
Abstract
Introduction
Inflammation is believed to be a contributing factor to many chronic diseases. The influence of vitamin D deficiency on inflammation is being explored but studies have not demonstrated a causative effect.
Methods
Low serum 25(OH)D is also found in healthy persons exposed to adequate sunlight. Despite increased vitamin D supplementation inflammatory diseases are increasing. The current method of determining vitamin D status may be at fault. The level of 25(OH)D does not always reflect the level of 1,25(OH)2D. Assessment of both metabolites often reveals elevated 1,25(OH)2D, indicating abnormal vitamin D endocrine function.
Findings
This article reviews vitamin D's influence on the immune system, examines the myths regarding vitamin D photosynthesis, discusses ways to accurately assess vitamin D status, describes the risks of supplementation, explains the effect of persistent infection on vitamin D metabolism and presents a novel immunotherapy which provides evidence of an infection connection to inflammation.
Conclusion
Some authorities now believe that low 25(OH)D is a consequence of chronic inflammation rather than the cause. Research points to a bacterial etiology pathogenesis for an inflammatory disease process which results in high 1,25(OH)2D and low 25(OH)D. Immunotherapy, directed at eradicating persistent intracellular pathogens, corrects dysregulated vitamin D metabolism and resolves inflammatory symptoms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammation is involved in many chronic diseases and concern has been raised about the influence of vitamin D deficiency on inflammatory processes. When studies found an association between inflammatory diseases and low serum 25-hydroxyvitamin D (25(OH)D), further research found evidence of low vitamin D in a large segment of the general population. This led some authorities to declare a world-wide epidemic of vitamin D deficiency and to recommend vitamin D supplementation. Experts are debating the definition of vitamin D deficiency and the appropriate vitamin D doses, while further research is being done to determine if vitamin D supplementation has the intended effect.
According to some current definitions of vitamin D deficiency, even healthy persons, exposed to adequate sunlight, are unable to acquire enough vitamin D without supplementation. Often reiterated causes of vitamin D deficiency can be disputed in the light of more current research. In the absence of definitive studies, authorities are questioning the wisdom of supplementing the general population with vitamin D. The definition of Vitamin D deficiency needs re-evaluation in view of the fact that low 25(OH)D is found in both healthy and sick individuals. Concerns about vitamin D deficiency merit a closer look at the current method of determining vitamin D status because the level of 25(OH)D does not always reflect the level of 1,25-dihydroxyvitamin-D (1,25(OH)2D). Analysis of this active metabolite may reveal elevated 1,25(OH)2D) in the presence of low 25(OH)D and lead to a diagnosis of abnormal vitamin D endocrine system function.
An infectious pathogenesis posits that intracellular bacteria disrupt the vitamin D regulated immune system, resulting in persistent infection and chronic inflammation. In the clinical setting, a novel immunotherapy is demonstrating the ability to resolve vitamin D metabolism dysfunction, restore immune function, and thus, eliminate infection and reduce inflammation. This review ponders the question, “Is low 25(OH)D a cause of, or a consequence of inflammation?” The answer is found in the evidence that adds persistent intracellular infection to the equation.
Vitamin D metabolism
The sequential metabolic processes that convert biologically inactive, parental vitamin D into active metabolites begin when vitamin D3 is photosynthesized in the skin or when vitamin D2 or D3 is ingested. Vitamin D is transported to the liver where it is hydroxylated by an enzyme (CYP2R1, also known as cytochrome P450 2R1) to produce 25(OH)D [1]. 25(OH)D is then transported to the kidneys where it is hydroxylated by another enzyme (CYP27B1, formerly 1a-hydroxylase) to produce 1,25(OH)2D. 1,25(OH)2D (also known as calcitriol), the active metabolite, is the most potent steroid hormone in the human body [2]. Feedback mechanisms regulate production of 1,25(OH)2D in the kidneys via serum levels of parathyroid hormone (PTH), fibroblast-like growth factor-23 (FGF23) calcium, and phosphate [3]. 1,25(OH)2D is also produced in many other tissues (e.g., skin, macrophages, colon, pancreas, blood vessels, etc.) by enzymatic actions [4]. The vitamin D binding protein (VDBP) transports 1,25(OH)2D to the vitamin D receptor (VDR) in the cell nucleus [5]. The VDR is a member of the nuclear receptor family of ligand-regulated transcription factors. 1,25(OH)2D binds to the VDR and mediates the transcription of DNA, triggered by signaling proteins, like nuclear factor kappa-B (NFk-B) [6] (Fig. 1).

