
Abstract.
Glucocorticoids (GCs) are a class of steroid hormones which regulate a variety of essential biological functions. The profound anti-inflammatory and immunosuppressive activity of synthetic GCs, combined with their power to induce lymphocyte apoptosis place them among the most commonly prescribed drugs worldwide. Endogenous GCs also exert a wide range of immunomodulatory activities, including the control of T cell homeostasis. Most, if not all of these effects are mediated through the glucocorticoid receptor, a member of the nuclear receptor superfamily. However, the signaling pathways and their cell type specificity remain poorly defined. In this review, we summarize our present knowledge on GC action, the mechanisms employed to induce apoptosis and the currently discussed models of how they may participate in thymocyte development. Although our knowledge in this field has substantially increased during recent years, we are still far from a comprehensive picture of the role that GCs play in T lymphocytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
T cells form a major branch of the acquired immune system [1]. They derive their name from the thymus, where they develop to immunocompetent T lymphocytes through a series of lineage and selection steps. This ensures that all cells which lack a functional T cell receptor (TCR) or bear a TCR with useless or dangerous specificity are deleted [2]. In the periphery, mature T cells undergo clonal expansion upon encountering their cognate antigen. Once the invading pathogen is cleared, the superfluous lymphocytes are removed in a process known as activation-induced cell death (AICD) [3]. Thus, both during thymocyte development and T cell-mediated immune responses, it is essential that cells are deleted in a controlled manner to prevent immunopathologies such as autoimmunity and cancer. This is achieved by a process called apoptosis [4] which, among other things, involves a series of proteolytic events that result in characteristic morphological alterations and ultimately lead to cell death.
Glucocorticoids (GCs) are a class of steroid hormones which display potent immunomodulatory activities including the ability to induce T lymphocyte apoptosis [5]. Although this was one of the first recognized forms of programmed cell death, it is still poorly understood [6]. GCs exert most, if not all, of their effects through binding to the glucocorticoid receptor (GR), a ligand-activated transcription factor [7]. While GR-mediated gene activation is clearly an essential component of the apoptotic pathway [8], the downstream events are debatable. Although GC-induced cell death does not directly proceed via one of the two classical apoptotic pathways, Bcl-2 proteins and caspases still appear to be involved in this process [5, 9, 10]. Furthermore, the physiological role of GC-induced T cell apoptosis is also not fully resolved. GCs have been hypothesized to direct positive and negative selection in the thymus [11], to limit AICD during the contraction phase of an adaptive immune response [12] and to induce generalized thymocyte apoptosis after polyclonal T cell activation [13]. This has been assigned to effects of both adrenally derived hormones as well as GCs synthesized within the thymus itself. Thus, not only the mechanism but also the physiological function of GC-induced apoptosis requires further investigation. Owing to their ability to mediate apoptosis in leukemia, lymphoma and myeloid cells, synthetic GCs are among the most widely prescribed drugs for the treatment of hematological maligancies [10, 14]. Consequently, a better understanding of GC-induced apoptosis is not only of theoretical interest but is also urgently required to improve the suitability of these drugs for clinical therapy. In this review we try to provide a broad picture of how GCs contribute to the development and apoptosis of thymocytes and mature T cells and the molecular mechanisms, signaling pathways and enzymatic systems that may be utilized during their action. Furthermore, we summarize how the generation and analysis of transgenic, knock-out and knock-in mice has contributed to our present knowledge about these processes [15]. We hope this provides the basis for a better understanding of this important class of steroid hormones and their role in T cell immunology.
The molecular and physiological basis of GC action
The glucocorticoid receptor
The GR belongs to the nuclear receptor superfamily which are characterized by their common arrangement of functionally distinct domains mediating transactivation, DNA binding, nuclear localization, dimerization and ligand recognition [16]. In the absence of hormone, the GR is found within a multimeric complex of heat shock proteins and immunophilins in the cytoplasm [17] (fig. 1). GCs such as cortisol in humans and corticosterone in rodents diffuse passively into the cell, where they bind to the GR [7]. Following dissociation from the heat shock protein complex, the GR translocates into the nucleus. There it recognizes imperfect palindromic sequences, so-called glucocorticoid response elements (GREs), present in the promoter and enhancer regions of a variety of genes [18]. The GR can drive transcription from these response elements by using its surfaces as platforms for the docking of transcriptional coactivators that are capable of altering the local chromatin or recruiting and stabilizing the transcription machinery [19].

Molecular modes of GR action. GCs passively diffuse into the cell and bind to the GR (1). This results in the dissociation of the heat shock protein complex (Hsps) (2) and translocation of the ligand-bound GR into the nucleus. There the GR modulates transcription either by binding to DNA (3) or via interaction with other transcription factors (4). Non-genomic mechanisms of GR action include interference with cytosolic signaling molecules (5).
Although many genes are known to be upregulated in response to GCs, including some important to the activation, function and apoptosis of immune cells, such as Bcl-xL [20], IkB [21], GILZ [22] and GITR [23], there is scant evidence that they are directly upregulated through GREs. Genes which have been shown to be positively regulated through well-defined GREs in vivo include tyrosine amino transferase [24] and phosphoenolpyruvate carboxykinase [25]. In contrast to these DNA binding-dependent activities, the GR also regulates gene expression by interfering with other transcription factors such as NF-kB [26], AP-1 [27], NF-AT [28], CREB [29] and Stat5 [30]. Intriguingly, the GR can control gene expression in this way, mostly in a transrepressing manner, without binding to DNA itself (fig. 1). This was most convincingly demonstrated by introducing a point mutation into the GR which prevents it from both dimerizing and binding DNA [8]. When present in the germline of knock-in mice (GRdim), the mutated GR was unable to mediate transcription from GREs but its ability to regulate transcription from AP-1-, NF-kB- and Stat5-driven promoters was not compromised [31–34]. In addition to its established effects on transcription, evidence is growing that the GR can also play a role in cytosolic signaling pathways, including the activation of PI3 kinase [35].
It has also been recently recognized that multiple N-terminal isoforms are generated from the GR gene by translational mechanisms [36]. Importantly, these isoforms appear to regulate different sets of genes, suggesting that they are not functionally equivalent. Given the tissue-specific distribution of the various isoforms, it will be intriguing to see whether they are differentially involved in immunoregulatory functions in the same way that the use of alternative promoters of the GR has been shown to correlate with the sensitivity of T lymphocytes to apoptosis [37, 38]. Taken together, differential transcription and translation events give rise to a plethora of GR species which control cellular processes by several different mechanisms [39]. This in turn forms the basis for the variety of activities that GCs exert in the immune system.
Control of GC secretion and synthesis
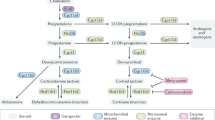
The primary site of GC synthesis is the adrenal gland, which was first described in the 16th century by the Italian anatomist Eustachi [40]. However, only in 1855 did Addison realize the importance of this organ for survival. In the first half of the 20th century, scientists finally discovered the importance of cortical hormones which, in the 1940s, were shown not only to be involved in the stress response but also to exert potent anti-inflammatory activity [41].
