 20, 1848–1867.
Wutz, A. and Gribnau, J. (2007) X inactivation Xplained. Curr. Opin. Genet. Dev. 17, 387–393.
Augui, S., Filion, G. J., Huart, S., Nora, E., Guggiari, M., Maresca, M., Stewart, A. F. and Heard, E. (2007) Sensing X chromosome pairs before X inactivation via a novel X-pairing region of the Xic. Science 318, 1632–1636.
Monkhorst, K., Jonkers, I., Rentmeester, E., Grosveld, F. and Gribnau, J. (2008) X inactivation counting and choice is a stochastic process: Evidence for involvement of an X-linked activator. Cell 132, 410–421.
Lee, J. T., Davidow, L. S. and Warshawsky, D. (1999) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 21, 400–404.
Keohane, A.M., O’Neill L, P., Belyaev, N. D., Lavender, J. S. and Turner, B. M. (1996) X-Inactivation and histone H4 acetylation in embryonic stem cells. Dev. Biol. 180, 618–630.
Heard, E., Rougeulle, C., Arnaud, D., Avner, P., Allis, C. D. and Spector, D. L. (2001) Methylation of histoneH3at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 107, 727–738.
Plath, K., Fang, J., Mlynarczyk-Evans, S. K., Cao, R., Worringer, K. A., Wang, H., de la Cruz, C. C., Otte, A. P., Panning, B. and Zhang, Y. (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135.
Kohlmaier, A., Savarese, F., Lachner, M., Martens, J., Jenuwein, T. and Wutz, A. (2004) A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol. 2, E171.
de Napoles, M., Mermoud, J. E., Wakao, R., Tang, Y. A., Endoh, M., Appanah, R., Nesterova, T. B., Silva, J., Otte, A. P., Vidal, M., Koseki, H. and Brockdorff, N. (2004) Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 7, 663–676.
Costanzi, C. and Pehrson, J. R. (1998) Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393, 599–601.
Hellman, A. and Chess, A. (2007) Gene body-specific methylation on the active X chromosome. Science 315, 1141–1143.
Sado, T., Fenner,M. H., Tan, S. S., Tam, P., Shioda, T. and Li, E. (2000) X inactivation in the mouse embryo deficient for Dnmt1: Distinct effect of hypomethylation on imprinted and random X inactivation. Dev. Biol. 225, 294–303.
Takagi, N., Sugawara, O. and Sasaki, M. (1982) Regional and temporal changes in the pattern of X-chromosome replication during the early post-implantation development of the female mouse. Chromosoma 85, 275–286.
Chaumeil, J., Le Baccon, P., Wutz, A. and Heard, E. (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev. 20, 2223–2237.
Barlow, D. and Bartolomei, M. (2007) Genomic imprining in mammals. In: Epigenetics, pp. 357–375, Allis, C., Jenuwein, T., Reinberg, D. and Caparros, M. (eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
Mouse imprinting Data and References, http://www.mgu.-har.mrc.ac.uk/research/imprinting/function.html.
Edwards, C. A. and Ferguson-Smith, A. C. (2007) Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 19, 281–289.
Lyle, R., Watanabe, D., te Vruchte,D., Lerchner, W., Smrzka, O. W., Wutz, A., Schageman, J., Hahner, L., Davies, C. and Barlow, D. P. (2000) The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nat. Genet. 25, 19–21.
Zwart, R., Sleutels, F., Wutz, A., Schinkel, A. H. and Barlow, D. P. (2001) Bidirectional action of the Igf2r imprint control element on upstream and downstream imprinted genes. Genes Dev. 15, 2361–2366.
Spahn, L. and Barlow,D. P. (2003)AnICE pattern crystallizes. Nat. Genet. 35, 11–12.
Kaneda, M., Okano, M., Hata, K., Sado, T., Tsujimoto,N., Li, E. and Sasaki, H. (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903.
Bourc’his, D., Xu,G. L., Lin, C. S., Bollman, B. and Bestor, T. H. (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294, 2536–2539.
117 Bourc’his, D. and Bestor, T.H. (2004) Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431, 96–99.
Li, E., Beard, C. and Jaenisch, R. (1993) Role for DNA methylation in genomic imprinting. Nature 366, 362–365.
Howell, C. Y., Bestor, T. H., Ding, F., Latham, K. E., Mertineit,C., Trasler, J. M. and Chaillet, J. R. (2001) Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104, 829–838.
Mager, J., Montgomery, N.D., de Villena, F. P. and Magnuson, T. (2003) Genome imprinting regulated by the mouse Polycomb group protein Eed. Nat. Genet. 33, 502–507.
Wagschal, A., Sutherland, H. G., Woodfine, K., Henckel, A., Chebli, K., Schulz, R., Oakey, R. J., Bickmore, W.A. and Feil, R. (2008) G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol. Cell. Biol. 28, 1104–1113.
Pauler, F. M., Koerner, M. V. and Barlow, D. P. (2007) Silencing by imprinted noncoding RNAs: Is transcription the answer? Trends Genet. 23, 284–292.
Bell, A. C. and Felsenfeld, G. (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485.
Engel, N., Thorvaldsen, J. L. and Bartolomei, M. S. (2006) CTCF binding sites promote transcription initiation and prevent DNA methylation on the maternal allele at the imprinted H19/Igf2 locus. Hum. Mol. Genet. 15, 2945–2954.
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., McKernan, K., Meldrim, J., Mesirov, J. P., Miranda, C., Morris, W., Naylor, J., Raymond, C., Rosetti, M., Santos, R., Sheridan, A., Sougnez, C., Stange-Thomann, N., Stojanovic, N., Subramanian, A., Wyman, D., Rogers, J., Sulston, J., Ainscough, R., Beck, S. et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921.
Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., Antonarakis, S. E., Attwood, J., Baertsch, R., Bailey, J., Barlow,K., Beck, S., Berry, E., Birren, B., Bloom, T., Bork, P., Botcherby, M., Bray, N., Brent, M. R., Brown, D. G., Brown, S. D., Bult, C., Burton, J. et al. (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562.
Brouha,B., Schustak, J., Badge, R.M., Lutz-Prigge, S., Farley, A. H., Moran, J. V. and Kazazian, H. H. Jr. (2003) Hot L1 s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 100, 5280–5285.
Deininger, P. L., Moran, J. V., Batzer, M. A. and Kazazian, H. H. Jr. (2003) Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 13, 651–658.
Hasler, J. and Strub, K. (2006) Alu elements as regulators of gene expression. Nucleic Acids Res. 34, 5491–5497.
Rogozin, I.B., Mayorov, V. I., Lavrentieva, M.V., Milanesi, L. and Adkison, L. R. (2000) Prediction and phylogenetic analysis of mammalian short interspersed elements (SINEs). Brief. Bioinform. 1, 260–274.
Yoder, J. A., Walsh, C. P. and Bestor, T. H. (1997) Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13, 335–340.
Collier, L. S. and Largaespada, D. A. (2007) Transposable elements and the dynamic somatic genome. Genome Biol. 8Suppl 1, S5.
Kazazian, H. H. Jr. (1999) An estimated frequency of endogenous insertional mutations in humans. Nat. Genet. 22, 130.
Lavie, L., Kitova, M., Maldener, E., Meese, E. and Mayer, J. (2005) CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERVK(HML-2). J. Virol. 79, 876–883.
Martens, J. H., O’sullivan, R. J., Braunschweig, U., Opravil, S., Radolf, M., Steinlein, P. and Jenuwein, T. (2005) The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 24, 800–812.
Rae, P. M. and Steele, R. E. (1979) Absence of cytosine methylation at C-C-G-G and G-C-G-C sites in the rDNA coding regions and intervening sequences of Drosophila and the rDNA of other insects. Nucleic Acids Res. 6, 2987–2995..
Walsh, C. P., Chaillet, J. R. and Bestor, T. H. (1998) Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20, 116–117..
Wilson, A. S., Power, B. E. and Molloy, P. L. (2007) DNA hypomethylation and human diseases. Biochim. Biophys. Acta 1775, 138–162..
Steinhoff, C. and Schulz, W. A. (2003) Transcriptional regulation of the human LINE-1 retrotransposon L1.2B. Mol. Genet. Genomics 270, 394–402..
Takai, D., Yagi, Y., Habib, N., Sugimura, T. and Ushijima, T. (2000) Hypomethylation of LINE1 retrotransposon in human hepatocellular carcinomas, but not in surrounding liver cirrhosis. Jpn. J. Clin. Oncol. 30, 306–309..
Roman-Gomez, J., Jimenez-Velasco, A., Agirre, X., Cervantes, F., Sanchez, J., Garate, L., Barrios, M., Castillejo, J. A., Navarro, G., Colomer, D., Prosper, F., Heiniger, A. and Torres, A. (2005) Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene 24, 7213–7223..
Kato, M., Takashima, K. and Kakutani, T. (2004) Epigenetic control of CACTA transposon mobility in Arabidopsis thaliana. Genetics 168, 961–969..
Yan, Q., Cho, E., Lockett, S. and Muegge, K. (2003) Association of Lsh, a regulator of DNA methylation, with pericentromeric heterochromatin is dependent on intact heterochromatin. Mol. Cell. Biol. 23, 8416–8428..
144 miRBase:: Sequences, Sanger Institute, Wellcome Trust. http://microrna.sanger.ac.uk/sequences/index.shtml..
Qi, Y., He, X., Wang, X. J., Kohany, O., Jurka, J. and Hannon, G. J. (2006) Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature 443, 1008–1012..
Weinberg,M. S., Villeneuve, L.M., Ehsani, A., Amarzguioui, M., Aagaard, L., Chen, Z. X., Riggs, A. D., Rossi, J. J. and Morris, K. V. (2006) The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA 12, 256–262..
Sarraf, S. A. and Stancheva, I. (2004) Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 15, 595–605.
Rountree, M. R., Bachman, K. E. and Baylin, S. B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277.
Schermelleh, L., Haemmer, A., Spada, F., Rosing, N., Meilinger, D., Rothbauer, U., Cardoso, M. C. and Leonhardt, H. (2007) Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 35, 4301–4312.
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., Heine-Suner, D., Cigudosa, J. C., Urioste, M., Benitez, J., Boix-Chornet, M., Sanchez-Aguilera, A., Ling, C., Carlsson, E., Poulsen, P., Vaag, A., Stephan, Z., Spector, T. D., Wu, Y. Z., Plass, C. and Esteller, M. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 102, 10604–10609.
Acknowledgement
We are grateful to Drs. D. G. Comb and Rich Roberts at New England Biolabs, Inc. for their support and encouragement. The authors would like to apologize, as a vast expanse of exciting works could not be cited due to the lack of space. Constructive comments from Drs. Pierre-Olivier Estève and Jim Samelson are highly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kim, J.K., Samaranayake, M. & Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 66, 596 (2009). https://doi.org/10.1007/s00018-008-8432-4
Published:
DOI: https://doi.org/10.1007/s00018-008-8432-4