Synthesis and metabolism of vitamin D. Sequential metabolic processes convert biologically inactive, parental vitamin D into active metabolites
The influence of 1,25(OH)2D on the immune system is one of its most important roles. 1,25(OH)2D regulates the immune system via the VDR which is present in most immune cell types, particularly in antigen-presenting cells (APCs) such as monocytes, macrophages and dendritic cells [7]. 1,25(OH)2D activates the VDR to express antimicrobial peptides (AMPs) such as cathelicidin and beta defensins which attack pathogens [8, 9]. In general, the innate immune system is enhanced and the adaptive immune system is inhibited by 1,25(OH)2D [10, 11]. Thus, an effective immune response is heavily dependent on the vitamin D endocrine system which performs a balancing act of inflammation versus anti-inflammation.
Vitamin D deficiency
Concerns about vitamin D deficiency arose when studies showed patients with autoimmune diseases have lower levels of serum 25(OH)D and study subjects given vitamin D had lower rates of autoimmune diseases and fewer markers of inflammation [12, 13]. However, authorities have not agreed on the significance of low 25(OH)D and without a consistent normal range for serum 25(OH)D, the definitions of vitamin D insufficiency and deficiency from the Vitamin D Council, the Endocrine Society and the Institute of Medicine vary significantly.
The Vitamin D Council definition [14]:
-
Deficient: 0–40 ng/ml
-
Sufficient: 40–80 ng/ml
-
High normal: 80–100 ng/ml
The Endocrine Society definition [15]:
-
Deficiency ≤ 20 ng/ml
-
Insufficiency = 20–29 ng/ml
The Institute of Medicine definition [16]:
-
Risk/deficiency ≤12 ng/ml
-
Risk/insufficiency = 12–20 ng/ml
-
Sufficient = 20 ng/ml
In 2006, the Merck Manual listed 25–40 ng/ml as the normal 25(OH)D range [17]. Recently, this range has skyrocketed to 30–74 ng/ml [18]. Quest Diagnostics now lists the upper limit of normal 25(OH)D as 100 ng/ml [19]. Laboratory reference ranges for serum 25(OH)D levels have long been based upon average values from populations of healthy individuals but many people are now supplementing with vitamin D. In the US, the leading authority regarding medical research is the prestigious Institute of Medicine (IOM). The 2010 IOM report on vitamin D emphasized that the current measurements, or cut-off points, of sufficiency and deficiency of 25(OH) D in use by laboratories have not been set using rigorous scientific studies. It suggests that since no central authority has determined which cut-off points to use, reports of deficiency and lab ranges may be skewed and numbers overestimated [16]. Therefore, it would be prudent to use the IOM vitamin D deficiency guideline in the clinical setting, for clinical studies and when evaluating research results.
Purported reasons for vitamin D deficiency
Is low 25(OH)D among the general population an accurate assessment of vitamin D deficiency? Many reasons are cited for the current ‘epidemic’ of vitamin D ‘deficiency’ but closer examination reveals these beliefs are based on outdated or limited studies and can be challenged with more recent research.
Melanin pigmentation is only one factor that determines the amount of vitamin D3 which is photosynthesized [20, 21]. Bogh et al. [22] measured the baseline serum 25(OH)D and total cholesterol levels of 182 fair-skinned and dark-skinned subjects; and studied the effect of UV radiation on their serum 25(OH)D levels. They found the amount of serum 25(OH)D produced was determined by the amount of cholesterol in the skin, not on skin pigmentation. Matsuoka et al. [23] investigated the effect of racial pigmentation on vitamin D3 formation, simulating the process with a fixed dose of UVB radiation and concluded that while racial pigmentation has a photo-protective effect, it does not prevent the generation of normal levels of active vitamin D metabolites. Persons with dark skin also compensate for low 25(OH)D by rapidly converting it to the active 1,25(OH)2D metabolite, thus allowing them to maintain adequate vitamin D status [24]. Skin pigmentation does not appear to negatively affect vitamin D status [25].