GC synthesis is under the control of the hypothalamus-pituitary-adrenal (HPA) axis, which controls hormone levels in the serum [42]. This mechanism involves the release of corticotrophin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) in response to stress and elevated cytokine levels which results in GC secretion from the adrenal gland. GCs ultimately limit the activity of the HPA axis at the level of the hypothalamus and the pituitary, thus establishing a negative feedback loop. Superimposed on that, GC production also follows a circadian rhythm, characterized in humans by high levels in the morning and low levels in the evening [43]. GCs have also been hypothesized to be produced in other tissues but the evidence for this remains scarce (see below). Collectively, GC action is controlled on multiple levels by the neuroendocrine as well as the immune system itself.
Animal models investigating GR function in T cells
Loss-of-function mutations
During the last decade, a considerable number of genetically modified mouse strains have been generated to study the role of the GR in various physiological settings and anatomical locations [15], including the development and function of T lymphocytes (table 1). The first approach of this kind was described in 1995 when Ashwell and colleagues expressed an antisense fragment to the GR under the control of the proximal lck promoter [44]. GR mRNA and protein expression were reduced by around half, resulting in a lower thymic cellularity and a partial resistance to GC-induced thymocyte apoptosis. Intriguingly, analysis of a second strain of transgenic mice which was generated by Okret and collegues using a largely similar construct arrived at exactly the opposite conclusion [45]. In particular, the total thymocyte number in this experiment was found to be elevated instead of diminished.
Analysis of knock-out mice is generally considered more reliable than using antisense transgenic animals since deletion should be complete and unspecific effects reduced. GR knock-out mice were first reported by Schütz and colleagues in 1995 [46]. The initial analysis of these mutants revealed that they die perinataly due to lung atelectasis. It took several years before the thymic phenotype of these mice was investigated using fetal mice and bone marrow chimeras. Thymocytes derived from these knock-out mice were found to be completely resistant to GC-induced apoptosis but otherwise no significant differences in cell number, subtype distribution and thymic selection were observed [47, 48]. Muglia and colleagues described another strain of GR-deficient mice, confirming the lack of any role for the GR in the thymus beyond inducing apoptosis [13]. Recently, two strains of T cell-specific GR knock-out mice were generated by independent laboratories. Like GR-deficient mice, these mutants turned out to be resistant to GC-induced apoptosis while having normal thymic cellularity and subtype distribution [12, 49]. As mentioned earlier, the GR modulates transcription through two principal modes of action. These can be dissected using the previously described GRdim mice which are impaired in their ability to transactivate genes from GRE-containing promoters [8]. While no effect on T cell development was detected, thymocytes from these knock-in mice were refractory to GC-induced cell death in vitro (table 1). This confirmed that the DNA-binding activity of the GR is dispensable for T cell development but essential for thymocyte apoptosis. Collectively, the analysis of all five GR mutant mouse strains has consistently failed to demonstrate a role for the receptor in T cell development and thymic selection which contrasts with the observations made in antisense transgenic mice (table 1). In conclusion, GCs, at least when acting via the classical GR, seem unlikely to be involved in these processes while their role in inducing thymocyte apoptosis is beyond question.
Gain-of-function mutations
Two mouse strains displaying increased GR signaling have been described in recent years (table 1). The first approach aimed at doubling the GR gene dosage by introducing a large DNA fragment covering the whole genomic locus into the germline of mice [50]. In accordance with the increased GR expression, transgenic thymocytes were more sensitive to GC-induced apoptosis than control cells. This indicates that apoptosis critically depends on the amount of GR protein in the cell. In the second transgenic mouse line, GR overexpression was targeted to developing thymocytes by expressing the GR under the control of the proximal lck promoter [45]. Overexpression was accompanied by increased sensitivity to GC-induced apoptosis, lower recovery of thymocytes and reduced numbers of peripheral T cells. The same group recently reported the generation of another mouse strain characterized by conditional GR expression in thymocytes and T cells [51]. Upon induction of the GR, thymic cellularity was reduced, both in the presence and absence of adrenally derived GCs, although no influence on subtype distribution could be demonstrated (table 1). This was interpreted as a proof for GCs of thymic origin, a conclusion which is not supported by our own studies [unpublished data].
Apoptosis
The concept of apoptosis and its function in the immune system
Apoptosis is defined by a series of molecular and morphological events involving the loss of mitochondrial membrane potential, chromatin condensation, DNA fragmentation, membrane blebbing and the generation of apoptotic vesicles (fig. 2). Mediated by executioner caspases, this process culminates in the orchestrated disassembly and phagocytosis of the dying cell [52].

The major pathways of lymphocyte apoptosis. The ‘intrinsic’ pathway involves the activation of ‘BH3-only’ molecules (Bad, Bim etc.) which in turn activate the ‘multidomain’ proteins Bax and Bak. This leads to the formation of the ‘apoptosome’ and activation of caspase-3, a process which is counteracted by the anti-apoptotic proteins Bcl-2 and Bcl-xL. The ‘extrinsic’ pathway is initiated by oligomerization of death receptors followed by caspase-8 activation and also converges on caspase-3. An alternative pathway induced by lysosomal stress involves the release of cathepsins.
Apoptosis is essential for the development and maintenance of the immune system [4]. It may be initiated because the developing lymphocytes fail to express a productively rearranged antigen receptor or alternatively because they produce a receptor displaying too weak or too strong of an affinity for MHC/self-peptide [1]. These selection steps ensure proper recognition of a broad range of foreign antigens and help to achieve tolerance and prevent autoimmune diseases. Apoptosis is not only critical to the establishment of the immune system but also plays an important role in the termination of adaptive immune responses [3]. During the contraction phase, clonally expanded antigen-specific lymphocytes are rapidly removed by AICD, a process induced by TCR triggering and cytokine withdrawl. This serves to prevent uncontrolled proliferation of activated lymphocytes and thereby avoids immunopathology and autoimmune disorders.
The pathways of apoptosis
Lymphocytes are known to undergo two distinct apoptotic pathways which have been well characterized in recent years [4] (fig. 2). One is initiated by cellular stress or high-affinity ligation of antigen receptors during negative selection of autoreactive lymphocytes. This so-called ‘intrinsic’ pathway is regulated by the interplay of pro- and anti-apoptotic members of the Bcl-2 family at the level of the mitochondria [53]. The second mechanism, also known as the ‘extrinsic’ apoptotic pathway, is activated by ligation of death receptors and occurs, for example, during AICD [3]. These two pathways share common downstream components such as certain caspases, a family of aspartate-specific proteases.
Initiation of the ‘intrinsic’ pathway involves the release of apoptotic mediators from the mitochondria following disruption of its outer membrane [54]. This is tightly regulated by the balance between pro- and anti-apoptotic Bcl-2 family members (fig. 2). Pro-apoptotic factors of the so-called ‘BH3-only’ family such as Bim, Bid, Bad, Puma and Noxa serve as conduits of apoptotic stimuli by activating the ‘multi-domain’ family members Bax and Bak [55, 56]. This process is opposed by anti-apoptotic Bcl-2 family members such as Bcl-2 and Bcl-xL which bind and neutralize their pro-apoptotic counterparts [53, 57–60]. Following formation of pores in the outer mitochondrial membrane by Bax and Bak, cytochrome c is released, resulting in the formation of the so-called ‘apoptosome’ (fig. 2). This multimeric complex containing caspase-9 and Apaf1 activates caspase-3 which finally leads to apoptosis [52].
