
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is capable of inducing the activation of NACHT, leucine-rich repeat, and pyrin domain-containing protein 3 (NLRP3) inflammasome, a macromolecular structure sensing the danger and amplifying the inflammatory response. The main product processed by NLRP3 inflammasome is interleukin (IL)-1β, responsible for the downstream production of IL-6, which has been recognized as an important mediator in coronavirus disease 2019 (COVID-19). Since colchicine is an anti-inflammatory drug with the ability to block NLRP3 inflammasome oligomerization, this may prevent the release of active IL-1β and block the detrimental effects of downstream cytokines, i.e. IL-6. To date, few randomized clinical trials and many observational studies with colchicine have been conducted, showing interesting signals. As colchicine is a nonspecific inhibitor of the NLRP3 inflammasome, compounds specifically blocking this molecule might provide increased advantages in reducing the inflammatory burden and its related clinical manifestations. This may occur through a selective blockade of different steps preceding NLRP3 inflammasome oligomerization as well as through a reduced release of the main cytokines (IL-1β and IL-18). Since most evidence is based on observational studies, definitive conclusion cannot be drawn and additional studies are needed to confirm preliminary results and further dissect how colchicine and other NLRP3 inhibitors reduce the inflammatory burden and evaluate the timing and duration of treatment.
Similar content being viewed by others
Introduction
Coronavirus disease 2019 (COVID-19)—the disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)—is caused by a dysregulated hyperinflammatory response of the host during the late phase of immune response against SARS-CoV-2 [1, 2]. This occurs in a small portion of patients with COVID-19, as the disease has an asymptomatic or paucisymptomatic course in most patients [3]. Progression towards severe and critical COVID-19 leading to multi-organ failure is mainly driven by increasing levels of pro-inflammatory mediators, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), IL-18, granulocyte–macrophage colony-stimulating factor (GM-CSF), among others [4,5,6,7,8]. In light of this, anti-cytokine agents targeting IL-6 and IL-1β have been tested with encouraging results [9,10,11,12,13]. Recently, SARS-CoV-2 has been shown to activate the NACHT, leucine-rich repeat, and pyrin domain-containing protein 3 (NLRP3) inflammasome both in vitro and in vivo [14,15,16,17].
Colchicine is an anti-inflammatory drug currently used for gout, Adamantiades-Behçet’s disease, acute and recurrent pericarditis, and other autoinflammatory diseases, such as familial Mediterranean fever [18, 19]. Apart from its effect on neutrophil activity, colchicine can indirectly block the NLRP3 inflammasome [20, 21], a macromolecular complex that senses danger and triggers a local or systemic inflammatory response by releasing pro-inflammatory cytokines, such as IL-1β [22]. Indeed, colchicine has been tested in COVID-19 both in the outpatient and inpatient setting [23,24,25,26,27,28,29,30,31].
We herein summarize currently available evidence about the involvement of the NLRP3 inflammasome in the pathophysiology of COVID-19 and studies testing colchicine as a targeted therapeutic approach.
Search strategy and selection criteria
A literature search has been performed using MEDLINE (Pubmed database) including original articles, reviews, systematic reviews, and meta-analyses published over the past months (from January 1st 2021 until December 31st 2021). Additional articles were identified from the reference list of the searched articles and from medRxiv and bioRxiv (i.e. pre-prints). For ongoing trials, we searched using ClinicalTrials.gov. Only articles published in English language were included. The search strategy included the following terms or their combination: “colchicine”, “NLRP3 inflammasome”, “NLRP3 inhibitors”, “IL-1β”, “COVID-19”, “SARS-CoV-2”, “hyperinflammation”, “cytokine storm”.
Inflammasomes as masters of inflammation
Inflammasomes are high-molecular-weight structures located in the cytosol of stimulated immune cells responsible for caspase-1 activation and processing of pro-inflammatory cytokines, such as IL-1β and IL-18 [32, 33]. Based on the activation of different pattern-recognition receptors (PRRs)—NLRP1, NLRP3, NLRC4, and absent in melanoma 2 (AIM2), —in response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), several inflammasomes have been recognized [33]. Following the recognition of ligands, the sensor protein activates, oligomerizes, and binds the scaffold protein—apoptosis-associated speck-like protein containing a carboxy-terminal containing a caspase recruiting domain (ASC)—and pro-caspase-1. Importantly, the presence of ASC through the visualization of micron-sized structures through immunofluorescence staining, namely puncta or specks, is considered as a read-out of active inflammasomes in the cells [34]. After the autocatalytic cleavage of pro-caspase-1 to active caspase-1 [35], the final step is the processing of pro-IL-1β, pro-IL-18, pro-IL-33, and pro-IL-37 to their mature, active forms [36]. Along with the abovementioned canonical pathway, additional caspases may be activated through a non-canonical pathway, i.e. caspase-4 and caspase-5 in humans and caspase-11 in mice [37,38,39,40].
Inflammasomes are now recognized to be crucially involved in host defense against pathogens [41] and also in sterile inflammation [22], however, a dysregulated activation is responsible for development of autoimmune, autoinflammatory, and metabolic diseases and cancer [42,43,44].
NLRP1, NLRC4, and AIM2 inflammasomes have been widely studied for their role in infections, but less in non-infectious, inflammatory conditions [33, 45]. The NLRP3 inflammasome has been found in most infectious and non-infectious diseases and largely studied in the cardiovascular field [22, 46,47,48].