Clothing is a barrier to vitamin D photosynthesis but this is an issue only for people who cover themselves from head to toe [26]. It takes relatively little sunlight exposure to acquire adequate stores of vitamin D and few people wear enough clothes to prevent that from happening. Ten to 15 min of sunlight or daylight exposure to a small area of skin (e.g., the forearm or face, etc.) twice a week, without sunscreen, supplies all the vitamin D necessary for health [27]. The belief that sunscreen lotion blocks vitamin D production is based on a 1987 study done by Matsuoka et al. [28] that was funded by the ultraviolet foundation, which is supported by the tanning bed industry. Contradictory information was provided by Diehl and Chiu [29] which concluded that although sunscreens are effective, many may not actually be blocking UV-B because they are improperly or inadequately applied [30]. Thus, sunscreen use may not actually diminish vitamin D synthesis in real world use. However, prolonged unprotected sun exposure should be avoided to reduce the risk of developing skin cancer.
Although pollution can block some ultraviolet radiation, even in urban areas of high pollution 50 % of UV rays reach the ground [31]. A significant amount of UV radiation exposure can be obtained in dense metropolitan areas; tall buildings provide shade but shade gives up to 50 % of UV rays. Indoor workers receive 10–20 % of outdoor workers’ yearly UV exposure [31]; and for many, this may be adequate, especially if sunlight exposure is higher when they are not working. UV radiation is reflected or scattered to varying extents by different surfaces. The scattering and absorption of light by clouds may not significantly reduce natural light exposure because over 90 % of UV rays may penetrate clouds [31]. Environmental factors are rarely an impediment to photosynthesis of adequate vitamin D.
As the skin ages, there is a decline in the cutaneous levels of 7-dehydrocholesterol, resulting in a marked reduction of the skin’s capacity to produce vitamin D3 [32]. However, despite the up to fourfold reduction in vitamin D3 production in a 70-year-old compared to a 20-year-old, the skin has such a high capacity to make vitamin D3 that elders exposed to sunlight will produce an adequate amount of vitamin D3 to satisfy their vitamin D requirement [33, 34].
A 1988 study by Webb et al. [35] is often cited to support the conviction that latitude dramatically influences the amount of solar radiation available to synthesize vitamin D3. However, other researchers who conducted more recent studies refute this hypothesis. Kimlin et al. [36] report, “It may no longer be correct to assume that vitamin D levels in populations follow latitude gradients”. And Lubin [37] states, “Geophysical surveys have shown that UV-B penetration over 24 h, during the summer months at Canadian north latitudes when there are many hours of sunlight, equals or exceeds UV-B penetration at the equator.” Ross et al. [16] report that ample opportunities exist to form vitamin D (and store it in the liver and fat for later use) from exposure to sunlight during the spring, summer, and fall months even in the far north latitudes.
Acceptance of these vitamin D myths regarding photosynthesis prevents consideration of the alternate hypothesis—low serum 25(OH)D, despite adequate photosynthesis of vitamin D3, is a result of an inflammatory process.
Low vitamin D is found in healthy subjects
Many studies of healthy subjects have found levels of 25(OH)D that, by some vitamin D definitions, are declared deficient (hypovitaminosis-D) [38, 39]. Vitamin D levels that are considered deficient have even been found in persons who are exposed to abundant sunlight [40]. Binkley et al. [41] showed a mean 25(OH)D level of 31.6 ng/ml among healthy young adult Hawaiian surfers. It is clear that low levels of 25(OH)D are found in both healthy persons and those with autoimmune or chronic inflammatory diseases. Opposing reasoning can be used to explain this contradiction. One explanation reasons that healthy persons with low 25(OH)D will become sick and sick people will develop lower 25(OH)D levels (Fig. 2); however, studies do not support this hypothesis. The correct explanation may be that, in the absence of disease, low 25(OH)D is normal.

Interpretation of vitamin D deficiency via calcitriol measurement. Since low 25(OH)D is found in both healthy persons and those with autoimmune or chronic inflammatory diseases, assessing vitamin D status with measurement of 1,25(OH)2D may be helpful
Low vitamin D in the presence of diseases
Since low 25(OH)D is found in both healthy persons and those with autoimmune or chronic inflammatory diseases, assessing vitamin D status with the measurement of an additional clinical marker may be helpful. It is asserted that low levels of 25(OH)D accurately reflect vitamin D status; however, measurement of 1,25(OH)2D often demonstrates a positive correlation of elevated 1,25(OH)2D to inflammatory diseases (Fig. 2). This is illustrated by Blaney et al. [42] in a study of 100 patients with autoimmune and chronic disease which found that 85 % of subjects had levels of 1,25(OH)2D higher than 46.2 pg/ml without hypercalcemia. Although this serum level may be considered normal by some, lab ranges for 1,25(OH)2D may have been skewed high by the presence of patients with unrecognized persistent intracellular infection and thus, dysregulated vitamin D metabolism. The Danish 1,25(OH)2D population data (from a large and reliable study) found the mean value for 1,25(OH)2D in a normal population was 29 pg/ml with a standard deviation of 9.5 [43]. More frequent measurement of both D-metabolites in the clinical and research settings, may shed light on the true meaning of low 25(OH)D.