The ‘extrinsic’ apoptotic pathway is initiated by the ligation, oligomerization and subsequent activation of death receptors as typified by Fas [61] and results in the activation of caspase-8 (fig. 2). Depending on the cell type, subsequent activation of caspase-3 occurs either directly or requires an additional amplification loop involving cleavage of the pro-apoptotic factor Bid and destabilization of the mitochondrial membrane. However, irrespective of which biochemical events are involved, the ‘intrinsic’ and the ‘extrinsic’ pathway both converge on the activation of caspase-3, the point of no return in most cell death cascades (fig. 2).
Although the two aforementioned pathways are considered to explain most forms of apoptosis, evidence for alternative pathways is accumulating [62, 63]. One postulated mechanism involves the destabilization of lysosomal membranes and subsequent release of cathepsins B and D [64] (fig. 2). This results not only in generalized proteolysis but also causes disruption of the outer mitochondrial membrane and caspase-3 activation [65–68]. A second unconventional pathway involves the release of death-inducing factors from the mitochondria such as AIF and endonuclease G, which are capable of initiating cell death independently of caspases [63].
The mechanism of GC-induced apoptosis
Initiation of GC-induced apoptosis. The current model implies that GC-induced apoptosis requires the presence of a functional GR and in particular its transactivating function [8,13, 15, 47, 50]. Although most data suggest that initiation of cell death by GCs is linked to de novo gene expression, it is noteworthy that some events may also involve so-called non-genomic actions of the GR (fig. 3).

Cellular processes involved in GC-induced apoptosis. Bcl- 2 family: transcription of Bim and Puma is upregulated; caspases: caspase-3, -8 and -9 are activated. lysosomes: cathepsin B is released. proteasomal degradation: c-IAP1 and XIAP are degraded at the protein level; H2O2 levels are increased; ceramides are produced, and Na+/ K+ levels altered. The IP3 receptor is engaged. Kinases: PKC, Raf and 14-3-3 proteins interact with the GR.
During the last couple of years several approaches to identify GC-induced genes have been undertaken, but only a few convincing candidates were identified. Genes described to be up- or downregulated during GC-induced apoptosis include c-myc [69], tdag8 [70, 71], dig2 [72], Bim [73, 74] and PUMA [75]. Furthermore, large-scale expression analysis of GC-regulated genes has only been reported in cell lines so far. Both Tonko et al. [76] and Medh et al. [77] have studied GC-induced gene regulation in leukemic lymphoid CEM cells, whereas Wang et al. [73] have performed a large-scale analysis examining the two murine lymphoma cell lines S49.A2 and WEHI 7.2 [73]. The most promising gene identified in these screens is the pro-apoptotic protein Bim, but its function in this process still requires further investigation (see below).
The role of Bcl-2 proteins in GC-induced apoptosis
A plethora of experiments have implicated pro- and anti-apoptotic Bcl-2 family members in GC-induced apoptosis. Mice reconstituted with fetal liver cells from Bcl-2 transgenic mice [78, 79] as well as Bcl-2-overexpressing murine lymphoma cells [80, 81] are protected from GC-induced cell death, indicating that the disruption of mitochondrial integrity is essential to GC-induced apoptosis [82]. Studies carried out in murine T hybridoma cells suggest that Bcl-2 overexpression also blocks the release of both cytochrome c and AIF in response to GCs [83]. The importance of Bcl-2 is further underscored by the analysis of Bcl-2-deficient mice which display fulminant lymphoid apoptosis in vivo and enhanced cell death of thymocytes in vitro after GC treatment [84]. Collectively, these data suggest that the level of Bcl-2 determines the sensitivity to GCs. Since Bcl-2 is known to localize and act in mitochondria as well as the endoplasmatic reticulum, both organelles might play an important role in GC-induced apoptosis [58].
The role of Bcl-xL in this process is less clear. Bcl-xL becomes redistributed to the mitochondria following GC treatment which can be prevented by inhibitors of translation [85, 86]. Overexpression of Bcl-xL in a T hybridoma cell line can block GC-induced apoptosis [80] and sustained Bcl-xL expression was found to be a prerequisite for the resistance of single-positive thymocytes to GC-induced apoptosis in vivo [87]. Thus, Bcl-xL contributes to determining the sensitivity of lymphocytes to GC-induced apoptosis although this might be limited to certain developmental stages of T lymphocytes. Since Bcl-xL-deficient mice die around embryonic day 13 [88] and neither mice reconstituted with fetal liver cells nor tissue-specific knock-out mice have been described, further analysis of Bcl-xL function in GC-induced apoptosis in vivo is not possible at this point.
The pro-apoptotic members of the Bcl-2 family have also been implicated in GC-induced apoptosis. The role of the ‘multidomain’ members Bax and Bak has been assessed by gene targeting [89–92]. While thymocytes from individual knock-out mice showed no difference in their sensitivity to GC-induced apoptosis, in the absence of both molecules, thymocytes were completely resistant. This suggests that Bax and Bak are involved in mediating GC effects but at the same time fulfill redundant roles. As to how GCs affect Bax and Bak, nothing is known so far. Bax is partially redistributed from the cytosol to the membrane fraction upon GC treatment [85, 86]. However, given the redundancy of Bax and Bak, GCs, possibly through ‘BH3-only’ proteins such as Bim, are more likely to induce a conformational change of these pro-apoptotic proteins which then leads to destabilization of the mitochondrial membrane.
The current model of the pro-apoptotic ‘BH3-only’ proteins implies that individual family members are responsible for mediating the response to different apoptotic stimuli [53, 93]. Bid, for example, is known for its involvement in Fas-mediated apoptosis in certain cells [94]. In keeping with this notion, GC-induced thymocyte apoptosis was unaffected in Bid-deficient mice, excluding an essential role in GC-mediated cell death [95]. The role of Bad, which is posttranscriptionally regulated by Akt/PKB-dependent phosphorylation, has been addressed in T cell-specific transgenic mice. Overexpression of Bad sensitized thymocytes to GC-induced apoptosis [96]. However, this may simply reflect its antagonistic effect on the death-repressing activity of Bcl-xL and thus Bad is not involved in this process under normal circumstances. Unfortunately, the role of Bad in GC-induced apoptosis has neither been investigated in the recently published Bad-deficient mice nor in a strain of knock-in mice in which BAD can not be phosphorylated due to three engineered point mutations [97, 98].