NLRP3 inflammasome and SARS-CoV-2
Previous reports about SARS-CoV described viral proteins ORF3a, ORF8b, and viroporin 3a to trigger the activation of the NLRP3 inflammasome [49,50,51,52]. As SARS-Co-V and SARS-CoV-2 appear similar (they share 82% of nucleotide sequence homology [53] and the same receptor—angiotensin-converting enzyme 2 [ACE2] [54]), evidence is being accumulated about the ability of SARS-CoV-2 to activate the NLRP3 inflammasome [55]. Indeed, the observation of cell death and increased levels of IL-1β and IL-18 and lactate dehydrogenase (LDH)—a classic marker of cell death—in the serum of COVID-19 patients prompted the attention on the potential involvement of the inflammasome [4,5,6,7, 56]. A clear evidence, therefore, is needed as some of these products may derive from inflammasome-independent pathways [57,58,59,60].
Xu et al. showed that SARS-CoV-2 induces preferentially NLRP3 inflammasome through ORF3a, but neither NLRP1 nor NLRC4 inflammasomes [61]. In particular, ORF3a increased caspase-1 and pro-IL-1β levels, as occurs during inflammasome activation [61]. The inflammasome priming provided by ORF3a increases the expression of Il-1β mediated by nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and the activation of gasdermin D, the latter being responsible for pore formation leading to pyroptosis [61]. NIMA-related kinase 7 (NEK7) appears to be fundamental for inflammasome activation as NLRP3 associates with its catalytic domain. In fact, activated macrophages formed a NLRP3-NEK7 complex together with ASC oligomerization, that is abrogated in the absence of NEK7 [62]. In addition, Xu et al. found that blockade of potassium efflux blocks ORF3a-mediated caspase-1 cleavage [61].
Rodrigues et al. showed that SARS-CoV-2 is able to stimulate NLRP3 inflammasome activation in primary human monocytes, that was blocked by MCC950, a selective inhibitor of NLRP3 inflammasome [63]. In COVID-19 patients, authors found increased serum concentrations of active caspase-1 and IL-18 as well as an increased activation of NLRP3 inflammasome in peripheral blood mononuclear cells (PBMCs) through the visualization of ASC specks [14]. Rodrigues et al. also investigated whether NLRP3 inflammasome activation correlated with disease severity and clinical outcomes. They showed positive associations of caspase-1 and/or IL-18 levels with C-reactive protein (CRP), LDH, IL-6, and ferritin [14], that in turn correlated with COVID-19 severity [64,65,66,67,68,69,70]. With regard to clinical outcomes, IL-18 concentration was higher in patients needing mechanical ventilation compared with those who did not and in survivors compared with non-survivors. Considering 37 patients followed from the time of hospital admission up to 45 days, caspase-1 and IL-18 levels both decreased over time, but IL-18 levels remained higher in patients who died and never reached the levels of patients who recovered, irrespective of the need of mechanical ventilation [14]. The presence of the NLRP3 inflammasome has also been described in tissues from patients with COVID-19 [14, 15]. Importantly, the analysis of lung tissues from deceased COVID-19 patients showed the presence of active NLRP3 inflammasome in CD14+ cells (monocytes) [14] and was confirmed also by our group [15] (Fig. 1).

Reproduced with permission from Toldo et al., “Inflammasome formation in the lungs of patients with fatal COVID-19” [15]
NLRP3 inflammasome activation in lungs of patients who died of COVID-19 . Immunofluorescence stainings from patients with fatal COVID-19 (panels a, c, and e) and from individuals who died of cardiopulmonary arrest but without evidence of lung infection (panels b, d and f) are shown. In panel g, quantification of NLRP3 inflammasome activation, expressed as ASC specks per high-power fields, is provided with a significantly higher number of specks in COVID-19 patients compared with controls. ASC apoptosis-associated speck-like protein containing a caspase recruitment domain. NLRP3 NACHT, leucine-rich repeat, and pyrin domain-containing protein 3.
These findings strongly support a role for NLRP3 inflammasome in driving early pathologic manifestations of COVID-19, leading to progressively worsening respiratory failure culminating with ARDS. According to this evidence, it appears reasonable to therapeutically target the NLRP3 inflammasome to reduce the burden of hospitalization and morbidity .
Therapeutic effects of colchicine
Colchicine is an old anti-inflammatory drug derived from the plant Colchicum autumnale [18]. Currently, colchicine is approved for acute flares of gout and familial Mediterranean fever and used off-label for several diseases, like Adamantiades-Behçet’s disease, pericarditis, and other inflammatory and auto-inflammatory conditions [18].
The main anti-inflammatory effect of colchicine is mediated by its ability to dissolve microtubules within immune cells [71]. By intercalating into free α/β tubulin and inhibiting microtubule extension, the movement of adhesion molecules on cell surface is limited and this is especially seen in neutrophils, where colchicine concentration is much higher as compared with other immune cells. This latter effect depends on a reduction in the activity of P-glycoprotein membrane efflux pump that is responsible for energy-dependent colchicine efflux [72]. Colchicine is also able to reduce expression of L- and E-selectin, that mediate rolling and adhesion of neutrophils on the endothelium [73]. Microtubule breakdown also blocks neutrophil movements through blood vessels [74]. In addition, colchicine was found to negatively impact on neutrophil signaling and response during phagocytosis [75, 76].
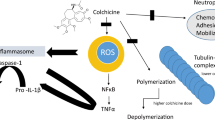
In recent years, colchicine was found to inhibit NLRP3 inflammasome, although the exact mechanism of action is not fully understood yet. The putative step through which colchicine blocks NLRP3 inflammasome formation may rely on microtubule breakdown, that interferes with the oligomerization of the inflammasome [20, 21]. The result is a reduced production of IL-1β, that in turn decreases the production of IL-6 and tumor necrosis factor (TNF), leading to a reduction in the recruitment of neutrophils and macrophages [77, 78] (Fig. 2).