Rickets is often cited as proof of the need for vitamin D supplementation. However, a review of the metabolic processes involved provides some prospective. Adequate vitamin D is essential to prevent rickets, but adequate calcium is equally important; if either calcium or vitamin D is deficient, bone health suffers. Hypophosphatemia is the common denominator of all rickets; low calcium intake leads to hyperparathyroidism, which leads to high phosphorus excretion and, thus, phosphorus deficiency [44]. Rickets is rare in the developed world; however, children in developing countries, who usually photosynthesize enough vitamin D from sunlight, develop rickets if poverty prevents them from eating enough calcium-rich food [45, 46]. Studies have found that rickets occurs in sunny countries due to poor calcium intake and is cured with increased calcium ingestion [47, 48].
Osteoporosis is another disease which is closely linked with vitamin D. Adequate vitamin D is an important factor in maintaining bone health to avoid osteoporosis but a study by Reid et al. [49] published in The Lancet found little evidence supporting the use of vitamin D supplements by seniors hoping to improve bone density and ward off potential fractures. 1,25(OH)2D maintains calcium homeostasis between blood, cells and bones by stimulating calcium absorption from the intestines, reabsorption in the kidneys, and resorption in bones [50]. 1,25(OH)2D up-regulates the VDR in the small intestine, which then transcribes genes that shuttle calcium and phosphorus through the intestinal epithelium. However, mucosal response and calcium/phosphorus absorption are dependent on a competent VDR and elevated 1,25(OH)2D reduces VDR competence [51]. Thus, calcium and phosphorus absorption may be inhibited if VDR function is impaired by elevated 1,25(OH)2D. This is illustrated by Abreu et al. [52] in a study of Crohn’s patients with elevated 1,25(OH)2D and low bone mineral density which concluded that treatment of the underlying inflammation would improve metabolic bone disease. In fact, there is ample evidence that elevated 1,25(OH)2D leads to bone loss. Brot et al. [53] found that elevated levels of 1,25(OH)2D were strongly associated with decreased bone mineral density and content, and increased bone turnover. When levels are above 42 pg/ml 1,25(OH)2D stimulates bone osteoclasts. This leads to osteoporosis, dental fractures and calcium deposition into the soft tissues [54]. Vanderschueren et al. [55] found that a combination of high 1,25(OH)2D and low 25(OH)D is associated with the poorest bone health.
Vitamin D supplementation
It is reasoned that if low 25(OH)D indicates a current or potential disease state, then increasing 25(OH)D by supplementing with vitamin D should provide some symptom relief and/or protection. So far, there is scant evidence for this hypothesis [56, 57]. According to Ross et al. [16] in the 2010 IOM report, “Outcomes related to autoimmune disorders, cancer, cardiovascular disease and hypertension, diabetes and metabolic syndrome, falls and physical performance, immune functioning, infections, neuropsychological functioning, and preeclampsia could not be linked reliably with calcium or vitamin D intake and were often conflicting.” Despite the recent increase in vitamin D supplementation, chronic diseases have increased and are expected to continue increasing [58, 59].
Consequently, more vitamin D experts are beginning to reconsider vitamin D supplementation among the general population [60]. Recommending higher vitamin D intake to large populations carries the potential risk of overdosing certain individuals [61]. It is difficult to ingest too much vitamin D from food, and natural mechanisms regulate the amount of vitamin D3 photosynthesized from sunlight [62]. However, elevated 25(OH)D and hypervitaminosis-D can occur due to vitamin D supplementation [63]. A study by Noordam et al. [65] cast doubt on the causal nature of previously reported associations between low levels of vitamin D and age-related diseases and mortality. A comprehensive review by Autier et al. [65] concluded that low concentrations of 25(OH)D are most likely an effect of health disorders and not a cause of illness. Commenting on the findings in a press statement, Autier et al. [64] advised against vitamin D supplementation and explained the observed discrepancy between observational and randomized trials:
Decreases in vitamin D levels are a marker of deteriorating health. Ageing and inflammatory processes involved in disease occurrence and clinical course reduce vitamin D concentrations, which would explain why vitamin D deficiency is reported in a wide range of disorders. We postulate that inflammation is the common factor between most non-skeletal health disorders and low 25(OH)D concentrations. Inflammatory processes involved in disease occurrence and clinical course would reduce 25(OH)D, which would explain why low vitamin D status is reported in a wide range of disorders. However, increases in 25(OH)D have no effect on inflammatory processes or on disorders at the origin of these processes.