As outlined above, Bim was identified in one of the large-scale screenings as being upregulated during GC-induced apoptosis in T lymphoma cell lines [73]. In keeping with this observation, reducing Bim levels by RNAi in pre-B ALL cell lines rendered them partially resistant to GC-induced apoptosis [74]. Furthermore, GC-induced apoptosis in Bim-deficient mice was partially impaired compared to wild-type animals [78, 79]. In this context, it is interesting that PUMA was also shown to be upregulated in response to GC treatment [75]. In addition, analysis of two distinct strains of PUMA knock-out mice by independent laboratories has revealed a partial resistance of thymocytes to GC-induced apoptosis [99, 100]. In contrast, Noxa knock-out mice do not appear to be compromised in this process. Taken together, Bim and PUMA are the most promising candidates for pro-apoptotic ‘BH3-only’ proteins induced by GCs during the initiation of apoptosis.
The role of caspases in GC-induced apoptosis
The involvement of caspases in GC-induced apoptosis is continually debated. Pharmacological inhibition of caspases can be achieved by application of chemically modified small-peptide derivatives but their specificity for individual family members is sometimes variable [101]. Taking such an approach, thymocytes were found to be refractory to GC treatment in the presence of the pan-caspase inhibitor z-VAD-fmk [82, 102]. Moreover, caspase-3 and -8 were proposed to mediate GC-induced apoptosis but whether this holds true for all cell types needs to be demonstrated [103]. In particular, our own unpublished results have revealed that the role of caspases in this process is cell type specific. In summary, experiments based on the pharmacological interference with caspase activity suggest some role for these enzymes in GC-induced thymocyte apoptosis.
In contrast, analysis of knock-out mice deficient in individual caspases led to contradictory conclusions. First, thymocytes and mature T cells derived from caspase-1- and caspase-2-deficient mice, as well as from mice with a T lymphocyte-specific deletion of caspase-8, show no differences from wild-type mice in their responsiveness to GC treatment [104–106]. In contrast, apoptosis in response to GCs was found to be reduced in both caspase-9- and Apaf-1-deficient mice [107, 108]. Since these animals suffer from severe developmental defects, the impaired apoptosis could also result from unspecific effects. This notion is supported by the analysis of animals reconstituted with fetal liver cells from Apaf-1- or caspase-9-deficient mice. Thymocytes and mature T cells from these mice undergo apoptosis in response to a large range of GC concentrations, suggesting that the intrinsic lack of caspase-9 and Apaf-1 in T lymphocytes does not impair GC-induced apoptosis [79]. Interestingly, thymocytes from caspase-3-deficient mice, which die within the first month after birth, show normal responsiveness to GC-induced apoptosis [109]. This is in sharp contrast to the pharmacological data which are in support of an important role for caspase-3 in this process [103]. However, other family members conceivably compensate for its lack under these circumstances.
Other mediators of GC-induced apoptosis
There is growing evidence that lysosomes may play an important role in both caspase-dependent and -independent forms of apoptosis [67]. We have recently shown that activation of cathepsin B precedes caspase activation during GC-induced cell death, suggesting an important role for this cellular compartment [unpublished data].
Several studies have implicated the proteasome complex in GC-induced thymocyte apoptosis [110]. Its pharmacological inhibition prevents disruption of the mitochondrial membrane potential and nuclear fragmentation in thymocytes. Furthermore, the apoptosis inhibitory proteins c-IAP1 and XIAP have been shown to be degraded by the proteasome following GC treatment [111]. Although good evidence exists for an important role for proteasomal degradation, our unpublished results suggest that this is limited to certain cell types rather than being part of a ubiquitious pathway of GC-induced apoptosis.
Hydrogen peroxide, which can act as a second messenger in cell signaling, is synthesized at modest levels following the ligation of cell surface receptors and in response to various apoptotic stimuli [112]. Evidence that this pathway can be initiated by GCs comes from the observation that treatment of thymocytes with GCs leads to an increase in the intracellular level of hydrogen peroxide [110]. This precedes the loss in mitochondrial membrane potential, cytochrome c release and caspase-3 activation. However, in our hands, attempts to repeat this experiment failed [unpublished data].
Additional events that have been hypothesized to be involved in GC-induced apoptosis include ceramide production [113], changes in the intracellular levels of sodium and potassium ions [114], activation of PI3K [35] and the inositol trisphosphate receptor [115], as well as interaction of the GR with signaling proteins such as protein kinase C, Raf and members of the 14-3-3 family [116]. Importantly, some of these events have been shown to occur in the cytosol independent of de novo gene expression [35] but their relative contribution to GR-mediated apoptosis remains elusive. For further information on the non-classical effects of GCs, the reader is directed to a review by Distelhorst [9].
The cell type specificity of GC-induced thymocyte apoptosis
It was recognized more than 20 years ago that thymocytes differ in their sensitivity to GC treatment dependent on their developmental stage [117]. When administered at pharmacological doses, CD4+CD8+ double-positve (DP) thymocytes undergo cell death in vivo whereas singlepositive (SP) thymocytes are resistant to GCs (fig. 4). We have recently proposed a model involving CD28-mediated protection of SP thymocytes to explain this difference [87]. Such a specific role for CD28 in mature thymocytes is also suggested by the finding that its expression in humans and rats increases during the course of thymocyte development [118] and that its principal ligands B7-1 and B7-2 are exclusively expressed by thymic medullary cells where the mature T cells reside [119].

The role of GCs in thymocyte development. At the double-positive (DP) stage developing thymocytes undergo positive and negative selection which is controlled by an interplay between GR- and T cell receptor (TCR)-derived signals. Mature single-positive (SP) thymocytes are protected from GC-induced apoptosis by CD28 signaling. cTEC, cortical thymic epithelial cell; DC, dendritic cells; mTEC, medullary thymic epithelial cell.
Although refractory to GC-induced cell death in vivo, SP thymocytes become sensitive when in culture. However, enforced CD28 signaling in the absence of TCR engagement restores their resistance to GC treatment, presumably by upregulating Bcl-xL [87]. This model is consistent with the previous finding that anti-apoptotic proteins such as Bcl-2 and Bcl-XL are developmentally regulated during thymocyte development [120–122]. Similarly, SP thymocytes in CD28 knock-out mice show increased sensitivity to GC-administration. This suggests that CD28 signaling is responsible for conferring resistance to GCs in vivo. The differential sensitivity of immature DP and mature SP thymocytes to apoptosis may form the basis for the postulated role GCs play in positive and negative selection [11] (see below). If this model holds true, DP thymocytes need to be sensitive to GCs during selection but loose responsiveness to the same stimulus upon maturation. This could be achieved by the translocation of selected SP thymocytes from the cortical region to the medulla, where they would come into close contact to B7-expressing epithelial cells. Subsequent ligation of CD28 on these cortical SP thymocytes would then afford them protection from GCs.
Glucocorticoids and T cell development
GCs and thymic selection
An important checkpoint of T cell development occurs at the DP stage where interaction of the TCR with MHC/self-peptides on thymic epithelial and stromal cells initiates selection processes that lead to death by neglect, positive or negative selection [1]. At this stage, the majority of DP thymocytes die due to their inefficient ability to interact with self-ligands in the context of MHC. According to the avidity model of selection, DP cells with TCRs that interact with moderate affinity to self-ligands will be positively selected and further differentiate into mature functional T cells [2]. High-affinity interaction with self-MHC, which would be equivalent to autoreactivity in the periphery, destines DP cells to apoptosis and thus results in negative selection.