NLRP3 inflammasome formation and therapeutic implications of colchicine in COVID-19. Panel A. The formation of the NLRP3 inflammasome is a finely tuned process, that in most cases depends on two parallel pathways, i.e. priming and triggering. The priming includes signals that regulate the expression/degradation of inflammasome components (NLRP3, ASC, and caspase-1) and cytokines (IL-1β and IL-18). DAMPs activate PRRs (i.e. toll-like receptors, IL-1 receptor) leading to the translocation of the NF-kB into the nucleus. This is responsible for gene transcription of a wealth of proinflammatory genes—including inflammasome components. Except for monocytes, priming signaling by itself is not sufficient to prompt NLRP3 inflammasome activation. The translation of all inflammasome proteins is essential for the formation of the inflammasome, but does not coincide with its activation. Indeed, extra-cellular ATP or intracellular DAMPs trigger NLRP3 activation through diverse mechanisms involving the potassium efflux, namely inflammasome triggering. Once active, NLRP3 oligomerizes into a platform for recruitment of ASC and pro-caspase-1. At this stage, the activation of caspase-1 mediates the cleavage of pro-IL-1β, pro-IL-18, and gasdermin D (GSDMD). The oligomerization of the N-terminal fragment of GSDMD into a plasma membrane pore allows for the secretion of active IL-1β and IL-18 that sustain further autocrine, paracrine, and endocrine amplification of the immune response. Caspase-1 and GSDMD mediate also a form of regulated cell death known as pyroptosis. Panel B. SARS-CoV-2 induces the expression of the NLRP3 inflammasome. Following its oligomerization, NLRP3 inflammasome processes pro-inflammatory cytokines, such as IL-1β and IL-18. The release of active IL-1β triggers the production of IL-6 and leads to a dysregulated hyperinflammatory response, that is responsible for immune cell recruitment, especially neutrophils and macrophages. These events lead to organ failure, primarily the lungs, but can progress to multiorgan failure, often fatal. The figure in Panel A has been reproduced with permission from “NLRP3 Inflammasome in Acute Myocardial Infarction” by Mauro et al. [48]. The figure in Panel B has been partially created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com. DAMPs damage-associated molecular patterns. eATP extra-cellular ATP. G-CSF granulocyte colony-stimulating factor. GM-CSF granulocyte–macrophage colony-stimulating factor. GSDMD gasdermin D. GSDMD-Nt N-terminal fragment of gasdermin D. IFN interferon. IL interleukin. K+ potassium. NF-kB nuclear factor kappa-light-chain-enhancer of activated B cell. NLRP3 NACHT, leucine-rich repeat, and pyrin domain-containing protein 3. PRR pattern recognition receptor. SARS-CoV-2 severe acute respiratory syndrome coronavirus 2. TNF tumor necrosis factor.
Colchicine was also demonstrated to play a role in the crosstalk between inflammation and thrombosis, although most evidence on this derives from pre-clinical studies. Colchicine can inhibit the release of α-defensin from neutrophils in mice, thus reducing the size of thrombi [79, 80]. Colchicine was also found to deform platelets through its effect on microtubules and block calcium entry into platelets [81], thus reducing platelet activity. An in vitro study showed that colchicine may blunt neutrophil-platelet interactions at physiological doses [82], suggesting a role in interfering with the inflammation/thrombosis interface.
Colchicine to treat COVID-19: what is the rationale?
Based on the pathophysiological premises previously mentioned, colchicine might have a positive impact on the natural history of SARS-CoV-2 infection [83, 84]. Since SARS-CoV-2 activates the NLRP3 inflammasome, this leads to an increased production of pro-inflammatory cytokines [1]. Differently from other repurposed anti-rheumatic agents used in COVID-19 patients (anakinra, tocilizumab, or sarilumab targeting either IL-1β or IL-6), colchicine has a pleiotropic mechanism of action with diverse effects on the inflammatory cascade. Although less potent when compared with glucocorticoids (dexamethasone in particular), colchicine has the great advantage to be available for oral administration. Moreover, colchicine has an extremely favorable safety profile and since it does not exert any overt immunosuppressive activity, it does not interfere with the effective viral clearing nor is associated with the occurrence of secondary infections [83]. Additionally, compared with anti-cytokine agents, colchicine is less immunosuppressive and cheaper.
By taking in mind the pathophysiology of SARS-CoV-2, the use of colchicine should be considered in the early inflammatory phase (i.e. phase 2 according to the clinical-therapeutic staging proposal [85]) in order to prevent progression towards phase 3 or hyperinflammatory stage. This may be the case of non-hospitalized patients with symptoms dating back a few days or also hospitalized patients not progressing to critical disease. As for other agents, the optimal timing of administration of colchicine is still a matter of debate and warrants future investigation.