A 2014 meta-analysis by Bolland et al. [66] on the effects of vitamin D supplementation on skeletal, vascular, or cancer outcomes concludes that vitamin D supplementation with or without calcium does not reduce skeletal or non-skeletal outcomes in unselected community-dwelling individuals by more than 15 %. The authors further state that future trials with similar designs are unlikely to alter these conclusions. Because of emerging concerns about elevated 25(OH)D, the IOM has shifted the paradigm from thinking about ‘more is better’ to a more risk-averse approach [67]. It has also challenged the notion that harm should be viewed in terms of vitamin D toxicity such as hypercalcemia, hypercalciuria, or metastatic calcification and has advanced the concept of ‘harm’ in terms of chronic disease outcomes and mortality [16]. Because adverse effects of vitamin D supplementation may take decades to be realized, clinicians (mindful of the medical ethics precept “First, do no harm”) should err on the side of caution; follow the IOM guideline and wait for the results of long-term vitamin D studies.
Bacterial pathogenesis of low vitamin D hypothesis
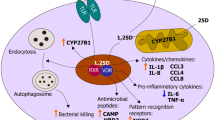
If evidence indicates that most people get adequate vitamin D from sunlight exposure but healthy persons are found to be ‘deficient’ by recent standards, what is the explanation for this phenomenon? Vitamin D proponents use a disease deficiency model to explain low levels of 25(OH)D. Their hypothesis states low 25(OH)D causes chronic diseases; however, a pathogenesis has not been elucidated [68]. Low serum 25(OH)D in the presence of disease can also be explained with a dysregulated vitamin D metabolism model [69]. This hypothesis proposes that low vitamin D is the consequence of a chronic inflammatory process caused by persistent infection. The bacterial pathogenesis theorizes that intracellular (cell wall deficient) bacteria invade nucleated cells, use strategies to avoid destruction and cause abnormal vitamin D endocrine function, resulting in low vitamin D. Excess 1,25(OH)2D is produced in an effort to up-regulate the VDR to transcribe AMPs; and 25(OH)D is rapidly metabolized in the process, resulting in a low serum level. The resulting elevated 1,25(OH)2D causes chronic, systemic inflammation and its accompanying symptoms (Fig. 3).

Proposed hypothesis for chronic inflammation caused by persistent intracellular infection. Intracellular bacteria invade nucleated cells and use strategies to avoid destruction. Excess 1,25(OH)2D is produced in an effort to up-regulate the vitamin D receptor to transcribe AMPs; and 25(OH)D is rapidly metabolized in the process, resulting in a low serum level. The resulting elevated 1,25(OH)2D causes chronic, systemic inflammation and its accompanying symptoms
The existence of bacteria which are capable of invading human cells has been known for over a century and are described by many authors [70, 71]. The lack of a cell wall enables them to enter human cells and proliferate because they fail to elicit an appropriate response when the immune system is compromised. In particular, they enter the macrophages—the very immune cells deployed to kill invading pathogens. The inability of most research labs to culture cell wall deficient (CWD) bacteria has been an obstacle to their acceptance, and reliance on Koch’s postulates has made it difficult to correlate CWD bacteria to specific diseases [72]. But some researchers believe Koch’s postulates may have to be redefined in terms of molecular data when dormant and non-culturable bacteria are implicated as causative agents of mysterious diseases [73]. O´Connor et al. [74] (in their article entitled Emerging Infectious Determinants of Chronic Diseases) report, “microbes can now be irrefutably linked to pathology without meeting Koch’s postulates” and “…powerful tools of molecular biology have exposed new causal links by detecting difficult-to-culture and novel agents in chronic illness settings.”