Most models of positive and negative selection postulate that the strength of TCR signaling determines thymocyte fate. In the ‘mutual antagonism’ model, this is assigned to an interplay between GC- and TCR-induced apoptosis [11]. Alone, signaling through the TCR or GR induces apoptosis, but when both receptors are coordinately stimulated they oppose each other (fig. 4). Thus, thymocytes expressing a TCR with high affinity for MHC/self-peptide undergo negative selection since GR signaling is not sufficient to overcome the strong signal originating from the TCR. In contrast, thymocytes with low-avidity TCRs undergo GC-induced cell death, since in this case the TCR signal cannot override GR signaling. Finally, in thymocytes with moderate-avidity TCRs, the two signals neutralize each other and thereby rescue the cells from apoptosis.
Since the ‘mutual antagonism’ model was proposed, arguments both in favour and against it have been put forward. Analysis of mice expressing an antisense GR in the thymus were found to possess a T cell repertoire which is shifted toward cells with lower affinity for MHC/self-peptide and thus have a reduced number of potentially autoreactive cells [44, 123, 124]. In contrast, analysis of several strains of GR knock-out mice failed to provide any support for the ‘mutual antagonism’ model [13, 47–49]. However, most of these strains have not been analyzed for more subtle effects on the antigen receptor repertoire. Thus, the idea that GC-induced apoptosis participates in thymic selection remains attractive, although it awaits further support by more refined approaches.
The role of thymus-derived GCs
There has been a long-standing debate as to whether the thymus synthesizes GCs itself or whether this is an exclusive characteristic of the adrenal gland [125] (fig. 4). The presence of the necessary biosynthetic machinery was demonstrated in the thymus [126]. In agreement with this finding, thymic epithelial cells (TECs) were shown to convert GC precursors to deoxycorticosterone in vitro [127, 128]. In addition, TECs also make metabolites which are able to activate GR-dependent transcription [127]. In fetal thymus organ culture, inhibition of GC synthesis by metyrapone turned out to impact on apoptosis, suggesting that steroid hormones might indeed be produced in the thymus [126]. However, another group could show that metyrapone impairs thymocyte development regardless of the presence or absence of a functional GR, indicating that this reagent inhibits thymocyte development in an unspecific fashion [48]. Recently, Okret and colleagues reported a new transgenic mouse model in which overexpression of the GR in thymocytes was controlled by the tetracycline-inducible system [51]. Increased expression of the GR in mice lacking adrenally derived GCs caused a slight reduction in thymic cellularity while subtype composition was unaltered. This observation was assigned to increased apoptosis following induction of GR overexpression. The finding that application of the GR antagonist RU486 was able to increase thymic cellularity even in adrenalectomized mice was taken as additional evidence of a role for intrathymically-synthesized GCs. However, our own unpublished results argue against extra-adrenal GC synthesis. Thymocyte numbers in transgenic rats overexpressing a mutant GR with increased ligand affinity are dramatically reduced while adrenalectomy restored cellularity within 3 weeks after surgery [unpublished data]. Thus, on the basis of our own findings, a major role for thymic GCs in T cell apoptosis and development is unlikely. However, more subtle influences on thymocyte selection, such as those suggested by the mutual antagonism model still remain possible.
Concluding remarks
Since the cloning of the GR [129] and the observation that GCs induce the death of T cells [6], we have come a long way in understanding the mechanisms underlying the actions of these hormones in T cell apoptosis and development. However, we are still far from having a coherent picture of either the physiological role or the molecular mechanisms of GCs in these processes. For example, we have limited knowledge of which genes are essential for T cell apoptosis. The issue of thymusderived GCs and the involvement of these hormones in thymocyte selection also remains debatable. Transgenesis and gene targeting have contributed enormously to our current understanding of GC action but new animal models carrying more refined mutations and transgenes are required to adequately address these questions. This is not only essential to extend our theoretical knowledge but is also of clinical relevance, since GCs are still among the most widely prescribed drugs for the treatment of hematological malignancies owing to their ability to mediate apoptosis in leukemia, lymphoma and myeloma cells [10, 14]. Thus, despite the long history of research on GC-induced apoptosis, it will continue to be a fertile and fascinating ground for basic and applied biomedical studies.
References
Bommhardt U., Beyer M., Hünig T. and Reichardt H. M. (2004) Molecular and cellular mechanisms of T cell development. Cell. Mol. Life Sci. 61: 263–280
Bevan M. J. (1997) In thymic selection, peptide diversity gives and takes away. Immunity 7: 175–178
Budd R. C. (2001) Activation-induced cell death. Curr. Opin. Immunol. 13: 356–362
Opferman J. T. and Korsmeyer S. J. (2003) Apoptosis in the development and maintenance of the immune system. Nat. Immunol. 4: 410–415
Tuckermann J. P., Kleiman A., McPherson K. G. and Reichardt H. M. (2005) Molecular mechanisms of glucocorticoids in the control of inflammation and lymphocyte apoptosis. Crit. Rev. Clin. Lab. Sci. 42: 71–104
Wyllie A. H. (1980) Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284: 555–556
Beato M., Herrlich P. and Schütz G. (1995) Steroid hormone receptors: many actors in search of a plot. Cell 83: 851–857
Reichardt H. M., Kaestner K. H., Tuckermann J., Kretz O., Wessely O., Bock R. et al. (1998) DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93: 531–541
Distelhorst C. W. (2002) Recent insights into the mechanism of glucocorticosteroid-induced apoptosis. Cell Death Differ. 9: 6–19
Frankfurt O. and Rosen S. T. (2004) Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr. Opin. Oncol. 16: 553–563
Vacchio M. S. and Ashwell J. D. (2000) Glucocorticoids and thymocyte development. Semin. Immunol. 12: 475–485
Baumann S., Dostert A., Novac N., Bauer A., Schmid W., Fas S. C. et al. (2005) Glucocorticoids inhibit activation-induced cell death (AICD) via direct DNA-dependent repression of the CD95 ligand gene by a glucocorticoid receptor dimer. Blood 106: 617–625
Brewer J. A., Kanagawa O., Sleckman B. P. and Muglia L. J. (2002) Thymocyte apoptosis induced by T cell activation is mediated by glucocorticoids in vivo. J. Immunol. 169: 1837–1843