To date, few randomized clinical trials (RCTs) have been conducted with colchicine given on top of standard-of-care [23, 26, 30, 31, 86, 87] (Table 1). Recruitment to the colchicine arm of the RECOVERY trial was interrupted for futility, as preliminary data showed no convincing evidence for a mortality benefit [31, 88]. This might either depend on a limited anti-inflammatory power of colchicine in the advanced stages of COVID-19. In addition, the concurrent administration of dexamethasone in the majority of patients should be considered as its anti-inflammatory property is much larger compared with that of colchicine. However, the latter is not fully supported by data from this trial, as colchicine was not found beneficial in patients not receiving glucocorticoids [31], probably suggesting a limited utility of colchicine in the hyperinflammatory phase of COVID-19. In the trial by Deftereos et al., colchicine reduced the primary clinical endpoint (time from baseline to clinical deterioration defined as a 2-grade increase on World Health Organization ordinal clinical scale) compared with control (odds ratio [OR] 0.11, 95% confidence interval [CI] 0.01–0.96. p = 0.046) in hospitalized COVID-19 patients [23]. Lopes et al. found that among hospitalized patients, those treated with colchicine suspended supplemental oxygen use before those treated with placebo (median 4 vs. 6.5 days, p < 0.001) and reduced their hospital stay (median 7 vs. 9 days, p = 0.003). In addition, after 1 week of treatment, colchicine markedly reduced C-reactive protein levels compared with placebo [26]. The ColCORONA trial was conducted among non-hospitalized patients in Canada treated with colchicine or placebo. The primary endpoint (a composite of death or hospitalization due to COVID-19 in the 30 days after randomization) was not met (OR 0.79, 95% CI 0.61–1.03, p = 0.08) [30]. In the pre-specified analysis of the study including 4,159 patients with a polymerase chain reaction-confirmed diagnosis, the primary endpoint occurred less frequently in colchicine-treated than in placebo-treated patients (OR 0.75, 95% CI 0.57–0.99, p = 0.04) and the risk for hospitalization was decreased (OR 0.75, 95% CI 0.57–0.99) [30]. For all of these trials, the most common adverse event was diarrhea, especially among patients treated with colchicine, while no safety concerns with regard to infections were recorded. Several other observational studies reported encouraging results, with few, tolerable side effects [24, 25, 27,28,29, 89,90,91,92] (Table 2). Piantoni et al. also reported about long-term results, describing an improved survival in patients treated with colchicine compared with standard-of-care (mortality rate at 270 days: 20% vs. 44%, p = 0.0001) [91].
There are ongoing clinical trials testing whether colchicine may reduce the rate of clinical worsening in hospitalized patients with moderate disease (Treatment with COLchicine of patients affected by COVID-19: a Pilot Study [COLVID- 19], EudraCT number: 2020–001,475-33; Colchicine Counteracting Inflammation in COVID-19 Pneumonia [ColCOVID-19], NCT04322565) or hospitalization in outpatients (CHOICE-19, EudraCT number: 2020-001,806-42). A complete list can be found online (https://clinicaltrials.gov/ct2/results?cond=Covid19&term=colchicine&cntry=&state=&city=&dist =).
Recent meta-analyses, although based on few available studies, have shown encouraging signals of benefit for mortality [84, 94,95,96] and reduced progression towards severe COVID-19 [95]. However, the benefit was not confirmed in the subgroup analysis including RCTs [97]. A Cochrane meta-analysis including three RCTs (11,525 hospitalized patients and 1 RCT with 4,488 non-hospitalized subjects) concluded that colchicine has little or no effect on mortality or clinical progression compared with placebo or standard-of-care alone in patients hospitalized with moderate-to-severe COVID-19 [98]. Indeed, available findings from RCTs are certainly influenced by different sample size, different endpoints, and different follow-up. Importantly, a variable time of administration might represent an additional bias as the only common evidence for colchicine starting was hospitalization for COVID-19, except for the ColCORONA trial. It is then plausible that administration in a later phase of disease may not be as useful as when administered earlier. However, this is not fully understood based on current studies. It appears that, at present time, colchicine cannot be recommended as a first agent to treat hospitalized COVID-19 patients. However, it could be considered on a case-by-case basis for home patients with early presentation of mild symptoms.
Future perspectives
Colchicine has the ability to block NLRP3 inflammasome oligomerization and prevent the processing and release of active IL-1β, thus blocking the subsequent upregulation of downstream cytokines, that are responsible for detrimental effects (Fig. 2). Colchicine, however, is a nonspecific inhibitor of the NLRP3 inflammasome as it has multiple mechanisms of inhibition, either upstream and downstream of NLRP3 inflammasome [99]. For this reason, a specific inhibition might have increased advantages in reducing the progression of the inflammatory burden and its related clinical manifestations through a selective blockade of different steps preceding NLRP3 inflammasome oligomerization and reduced release of IL-1β and IL-18. To this end, different experimental NLRP3 inflammasome inhibitors (BAY 11–7082, MCC950, OLT1177 [dapansutrile], INF4E, 16,673-34-0) were tested in animal models of acute myocardial infarction and are summarized in detail elsewhere [99]. Among these compounds, OLT1177 (dapansutrile) is the only orally active specific NLRP3 inhibitor [100] that has been studied in patients with acute flares of gout [101] and heart failure [102], showing positive results. An RCT is currently ongoing to test the safety and efficacy of dapansutrile in moderate COVID-19 (NCT04540120). Based on current evidence, it is reasonable that the downregulation of NLRP3 inflammasome might blunt the inflammatory cascade elicited by SARS-CoV-2 infection, especially in high-risk patients (i.e., those with obesity, diabetes, cardiovascular or pulmonary diseases). This could depend by the fact that most of these patients have an IL‑1β-mediated inflammation related to their underlying conditions, further exacerbated by viral infection.