Domingue [75] commented, “This might translate into an etiology for chronic inflammatory diseases, when the stressed bacteria increase in numbers and overwhelm the normal biological functions of the host.” Nunez [76] was quoted in the University of Michigan Health System newsletter, “In our study, the presence of bacterial microbes inside the cell is what triggers the immune response.” Rolhion and Darfeuille-Michaud [77] observed that the presence of pathogenic invasive bacteria could be the link between an innate immune response to invasive bacteria and the development of inflammation. A number of studies suggest disease associations with CWD bacteria [78–83]. Verway et al. [79] report “…data suggest that at least a subset of the genetic predisposition to Crohn’s disease results from defects in the detection and/or processing of intracellular pathogens by the innate immune system.” O’Connor et al. [74] state, “The epidemiologic, clinical, and pathologic features of many chronic inflammatory diseases are consistent with a microbial cause. Infectious agents likely determine more cancers, immune-mediated syndromes, neurodevelopmental disorders, and other chronic conditions than currently appreciated.”
CWD bacteria are considered communicable but not contagious; protective immunity depends on an effective cell-mediated immune response [80]. It is now well appreciated that pathologic processes caused by infectious agents may only emerge clinically after an incubation of decades [81]. Among the speculated causes of the increase in chronic infections are overuse of beta-lactam antibiotics [82, 83] and immunosuppression via excess 25(OH)D production [84] or immunosuppressive medications [85]. Many microbiologists now believe at least some, if not all, of the inflammation which drives the chronic disease process is caused by the presence of these stealthy intracellular pathogens [86]. A considerable body of experimental and clinical evidence supports the concept that difficult-to-culture and dormant bacteria are involved in latency of infection and that these persistent bacteria may be pathogenic [73, 87, 88]. McDougal [89] states, “Evidence now confirms that non-communicable chronic diseases can stem from infectious agents.”
Effects of intracellular pathogens on the immune system
Pathogens gain many advantages by parasitizing immune cells and altering nuclear receptor activity. Tissue invasion provides a privileged niche with access to host protein and iron, sequestration from immune response, and a means for persistence [90]. In the arms race of host–microbe co-evolution, successful microbial pathogens have evolved innovative strategies to evade host immune responses. For example, ‘crosstalk manipulation’ undermines host defenses and contributes to microbial adaptive fitness [91, 92]. Pathogenic microbes also induce stress responses which protect the cell from lethal factors, express proteases that degrade AMPs, use biofilms as a shield and modulate host cell motility to facilitate establishment of an infection [93, 94]. Genetic foreign and host protein interactions alter gene transcription and translation mechanisms, and many species survive by horizontal gene transfer [95, 96].
It is theorized that bacteria have developed some of these strategies in order to invade host cells and remain undetected within cellular cytoplasm. Many bacterial pathogens form antibiotic-tolerant persister cells which can replicate within macrophages. In this form they can cause subclinical infection and have been associated with chronic diseases [95, 97, 98]. Intracellular bacteria can modulate cytokine production [99]; and in monocytes and macrophages, cytokine activation markedly inhibits 1,25(OH)2D/VDR gene transcription [100].
Macrophage microbicidal mechanisms are responsible for the control and elimination of pathogens. 1,25(OH)2D production and action in macrophages activates toll-like receptors to increase expression of the AMP cathelicidin which kills infectious invaders [101, 102]. When the immune system is fighting a persistent microbe, inflammatory molecules are continuously released in an effort to kill the pathogen [103]. Immune defenses stimulate Th17 cells and contribute to the development of chronic inflammatory conditions [104, 105]. An ineffective immunological response causes low-grade inflammation and phagocyte-inflicted tissue damage plays an important role in many chronic diseases [106]; autoimmune patients acquire a distinct pathogenic microbiota and multi-morbidity often results [107, 108]. Therefore, it is reasonable to infer that bacteria have evolved strategies which allow them to persist within host cells. The exact mechanisms are unknown and warrant further study.
The compromised immune system, infection and vitamin D
In an essay on the renin–angiotensin system (RAS) and immune response, Smith [109] postulated that unresolved cellular stress may be caused by infectious agents to avoid adaptive immune responses. The host immune response has developed many mechanisms to neutralize and remove pathogenic bacteria. In turn, pathogenic bacteria have developed mechanisms to alter and evade the host immune response [110, 111]. Regulation of the VDR is a common mechanism used in the host defense against pathogens but certain microbes have been shown to slow innate immune defenses by down-regulating the VDR:
-
Mycobacterium tuberculosis down-regulates VDR activity [112].
-
Mycobacterium leprae inhibits VDR activity through down-regulation of CYP27B1 in monocytes [113].
-
Aspergillus fumigatus secretes a toxin capable of down-regulating the VDR in macrophages [114].
-
Epstein–Barr virus lowers VDR activity [115].
-
HIV completely shuts down VDR activity [116].