Ploner C., Schmidt S., Presul E., Renner K., Schrocksnadel K., Rainer J. et al. (2005) Glucocorticoid-induced apoptosis and glucocorticoid resistance in acute lymphoblastic leukemia. J. Steroid Biochem. Mol. Biol. 93: 153–160
Reichardt H. M. (2004) Immunomodulatory activities of glucocorticoids: insights from transgenesis and gene targeting. Curr. Pharm. Des. 10: 2797–2805
Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K. et al. (1995) The nuclear receptor superfamily: the second decade. Cell 83: 835–839
Pratt W. B., Gehring U. and Toft D. O. (1996) Molecular chaperoning of steroid hormone receptors. EXS 11: 79–95
Luisi B. F., Xu W. X., Otwinowski Z., Freedman L. P., Yamamoto K. R. and Sigler P. B. (1991) Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 352: 497–505
Jenkins B. D., Pullen C. B. and Darimont B. D. (2001) Novel glucocorticoid receptor coactivator effector mechanisms. Trends Endocrinol. Metab. 12: 122–126
Gascoyne D. M., Kypta R. M. and Vivanco M. M. (2003) Glucocorticoids inhibit apoptosis during fibrosarcoma development by transcriptionally activating Bcl-xL. J. Biol. Chem. 278: 18022–18029
Deroo B. J. and Archer T. K. (2001) Glucocorticoid receptor activation of the I kappa B alpha promoter within chromatin. Mol. Biol. Cell. 12: 3365–3374
D'Adamio F., Zollo O., Moraca R., Ayroldi E., Bruscoli S., Bartoli A. et al. (1997) A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity 7: 803–812
Nocentini G., Giunchi L., Ronchetti S., Krausz L. T., Bartoli A., Moraca R. et al. (1997) A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA 94: 6216–6221
Schmid E., Schmid W., Jantzen M., Mayer D., Jastorff B. and Schutz G. (1987) Transcription activation of the tyrosine aminotransferase gene by glucocorticoids and cAMP in primary hepatocytes. Eur. J. Biochem. 165: 499–506
Hanson R. W. and Reshef L. (1997) Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu. Rev. Biochem. 66: 581–611
Heck S., Bender K., Kullmann M., Gottlicher M., Herrlich P. and Cato A. C. (1997) I kappaB alpha-independent downregulation of NF-kappaB activity by glucocorticoid receptor. EMBO J. 16: 4698–4707
Heck S., Kullmann M., Gast A., Ponta H., Rahmsdorf H. J., Herrlich P. et al. (1994) A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 13: 4087–4095
Vacca A., Felli M. P., Farina A. R., Martinotti S., Maroder M., Screpanti I. et al. (1992) Glucocorticoid receptor-mediated suppression of the interleukin 2 gene expression through impairment of the cooperativity between nuclear factor of activated T cells and AP-1 enhancer elements. J. Exp. Med. 175: 637–646
Imai E., Miner J. N., Mitchell J. A., Yamamoto K. R. and Granner D. K. (1993) Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J. Biol. Chem. 268: 5353–5356
Stoecklin E., Wissler M., Moriggl R. and Groner B. (1997) Specific DNA binding of Stat5, but not of glucocorticoid receptor, is required for their functional cooperation in the regulation of gene transcription. Mol. Cell. Biol. 17: 6708–6716
Tuckermann J. P., Reichardt H. M., Arribas R., Richter K. H., Schutz G. and Angel P. (1999) The DNA binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J. Cell. Biol. 147: 1365–1370
Reichardt H. M., Horsch K., Gröne H. J., Kolbus A., Beug H., Hynes N. et al. (2001) Mammary gland development and lactation are controlled by different glucocorticoid receptor activities. Eur. J. Endocrinol. 145: 519–527
Reichardt H. M., Tuckermann J. P., Göttlicher M., Vujic M., Weih F., Angel P. et al. (2001) Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 20: 7168–7173.
Tronche F., Opherk C., Moriggl R., Kellendonk C., Reimann A., Schwake L. et al. (2004) Glucocorticoid receptor function in hepatocytes is essential to promote postnatal body growth. Genes Dev. 18: 492–497
Limbourg F. P., Huang Z., Plumier J. C., Simoncini T., Fujioka M., Tuckermann J. et al. (2002) Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J. Clin. Invest. 110: 1729–1738
Lu N. Z. and Cidlowski J. A. (2005) Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell 18: 331–342
Diba F., Watson C. S. and Gametchu B. (2001) 5'UTR sequences of the glucocorticoid receptor 1A transcript encode a peptide associated with translational regulation of the glucocorticoid receptor. J. Cell. Biochem. 81: 149–161
Purton J. F., Monk J. A., Liddicoat D. R., Kyparissoudis K., Sakkal S., Richardson S. J. et al. (2004) Expression of the glucocorticoid receptor from the 1A promoter correlates with T lymphocyte sensitivity to glucocorticoid-induced cell death. J. Immunol. 173: 3816–3824
Zhou J. and Cidlowski J. A. (2005) The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids 70: 407–417
Yeager M. P., Guyre P. M. and Munck A. U. (2004) Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol. Scand. 48: 799–813
Hench P. S., Kendall E. C., Slocumb C. H. and Polley H. F. (1950) Effects of cortisone acetate and pituitray ACTH on rheumatoid arthritis, rheumatic fever and certain other conditions. Arch. Intern. Med. 85: 545–566
Webster J. I., Tonelli L. and Sternberg E. M. (2002) Neuroendocrine regulation of immunity. Annu. Rev. Immunol. 20: 125–163
Balsalobre A., Brown S. A., Marcacci L., Tronche F., Kellendonk C., Reichardt H. M. et al. (2000) Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289: 2344–2347
King L. B., Vacchio M. S., Dixon K., Hunziker R., Margulies D. H. and Ashwell J. D. (1995) A targeted glucocorticoid receptor antisense transgene increases thymocyte apoptosis and alters thymocyte development. Immunity 3: 647–656
Pazirandeh A., Xue Y., Prestegaard T., Jondal M. and Okret S. (2002) Effects of altered glucocorticoid sensitivity in the T cell lineage on thymocyte and T cell homeostasis. FASEB J. 16: 727–729
Cole T. J., Blendy J. A., Monaghan A. P., Krieglstein K., Schmid W., Aguzzi A. et al. (1995) Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 9: 1608–1621
Purton J. F., Boyd R. L., Cole T. J. and Godfrey D. I. (2000) Intrathymic T cell development and selection proceeds normally in the absence of glucocorticoid receptor signaling. Immunity 13: 179–186
Purton J. F., Zhan Y., Liddicoat D. R., Hardy C. L., Lew A. M., Cole T. J. et al. (2002) Glucocorticoid receptor deficient thymic and peripheral T cells develop normally in adult mice. Eur. J. Immunol. 32: 3546–3555
Brewer J. A., Khor B., Vogt S. K., Muglia L. M., Fujiwara H., Haegele K. E. et al. (2003) T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat. Med. 9: 1318–1322
Reichardt H. M., Umland T., Bauer A., Kretz O. and Schütz G. (2000) Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol. Cell. Biol. 20: 9009–9017
Pazirandeh A., Jondal M. and Okret S. (2005) Conditional expression of a glucocorticoid receptor transgene in thymocytes reveals a role for thymic-derived glucocorticoids in thymopoiesis in vivo. Endocrinology 146: 2501–2507
Hengartner M. O. (2000) The biochemistry of apoptosis. Nature 407: 770–776