Importantly, when considering colchicine for COVID-19 patients, it is important to consider the ground-breaking results provided by the RECOVERY trial testing dexamethasone [103]. Dexamethasone is more powerful compared with colchicine in terms of anti-inflammatory efficacy and suppression of the hyperinflammatory syndrome. The latter, indeed, is responsible for progression of COVID-19 towards severe and critical stages, often fatal. However, dexamethasone is also associated with poor prognosis when administered too early in the course of the disease [103]. For this reason, attention should be paid on the timing of administration of colchicine, that is still a matter of debate. Based on current, limited evidence, it might be hypothesized that colchicine should be administered (i) in very early phases of disease (when dexamethasone is contraindicated) or (ii) in patients with low-grade inflammation and on low-flow oxygen on top of glucocorticoids or (iii) as a long-term therapy (i.e. 1 to 3 months) after hyperinflammation has been controlled. For the latter case, colchicine may be considered as a good choice for patients with persistently increased levels of pro-inflammatory cytokines and persisting symptoms (i.e. Long COVID patients), allowing to switch off inflammation without the long-term side effects of glucocorticoids. This may also be the case of pericarditis, that has been described in patients with COVID-19 [104] and may benefit from colchicine [105]. However, dedicated studies testing colchicine, dapansutrile and dexamethasone in different phases of COVID-19 may help better understand the efficacy of these drugs and the best timing of administration .
While considering hyperinflammation and the role of NLRP3 inflammasome/IL-1β axis in COVID-19, results from the SAVE-MORE (suPAR-guided Anakinra treatment for Validation of the risk and Early Management Of seveRE respiratory failure by COVID-19) trial showed a significant reduction in mortality and hospital stay using anakinra, a recombinant human IL-1 receptor antagonist [106]. The SAVE-MORE study is a phase 3, double-blind RCT that investigated early start of anakinra in hospitalized patients with moderate-to-severe COVID-19 at risk for respiratory failure (defined by an elevated serum level of soluble urokinase-type plasminogen activator receptor [suPAR]). Importantly, nearly 90% of patients were receiving dexamethasone. After 28 days, anakinra reduced clinical deterioration by 64% as compared to placebo. In addition, 28-day mortality was reduced by 55% as well as hospital stay [106]. These findings definitely consolidate previous findings from observational studies about the use of anakinra in COVID-19 patients [10, 11, 107,108,109] and a recent meta-analysis [110]. It is likely that the positive effect of anakinra in COVID-19 patients might depend on a selective blockade of IL-1 differently from colchicine. Indeed, colchicine is an indirect NLRP3 inflammasome inhibitor and this may explain the limited efficacy observed in RCTs.
Conclusions
SARS-CoV-2 can induce the activation of the NLRP3 inflammasome that leads to the activation of several pro-inflammatory pathways. This suggests that pharmacological blockade of the NLRP3 inflammasome is of interest in COVID-19. Indeed, colchicine is known to block the NLRP3 inflammasome and has many advantages, such as a limited immunosuppression, oral administration, and few side effects. As few randomized studies have been published to date and our knowledge of COVID-19 is evolving, additional studies are warranted to unravel how colchicine and other NLRP3 inhibitors reduce the inflammatory burden caused by SARS-CoV-2 and to evaluate the best timing and duration of treatment, either in hospitalized patients and outpatients.
References
Bonaventura A, Vecchie A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021. https://doi.org/10.1038/s41577-021-00536-9.
Iba T, Connors JM, Levy JH. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm Res. 2020;69:1181–9.
Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324:782–93.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet. 2020;395:497–506.
Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–9.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130:2620–9.
Chi Y, Ge Y, Wu B, Zhang W, Wu T, Wen T, et al. Serum cytokine and chemokine profile in relation to the severity of coronavirus disease 2019 in China. J Infect Dis. 2020;222:746–54.
Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020;26:1636–43.
Investigators R-C, Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19. N Engl J Med. 2021;384:1491–502.
Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2:e325–31.
Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. 2020;2:e393–400.
Ucciferri C, Auricchio A, Di Nicola M, Potere N, Abbate A, Cipollone F, et al. Canakinumab in a subgroup of patients with COVID-19. Lancet Rheumatol. 2020;2:e457-ee458.
Group RC. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397:1637–45.
Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 2021;218:e20201707.
Toldo S, Bussani R, Nuzzi V, Bonaventura A, Mauro AG, Cannata A, et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Inflamm Res. 2021;70:7–10.
Junqueira C, Crespo A, Ranjbar S, Ingber J, Parry B, Ravid S, et al. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release. medRxiv. 2021 Mar 8;2021.03.06.21252796.
van den Berg DF, Te Velde AA. Severe COVID-19: NLRP3 inflammasome dysregulated. Front Immunol. 2020;11:1580.
Liantinioti G, Argyris AA, Protogerou AD, Vlachoyiannopoulos P. The role of colchicine in the treatment of autoinflammatory diseases. Curr Pharm Des. 2018;24:690–4.
Schattner A. Colchicine-new horizons for an ancient drug. Eur J Intern Med. 2021;S0953–6205(21):00332.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41.
Marques-da-Silva C, Chaves MM, Castro NG, Coutinho-Silva R, Guimaraes MZ. Colchicine inhibits cationic dye uptake induced by ATP in P2X2 and P2X7 receptor-expressing cells: implications for its therapeutic action. Br J Pharmacol. 2011;163:912–26.
Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. 2020;126:1260–80.
Deftereos SG, Giannopoulos G, Vrachatis DA, Siasos GD, Giotaki SG, Gargalianos P, et al. Effect of colchicine vs standard care on cardiac and inflammatory biomarkers and clinical outcomes in patients hospitalized with coronavirus disease 2019: the GRECCO-19 randomized clinical trial. JAMA Netw Open. 2020;3:2013136.
Brunetti L, Diawara O, Tsai A, Firestein BL, Nahass RG, Poiani G, et al. Colchicine to weather the cytokine storm in hospitalized patients with COVID-19. J Clin Med. 2020. https://doi.org/10.3390/jcm9092961.