-
In VDR knockout mice, a circumstance that closely mimics extreme VDR dysregulation, 1,25(OH)2D levels increase by a factor of ten [117].
Studies also point to immune system depression and elevated 1,25(OH)2D in chronic diseases [118]:
-
Sarcoidosis patients are deficient in cathelicidin despite healthy vitamin D3 levels [119].
-
1,25(OH)2D is high (>60 pg/ml) in 42 % of Crohn’s patients and the source of the active vitamin D may be the inflamed intestine [52].
-
1,25(OH)2D is elevated in the synovial fluid of patients with RA (rheumatoid arthritis) [120].
-
Crohn’s disease decreases expression of cathelicidin [121].
High levels of 1,25(OH)2D may result when down-regulation of the VDR by bacterial ligands prevents the receptor from expressing enzymes necessary to keep 1,25(OH)2D in a normal range [42]. Elevated 1,25(OH)2D further reduces VDR competence, suppresses macrophage function, and blocks the nuclear factor kappa-B pathway; thus inhibiting immune system function [116, 122, 123]. Reducing the ability of the VDR to express elements of innate immune function allows intracellular bacteria to persist in the cytoplasm of nucleated cells and may account for the increased susceptibility to non-bacterial co-infections that are commonly found in patients with chronic illnesses [124, 125]. Theoretically, immune system suppression allows parasitic microbes to persist and proliferate in host phagocytes, successfully compete for nutritional resources, and displace commensal organisms from their niche [126]. Elevated 1,25(OH)2D appears to be evidence of a disabled immune system’s attempt to activate the VDR to combat infection.
Autoimmune disease
Numerous examples can be found in which pathogens express antigens that cross-react with host antigens or induce local inflammatory responses that can lead to autoimmune responses through a very complex set of circumstances [127]. The prevailing theory regarding the etiology of autoimmune disease states that an overactive immune system produces auto-antibodies against self, but infection as an environmental factor in autoimmunity has long been recognized. An alternate hypothesis posits a bacterial etiology in which a persistent intracellular infection causes a cytokine release that induces signals to T cells and B cells, and the antibodies they produce (to the intracellular invader) include some that attack human proteins, as well as target the pathogens [128, 129]. Christen et al. [130] explored this hypothesis, “In theory, a structural similarity or identity between the host and an invading pathogen might cause the immune system of the host to react not only to the pathogen but also to self-components.” Infections can act as environmental triggers inducing or promoting autoimmune disease in genetically predisposed individuals [131]; researchers have shown how antinuclear antibodies (ANA) are created in response to infectious agents [132, 133].
Vitamin D appears to have a positive effect on autoimmune disease due to immune system suppression [122, 134, 135] and immune suppression is considered therapeutically beneficial for autoimmune diseases [136, 137]. However, vitamin D proponents have failed to recognize that positive effects are due to the immunosuppressive effect of elevated 25(OH)D or to understand that immunosuppression is contraindicated because of the probable presence of intracellular infection. When the immune system is suppressed clinical disease markers and symptoms are reduced but immunosuppression does not address an underlying cause of persistent bacteria, thus relapse is common [138]. Verway et al. [79] wonder, “Is a specific pathogen responsible for disease or rather is a dysregulated immune response generated against a complex microbial population? Why would immune-suppressive drugs be efficacious if the primary defect is an immune deficiency?” Much of current research focuses on finding drugs to suppress inflammation but, according to Collins [139], 95 % of these studies have failed It seems clear a better direction is needed. Immunotherapy which restores VDR competence corrects dysregulated vitamin D metabolism and eliminates intracellular bacteria could be the answer (as discussed in the section titled Restoring VDR Competence).
Dysregulated vitamin D metabolism
In a healthy individual, the complex interplay between innate and adaptive immunity cooperates to mount an appropriate response to infection through regulation of the vitamin D endocrine system [140]. The immune system detects and responds to the presence of intracellular bacteria by producing more 1,25(OH)2D to activate the VDR and express the crucial endogenous AMPs which enable the innate immune system to target intracellular pathogens [141]. Renal production of 1,25(OH)2D is tightly self-regulated, with the end product down-regulating its own further production. In contrast, extra-renal tissues (e.g., uterine decidua and placenta, colon, breast, prostate, spleen, bone, keratinocytes, melanoma and synovial cells, pulmonary monocytes and macrophages, etc.) which produce 1,25(OH)2D are regulated by cytokines (e.g., interferon-gamma), lipopolysaccharide, nitric oxide and intracellular VDBP, which activate the enzyme CYP27B1 to stimulate conversion of 25(OH)D to 1,25(OH)2D [142]. This extra-renal production of 1,25(OH)2D in tissues infected with intracellular bacteria can result in an excess in production of 1,25(OH)2D which may contribute to depletion and low levels of 25(OH)D [143] (Fig. 4).