Strasser A. (2005) The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5: 189–200
Wang X. (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15: 2922–2933
Cheng E. H., Wei M. C., Weiler S., Flavell R. A., Mak T. W., Lindsten T. et al. (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8: 705–711
Green D. R. and Kroemer G. (2004) The pathophysiology of mitochondrial cell death. Science 305: 626–629
Zong W. X., Edelstein L. C., Chen C., Bash J. and Gelinas C. (1999) The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 13: 382–387
Kaufmann T., Schlipf S., Sanz J., Neubert K., Stein R. and Borner C. (2003) Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J. Cell. Biol. 160: 53–64
Bassik M. C., Scorrano L., Oakes S. A., Pozzan T. and Korsmeyer S. J. (2004) Phosphorylation of BCL-2 regulates ER Ca(2+) homeostasis and apoptosis. EMBO J. 23: 1207–1216
Herrant M., Jacquel A., Marchetti S., Belhacene N., Colosetti P., Luciano F. et al. (2004) Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene 23: 7863–7873
Barnhart B. C., Alappat E. C. and Peter M. E. (2003) The CD95 type I/type II model. Semin. Immunol. 15: 185–193
Chipuk J. E. and Green D. R. (2005) Do inducers of apoptosis trigger caspase-independent cell death? Nat. Rev. Mol. Cell Biol. 6: 268–275
Kroemer G. and Martin S. J. (2005) Caspase-independent cell death. Nat. Med. 11: 725–730
Michallet M. C., Saltel F., Preville X., Flacher M., Revillard J. P. and Genestier L. (2003) Cathepsin-B-dependent apoptosis triggered by antithymocyte globulins: a novel mechanism of Tcell depletion. Blood 102: 3719–3726
Bidere N., Lorenzo H. K., Carmona S., Laforge M., Harper F., Dumont C. et al. (2003) Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J. Biol. Chem. 278: 31401–31411
Canbay A., Guicciardi M. E., Higuchi H., Feldstein A., Bronk S. F., Rydzewski R. et al. (2003) Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J. Clin. Invest. 112: 152–159
Cuervo A. M. (2004) Autophagy: many paths to the same end. Mol. Cell. Biochem. 263: 55–72
Liu N., Phillips T., Zhang M., Wang Y., Opferman J. T., Shah R. et al. (2004) Serine protease inhibitor 2A is a protective factor for memory T cell development. Nat. Immunol. 5: 919–926
Forsthoefel A. M. and Thompson E. A. (1987) Glucocorticoid regulation of transcription of the c-myc cellular protooncogene in P1798 cells. Mol. Endocrinol. 1: 899–907
Tosa N., Murakami M., Jia W. Y., Yokoyama M., Masunaga T., Iwabuchi C. et al. (2003) Critical function of T cell death-associated gene 8 in glucocorticoid-induced thymocyte apoptosis. Int. Immunol. 15: 741–749
Malone M. H., Wang Z. and Distelhorst C. W. (2004) The glucocorticoid-induced gene tdag8 encodes a pro-apoptotic G protein-coupled receptor whose activation promotes glucocorticoid-induced apoptosis. J. Biol. Chem. 279: 52850–52859
Wang Z., Malone M. H., Thomenius M. J., Zhong F., Xu F. and Distelhorst C. W. (2003) Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J. Biol. Chem. 278: 27053–27058
Wang Z., Malone M. H., He H., McColl K. S. and Distelhorst C. W. (2003) Microarray analysis uncovers the induction of the proapoptotic BH3-only protein Bim in multiple models of glucocorticoid-induced apoptosis. J. Biol. Chem. 278: 23861–23867
Abrams M. T., Robertson N. M., Yoon K. and Wickstrom E. (2004) Inhibition of glucocorticoid-induced apoptosis by targeting the major splice variants of BIM mRNA with small interfering RNA and short hairpin RNA. J. Biol. Chem. 279: 55809–55817
Han J., Flemington C., Houghton A. B., Gu Z., Zambetti G. P., Lutz R. J. et al. (2001) Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 98: 11318–11323
Tonko M., Ausserlechner M. J., Bernhard D., Helmberg A. and Kofler R. (2001) Gene expression profiles of proliferating vs. G1/G0 arrested human leukemia cells suggest a mechanism for glucocorticoid-induced apoptosis. FASEB J. 15: 693–699
Medh R. D., Webb M. S., Miller A. L., Johnson B. H., Fofanov Y., Li T. et al. (2003) Gene expression profile of human lymphoid CEM cells sensitive and resistant to glucocorticoid-evoked apoptosis. Genomics 81: 543–555
Bouillet P., Metcalf D., Huang D. C., Tarlinton D. M., Kay T. W., Kontgen F. et al. (1999) Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286: 1735–1738
Marsden V. S., O'Connor L., O'Reilly L. A., Silke J., Metcalf D., Ekert P. G. et al. (2002) Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419: 634–637
Memon S. A., Moreno M. B., Petrak D. and Zacharchuk C. M. (1995) Bcl-2 blocks glucocorticoid- but not Fas- or activation-induced apoptosis in a T cell hybridoma. J. Immunol. 155: 4644–4652
Huang S. T. and Cidlowski J. A. (2002) Phosphorylation status modulates Bcl-2 function during glucocorticoid-induced apoptosis in T lymphocytes. FASEB J. 16: 825–832
Mann C. L., Hughes F. M. Jr and Cidlowski J. A. (2000) Delineation of the signaling pathways involved in glucocorticoid-induced and spontaneous apoptosis of rat thymocytes. Endocrinology 141: 528–538
Susin S. A., Lorenzo H. K., Zamzami N., Marzo I., Snow B. E., Brothers G. M. et al. (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446
Veis D. J., Sorenson C. M., Shutter J. R. and Korsmeyer S. J. (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75: 229–240
Hsu Y. T., Wolter K. G. and Youle R. J. (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. USA 94: 3668–3672
Jia L., Macey M. G., Yin Y., Newland A. C. and Kelsey S. M. (1999) Subcellular distribution and redistribution of Bcl-2 family proteins in human leukemia cells undergoing apoptosis. Blood 93: 2353–2359
Brandt J. van den, Wang D. and Reichardt H. M. (2004) Resistance of single-positive thymocytes to glucocorticoid-induced apoptosis is mediated by CD28 signaling. Mol. Endocrinol. 18: 687–695
Motoyama N., Wang F., Roth K. A., Sawa H., Nakayama K., Negishi I. et al. (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267: 1506–1510
Lindsten T., Ross A. J., King A., Zong W. X., Rathmell J. C., Shiels H. A. et al. (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6: 1389–1399
Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J. et al. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730
Zong W. X., Lindsten T., Ross A. J., MacGregor G. R. and Thompson C. B. (2001) BH3-only proteins that bind prosurvival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15: 1481–1486
Rathmell J. C., Lindsten T., Zong W. X., Cinalli R. M. and Thompson C. B. (2002) Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat. Immunol. 3: 932–939
Marsden V. S. and Strasser A. (2003) Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 21: 71–105
Yin X. M. (2000) Bid, a critical mediator for apoptosis induced by the activation of Fas/TNF-R1 death receptors in hepatocytes. J. Mol. Med. 78: 203–211
Yin X. M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B. et al. (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891