Della-Torre E, Della-Torre F, Kusanovic M, Scotti R, Ramirez GA, Dagna L, et al. Treating COVID-19 with colchicine in community healthcare setting. Clin Immunol. 2020;217:108490.
Lopes MI, Bonjorno LP, Giannini MC, Amaral NB, Menezes PI, Dib SM, et al. Beneficial effects of colchicine for moderate to severe COVID-19: a randomised, double-blinded, placebo-controlled clinical trial. RMD Open. 2021. https://doi.org/10.1136/rmdopen-2020-001455.
Manenti L, Maggiore U, Fiaccadori E, Meschi T, Antoni AD, Nouvenne A, et al. Reduced mortality in COVID-19 patients treated with colchicine: results from a retrospective, observational study. PLoS ONE. 2021;16:e0248276.
Sandhu T, Tieng A, Chilimuri S, Franchin G. A case control study to evaluate the impact of colchicine on patients admitted to the hospital with moderate to severe COVID-19 infection. Can J Infect Dis Med Microbiol. 2020;2020:8865954.
Scarsi M, Piantoni S, Colombo E, Airo P, Richini D, Miclini M, et al. Association between treatment with colchicine and improved survival in a single-centre cohort of adult hospitalised patients with COVID-19 pneumonia and acute respiratory distress syndrome. Ann Rheum Dis. 2020;79:1286–9.
Tardif J-C, Bouabdallaoui N, L’Allier PL, Gaudet D, Shah B, Pillinger MH, et al. Colchicine for community-treated patients with COVID-19 (COLCORONA): a phase 3, randomised, double-blinded, adaptive, placebo-controlled, multicentre trial. Lancet Respir Med. 2021;9:924–32.
Group RC. Colchicine in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet Respir Med. 2021;9:1419–26.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Hauenstein AV, Zhang L, Wu H. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr Opin Struct Biol. 2015;31:75–83.
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7.
Dinarello CA. Overview of the interleukin-1 family of ligands and receptors. Semin Immunol. 2013;25:389–93.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21.
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92.
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9.
Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–3.
Spel L, Martinon F. Detection of viruses by inflammasomes. Curr Opin Virol. 2021;46:59–64.
Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev. 2018;281:99–114.
Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. 2021. https://doi.org/10.1038/s41590-021-00886-5.
Bonaventura A, Vecchie A, Mauro AG, Brucato AL, Imazio M, Abbate A. An update on the pathophysiology of acute and recurrent pericarditis. Panminerva Med. 2020. https://doi.org/10.23736/S0031-0808.20.04205-6.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Anand PK, Malireddi RK, Kanneganti TD. Role of the nlrp3 inflammasome in microbial infection. Front Microbiol. 2011;2:12.
da Costa LS, Outlioua A, Anginot A, Akarid K, Arnoult D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019;10:346.
Mauro AG, Bonaventura A, Mezzaroma E, Quader M, Toldo S. NLRP3 inflammasome in acute myocardial infarction. J Cardiovasc Pharmacol. 2019;74:175–87.
Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50.
Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101.
Siu KL, Yuen KS, Castano-Rodriguez C, Ye ZW, Yeung ML, Fung SY, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33:8865–77.
Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–9.
Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221–36.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3.
Freeman TL, Swartz TH. Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol. 2020;11:1518.
Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020;6:31.
Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60:3642–50.
Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H, et al. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009;60:3651–62.
Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, et al. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol. 2001;167:6568–75.
Tsutsui H, Kayagaki N, Kuida K, Nakano H, Hayashi N, Takeda K, et al. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity. 1999;11:359–67.
Xu H, Chitre SA, Akinyemi IA, Loeb JC, Lednicky JA, McIntosh MT, et al. SARS-CoV-2 viroporin triggers the NLRP3 inflammatory pathway. bioRxiv. 2020;69:1450.
He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–7.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
Velavan TP, Meyer CG. Mild versus severe COVID-19: laboratory markers. Int J Infect Dis. 2020;95:304–7.
Huang I, Pranata R, Lim MA, Oehadian A, Alisjahbana B. C-reactive protein, procalcitonin, D-dimer, and ferritin in severe coronavirus disease-2019: a meta-analysis. Ther Adv Respir Dis. 2020;14:1753466620937175.
Lin Z, Long F, Yang Y, Chen X, Xu L, Yang M. Serum ferritin as an independent risk factor for severity in COVID-19 patients. J Infect. 2020;81:647–79.
Henry BM, Aggarwal G, Wong J, Benoit S, Vikse J, Plebani M, et al. Lactate dehydrogenase levels predict coronavirus disease 2019 (COVID-19) severity and mortality: a pooled analysis. Am J Emerg Med. 2020;38:1722–6.
Szarpak L, Ruetzler K, Safiejko K, Hampel M, Pruc M, Kanczuga-Koda L, et al. Lactate dehydrogenase level as a COVID-19 severity marker. Am J Emerg Med. 2020. https://doi.org/10.1016/j.ajem.2020.11.025.
Gorham J, Moreau A, Corazza F, Peluso L, Ponthieux F, Talamonti M, et al. Interleukine-6 in critically ill COVID-19 patients: a retrospective analysis. PLoS ONE. 2020;15:e0244628.
Mojtabavi H, Saghazadeh A, Rezaei N. Interleukin-6 and severe COVID-19: a systematic review and meta-analysis. Eur Cytokine Netw. 2020;31:44–9.
Andreu JM, Timasheff SN. Tubulin bound to colchicine forms polymers different from microtubules. Proc Natl Acad Sci USA. 1982;79:6753–6.