Mok C. L., Gil-Gomez G., Williams O., Coles M., Taga S., Tolaini M. et al. (1999) Bad can act as a key regulator of T cell apoptosis and T cell development. J. Exp. Med. 189: 575–586
Datta S. R., Ranger A. M., Lin M. Z., Sturgill J. F., Ma Y. C., Cowan C. W. et al. (2002) Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell 3: 631–643
Ranger A. M., Zha J., Harada H., Datta S. R., Danial N. N., Gilmore A. P. et al. (2003) Bad-deficient mice develop diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 100: 9324–9329
Jeffers J. R., Parganas E., Lee Y., Yang C., Wang J., Brennan J. et al. (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328
Villunger A., Michalak E. M., Coultas L., Mullauer F., Bock G., Ausserlechner M. J. et al. (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038
Talanian R. V., Quinlan C., Trautz S., Hackett M. C., Mankovich J. A., Banach D. et al. (1997) Substrate specificities of caspase family proteases. J. Biol. Chem. 272: 9677–9682
Hughes F. M. Jr and Cidlowski J. A. (1998) Glucocorticoid-induced thymocyte apoptosis: protease-dependent activation of cell shrinkage and DNA degradation. J. Steroid Biochem. Mol. Biol. 65: 207–217
Marchetti M. C., Di Marco B., Cifone G., Migliorati G. and Riccardi C. (2003) Dexamethasone-induced apoptosis of thymocytes: role of glucocorticoid receptor-associated Src kinase and caspase-8 activation. Blood 101: 585–593
Li P., Allen H., Banerjee S. and Seshadri T. (1997) Characterization of mice deficient in interleukin-1 beta converting enzyme. J. Cell. Biochem. 64: 27–32
O'Reilly L. A., Ekert P., Harvey N., Marsden V., Cullen L., Vaux D. L. et al. (2002) Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ. 9: 832–841
Salmena L., Lemmers B., Hakem A., Matysiak-Zablocki E., Murakami K., Au P. Y. et al. (2003) Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17: 883–895
Kuida K., Haydar T. F., Kuan C. Y., Gu Y., Taya C., Karasuyama H. et al. (1998) Reduced apoptosis and cytochrome cmediated caspase activation in mice lacking caspase 9. Cell 94: 325–337
Yoshida H., Kong Y. Y., Yoshida R., Elia A. J., Hakem A., Hakem R. et al. (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739–750
Kuida K., Zheng T. S., Na S., Kuan C., Yang D., Karasuyama H. et al. (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384: 368–372
Tonomura N., McLaughlin K., Grimm L., Goldsby R. A. and Osborne B. A. (2003) Glucocorticoid-induced apoptosis of thymocytes: requirement of proteasome-dependent mitochondrial activity. J. Immunol. 170: 2469–2478
Yang Y., Fang S., Jensen J. P., Weissman A. M. and Ashwell J. D. (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288: 874–877
Reth M. (2002) Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 3: 1129–1134
Cifone M. G., Migliorati G., Parroni R., Marchetti C., Millimaggi D., Santoni A. et al. (1999) Dexamethasone-induced thymocyte apoptosis: apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood 93: 2282–2296
Bortner C. D., Hughes F. M. Jr and Cidlowski J. A. (1997) A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 272: 32436–32442
Jayaraman T. and Marks A. R. (1997) T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis. Mol. Cell. Biol. 17: 3005–3012
Göttlicher M., Heck S. and Herrlich P. (1998) Transcriptional cross-talk, the second mode of steroid hormone receptor action. J. Mol. Med. 76: 480–489
Screpanti I., Morrone S., Meco D., Santoni A., Gulino A., Paolini R. et al. (1989) Steroid sensitivity of thymocyte subpopulations during intrathymic differentiation: effects of 17 beta-estradiol and dexamethasone on subsets expressing T cell antigen receptor or IL-2 receptor. J. Immunol. 142: 3378–3383
Tacke M., Clark G. J., Dallman M. J. and Hünig T. (1995) Cellular distribution and costimulatory function of rat CD28: regulated expression during thymocyte maturation and induction of cyclosporin A sensitivity of costimulated T cell responses by phorbol ester. J. Immunol. 154: 5121–5127
Reiser H. and Schneeberger E. E. (1994) The costimulatory molecule B7 is expressed in the medullary region of the murine thymus. Immunology 81: 532–537
Gratiot-Deans J., Ding L., Turka L. A. and Nunez G. (1993) bcl-2 proto-oncogene expression during human T cell development: evidence for biphasic regulation. J. Immunol. 151: 83–91
Veis D. J., Sentman C. L., Bach E. A. and Korsmeyer S. J. (1993) Expression of the Bcl-2 protein in murine and human thymocytes and in peripheral T lymphocytes. J. Immunol. 151: 2546–2554
Grillot D. A., Merino R. and Nunez G. (1995) Bcl-XL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J. Exp. Med. 182: 1973–1983
Tolosa E., King L. B. and Ashwell J. D. (1998) Thymocyte glucocorticoid resistance alters positive selection and inhibits autoimmunity and lymphoproliferative disease in MRL-lpr/lpr mice. Immunity 8: 67–76
Lu F. W., Yasutomo K., Goodman G. B., McHeyzer-Williams L. J., McHeyzer-Williams M. G., Germain R. N. et al. (2000) Thymocyte resistance to glucocorticoids leads to antigen-specific unresponsiveness due to holes in the T cell repertoire. Immunity 12: 183–192
Jondal M., Pazirandeh A. and Okret S. (2004) Different roles for glucocorticoids in thymocyte homeostasis? Trends Immunol. 25: 595–600
Vacchio M. S., Papadopoulos V. and Ashwell J. D. (1994) Steroid production in the thymus: implications for thymocyte selection. J. Exp. Med. 179: 1835–1846
Pazirandeh A., Xue Y., Rafter I., Sjovall J., Jondal M. and Okret S. (1999) Paracrine glucocorticoid activity produced by mouse thymic epithelial cells. FASEB J. 13: 893–901
Lechner O., Wiegers G. J., Oliveira-Dos-Santos A. J., Dietrich H., Recheis H., Waterman M. et al. (2000) Glucocorticoid production in the murine thymus. Eur. J. Immunol. 30: 337–346
Hollenberg S. M., Weinberger C., Ong E. S., Cerelli G., Oro A., Lebo R. et al. (1985) Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318: 635–641
Stephens G. L., Ashwell J. D. and Ignatowicz L. (2003) Mutually antagonistic signals regulate selection of the T cell repertoire. Int. Immunol. 15: 623–632
Finotto S., Krieglstein K., Schober A., Deimling F., Lindner K., Bruhl B. et al. (1999) Analysis of mice carrying targeted mutations of the glucocorticoid receptor gene argues against an essential role of glucocorticoid signalling for generating adrenal chromaffin cells. Development 126: 2935–2944
Acknowledgement
This work was supported by grants from Volkswagen Stiftung (I/75 403), Deutsche Forschungsgemeinschaft (Re1631/1) and Wilhelm Sander-Stiftung (2003.129.1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Received 20 August 2005; received after revision 27 September 2005; accepted 10 October 2005
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Herold, M.J., McPherson, K.G. & Reichardt, H.M. Glucocorticoids in T cell apoptosis and function. Cell. Mol. Life Sci. 63, 60–72 (2006). https://doi.org/10.1007/s00018-005-5390-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-005-5390-y