Ben-Chetrit E, Levy M. Does the lack of the P-glycoprotein efflux pump in neutrophils explain the efficacy of colchicine in familial Mediterranean fever and other inflammatory diseases? Med Hypotheses. 1998;51:377–80.
Cronstein BN, Molad Y, Reibman J, Balakhane E, Levin RI, Weissmann G. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J Clin Invest. 1995;96:994–1002.
Paschke S, Weidner AF, Paust T, Marti O, Beil M, Ben-Chetrit E. Technical advance: inhibition of neutrophil chemotaxis by colchicine is modulated through viscoelastic properties of subcellular compartments. J Leukoc Biol. 2013;94:1091–6.
Reibman J, Haines KA, Rich AM, Cristello P, Giedd KN, Weissmann G. Colchicine inhibits ionophore-induced formation of leukotriene B4 by human neutrophils: the role of microtubules. J Immunol. 1986;136:1027–32.
Rudolph SA, Greengard P, Malawista SE. Effects of colchicine on cyclic AMP levels in human leukocytes. Proc Natl Acad Sci USA. 1977;74:3404–8.
Fordham JN, Kirwan J, Cason J, Currey HL. Prolonged reduction in polymorphonuclear adhesion following oral colchicine. Ann Rheum Dis. 1981;40:605–8.
Slobodnick A, Shah B, Krasnokutsky S, Pillinger MH. Update on colchicine, 2017. Rheumatology (Oxford). 2018;57:i4–11.
Abu-Fanne R, Stepanova V, Litvinov RI, Abdeen S, Bdeir K, Higazi M, et al. Neutrophil alpha-defensins promote thrombosis in vivo by altering fibrin formation, structure, and stability. Blood. 2019;133:481–93.
Higazi M, Abdeen S, Abu-Fanne R, Heyman SN, Masarwy A, Bdeir K, et al. Opposing effects of HNP1 (alpha-defensin-1) on plasma cholesterol and atherogenesis. PLoS ONE. 2020;15:e0231582.
Menche D, Israel A, Karpatkin S. Platelets and microtubules. Effect of colchicine and D2O on platelet aggregation and release induced by calcium ionophore A23187. J Clin Invest. 1980;66:284–91.
Shah B, Allen N, Harchandani B, Pillinger M, Katz S, Sedlis SP, et al. Effect of colchicine on platelet-platelet and platelet-leukocyte interactions: a pilot study in healthy subjects. Inflammation. 2016;39:182–9.
Reyes AZ, Hu KA, Teperman J, Wampler Muskardin TL, Tardif JC, Shah B, et al. Anti-inflammatory therapy for COVID-19 infection: the case for colchicine. Ann Rheum Dis. 2020. https://doi.org/10.1136/annrheumdis-2020-219174.
Salah HM, Mehta JL. Meta-analysis of the effect of colchicine on mortality and mechanical ventilation in COVID-19. Am J Cardiol. 2021;145:170–2.
Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J Heart Lung Transplant. 2020;39:405–7.
Mareev VY, Orlova YA, Plisyk AG, Pavlikova EP, Akopyan ZA, Matskeplishvili ST, et al. Proactive anti-inflammatory therapy with colchicine in the treatment of advanced stages of new coronavirus infection. The first results of the COLORIT study. Kardiologiia. 2021;61:15–27.
Pascual-Figal DA, Roura-Piloto AE, Moral-Escudero E, Bernal E, Albendin-Iglesias H, Perez-Martinez MT, et al. Colchicine in recently hospitalized patients with COVID-19: a randomized controlled trial (COL-COVID). Int J Gen Med. 2021;14:5517–26.
RECOVERY trial closes recruitment to colchicine treatment for patients hospitalised with COVID-19. https://www.recoverytrial.net/news/recovery-trial-closes-recruitment-to-colchicine-treatment-for-patients-hospitalised-with-covid-19. Last accessed May 31 2021.
Mansouri N, Marjani M, Tabarsi P, von Garnier C, Mansouri D. Successful treatment of covid-19 associated cytokine release syndrome with colchicine. A case report and review of literature. Immunol Invest. 2020;50:1–7.
Rodriguez-Nava G, Trelles-Garcia DP, Yanez-Bello MA, Chung CW, Trelles-Garcia VP, Friedman HJ. Atorvastatin associated with decreased hazard for death in COVID-19 patients admitted to an ICU: a retrospective cohort study. Crit Care. 2020;24:429.
Piantoni S, Andreoli L, Colombo E, Mendeni M, Ghirardelli ML, Brucato A, et al. Response to: ‘Correspondence on “Association between treatment with colchicine and improved survival in a single-centre cohort of adult hospitalised patients with COVID-19 pneumonia and acute respiratory distress syndrome”’ by Kawada. Ann Rheum Dis. 2021. https://doi.org/10.1136/annrheumdis-2020-219787.
Pinzon MA, Cardona Arango D, Betancur JF, Ortiz S, Holguin H, Arias Arias C, et al. Clinical outcome of patients with COVID-19 pneumonia treated with corticosteroids and colchicine in Colombia. Ann Clin Microbiol Antimicrob. 2021;20:66.
Lombardy Section Italian Society I, Tropical D. Vademecum for the treatment of people with COVID-19. Infez Med. 2020;28:143–52.
Vrachatis DA, Giannopoulos GV, Giotaki SG, Raisakis K, Kossyvakis C, Iliodromitis KE, et al. Impact of colchicine on mortality in patients with COVID-19: a meta-analysis. Hellenic J Cardiol. 2021. https://doi.org/10.1016/j.hjc.2020.11.012.
Hariyanto TI, Halim DA, Jodhinata C, Yanto TA, Kurniawan A. Colchicine treatment can improve outcomes of coronavirus disease 2019 (COVID-19): a systematic review and meta-analysis. Clin Exp Pharmacol Physiol. 2021. https://doi.org/10.1111/1440-1681.13488.
Elshafei MN, El-Bardissy A, Khalil A, Danjuma M, Mubasher M, Abubeker IY, et al. Colchicine use might be associated with lower mortality in COVID-19 patients: a meta-analysis. Eur J Clin Invest. 2021. https://doi.org/10.1111/eci.13645.
Lien CH, Lee MD, Weng SL, Lin CH, Liu LY, Tai YL, et al. Repurposing colchicine in treating patients with COVID-19: a systematic review and meta-analysis. Life (Basel). 2021. https://doi.org/10.3390/life11080864.
Mikolajewska A, Fischer AL, Piechotta V, Mueller A, Metzendorf MI, Becker M, et al. Colchicine for the treatment of COVID-19. Cochrane Database Syst Rev. 2021;10:CD015045.
Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15:203–14.
Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A. 2018;115:E1530–9.
Kluck V, Jansen T, Janssen M, Comarniceanu A, Efde M, Tengesdal IW, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020;2:e270–80.
Wohlford GF, Van Tassell BW, Billingsley HE, Kadariya D, Canada JM, Carbone S, et al. Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J Cardiovasc Pharmacol. 2020;77:49–60.
Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in hospitalized patients with COVID-19. N Engl J Med. 2021;384:693–704.
Diaz-Arocutipa C, Saucedo-Chinchay J, Imazio M. Pericarditis in patients with coronavirus disease 2019: a systematic review. J Cardiovasc Med (Hagerstown). 2021. https://doi.org/10.2459/JCM.0000000000001202.
Imazio M, Brucato A, Lazaros G, Andreis A, Scarsi M, Klein A, et al. Anti-inflammatory therapies for pericardial diseases in the COVID-19 pandemic: safety and potentiality. J Cardiovasc Med (Hagerstown). 2020;21:625–9.
Kyriazopoulou E, Poulakou G, Milionis H, Metallidis S, Adamis G, Tsiakos K, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med. 2021;27:1752–60.
Cauchois R, Koubi M, Delarbre D, Manet C, Carvelli J, Blasco VB, et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. Proc Natl Acad Sci USA. 2020;117:18951–3.
Pontali E, Volpi S, Antonucci G, Castellaneta M, Buzzi D, Tricerri F, et al. Safety and efficacy of early high-dose IV anakinra in severe COVID-19 lung disease. J Allergy Clin Immunol. 2020;146:213–5.
Navarro-Millan I, Sattui SE, Lakhanpal A, Zisa D, Siegel CH, Crow MK. Use of anakinra to prevent mechanical ventilation in severe COVID-19: a case series. Arthritis Rheumatol. 2020;72:1990–7.
Kyriazopoulou E, Huet T, Cavalli G, Gori A, Kyprianou M, Pickkers P, et al. Effect of anakinra on mortality in patients with COVID-19: a systematic review and patient-level meta-analysis. Lancet Rheumatol. 2021;3:e690–7.
Acknowledgements
Figure 1 has been reproduced with permission from Toldo et al., “Inflammasome formation in the lungs of patients with fatal COVID-19” [15]. Figure 2, Panel A has been reproduced with permission from “NLRP3 Inflammasome in Acute Myocardial Infarction” by Mauro et al. [48]. Figure 2, Panel B has been partially created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
Funding
None.
Author information
Authors and Affiliations
Contributions
AB is responsible for conceptualization and first draft of the manuscript. AV contributed to first draft and figure design. LD, FT, AA, and FD critically reviewed the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Aldo Bonaventura received a travel grant from Kiniksa Pharmaceuticals Ltd. to attend the 2019 AHA Scientific Sessions and receive honoraria from Effetti s.r.l. (Milan, Italy) to collaborate on the medical website www.inflammology.org. Alessandra Vecchié received a travel grant from Kiniksa Pharmaceuticals Ltd. to attend the 2019 AHA Scientific Sessions and receive honoraria from Effetti s.r.l. (Milan, Italy) to collaborate on the medical website www.inflammology.org. Lorenzo Dagna received consultation honoraria from Abbvie, Amgen, Biogen, Bristol-Myers Squibb, Celltrion, Galapagos, GlaxoSmithKline, Kiniksa, Novartis, Pfizer, Roche, Sanofi-Genzyme, and SOBI. The Unit of Immunology, Rheumatology, Allergy and Rare Diseases (UnIRAR) received unrestricted research/educational grants from Abbvie, Bristol-Myers Squibb, Celgene, GlaxoSmithKline,Janssen, Kiniksa, Merk Sharp & Dohme, Mundipharma Pharmaceuticals, Novartis, Pfizer, Roche, Sanofi Genzyme, and SOBI. Antonio Abbate has received research support from Kiniksa, Novartis, Olatec, R-Pharm, Serpin Pharma, and has served as an advisor to Abiomed, Cromos Pharma, Effetti, Eli Lilly, Implicit Bioscience, Novo-Nordisk. The remaining authors have nothing to disclose related to this study.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bonaventura, A., Vecchié, A., Dagna, L. et al. Colchicine for COVID-19: targeting NLRP3 inflammasome to blunt hyperinflammation. Inflamm. Res. 71, 293–307 (2022). https://doi.org/10.1007/s00011-022-01540-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-022-01540-y