Proposed hypothesis for excess 1,25(OH)2D production in bacterially-stimulated extra-renal tissues. Extra-renal tissues, which produce 1,25(OH)2D, are regulated by cytokines, lipopolysaccharide, nitric oxide and intracellular VDBP, which activate the enzyme CYP27B1 to stimulate conversion of 25(OH)D to 1,25(OH)2D, resulting in low 25(OH)D
Because extra-renal production of 1,25(OH)2D is primarily dependent on the availability of 25(OH)D [144], supplementation with vitamin D to increase 25(OH)D may promote the production of 1,25(OH)2D in non-renal tissues that are sites of intracellular infection and result in hypervitaminosis-D. Sunlight appears to play a part in this process and many patients with autoimmune disease report sun sensitivity. The skin (dermal fibroblasts and keratinocytes possess VDR) has the capacity to synthesize 1,25(OH)2D, and represents an important target tissue for 1,25(OH)2D [145]. If keratinocytes in the skin are infected, natural regulation of photosynthesis may be thwarted and solar energy may overstimulate cellular activity, resulting in an increase in cutaneous production of vitamin D3, 25(OH)D and 1,25(OH)2D following sun exposure.
We hypothesize that when nucleated cells are parasitized by intracellular bacteria, extra-renal production of 1,25(OH)2D increases, the kidneys lose control of 1,25(OH)2D production, and pro-hormone 25(OH)D decreases due to rapid conversion to 1,25(OH)2D. The following mechanisms are thought to be responsible (Fig. 5):

Proposed hypothesis for dysregulated vitamin D metabolism caused by intracellular pathogens. Theoretically, when nucleated cells are parasitized by intracellular bacteria, extra-renal production of 1,25(OH)2D increases, the kidneys lose control of 1,25(OH)2D production, and pro-hormone 25(OH)D decreases due to rapid conversion to 1,25(OH)2D
-
Inflammatory cytokines activate CYP27B1, an enzyme that causes more 25(OH)D to be converted to 1,25(OH)2D [146].
-
The microbial-repressed VDR cannot transcribe CYP24A1 (formerly 24-hydroxylase), an enzyme that breaks down excess 1,25(OH)2D [147].
-
Excess 1,25(OH)2D binds the PXR (pregnane X receptor), to inhibit conversion of vitamin D3 to 25(OH)D so 25(OH)D is down-regulated [148].
-
1,25(OH)2D inhibits the hepatic synthesis of 25(OH)D [149].
Thus, low 25(OH)D may be a consequence of the inflammatory process. More studies are concluding that suboptimal circulating levels of vitamin D appear to be caused by the disease process. Waldronn et al. [150] found serum 25(OH)D was decreased following an acute inflammatory insult (i.e., orthopedic surgery) and concluded that hypovitaminosis-D may be the consequence rather than cause of chronic inflammatory diseases. Ferder et al. [151] state, “…there may be a relationship between inflammatory processes induced by chronic overstimulation of the renin angiotensin system (RAS) and the worldwide vitamin D deficiency. In fact, the pandemic of vitamin D deficiency could be the other face of increased RAS activity, which could potentially cause a lower activity or lower levels of Vitamin D.”
Diagnosis of dysregulated vitamin D metabolism
Assessing dysregulated vitamin D metabolism has the potential to guide clinical practice [152, 153]. Vitamin D status is currently determined by measuring the level of serum 25(OH)D which, presumably, reflects the serum levels of other vitamin D metabolites (e.g., vitamin D3, vitamin D2 and 1,25(OH)2D, etc.). This measurement may not, however, provide enough information to assess vitamin D endocrine function. The clinical utility of measuring 1,25(OH)D is not fully understood, but it is clear that associations are being made between this active metabolite of vitamin D and disease states [154]. 1,25(OH)2D is not being used as a measure associated with vitamin D nutritional status or as an intermediate marker related to health outcomes, or even routinely assessed in vitamin D research. In the context of solving the puzzle of low 25(OH)D, the reasons cited for this lapse fail to consider the possibility of abnormal levels in the presence of chronic inflammation:
-
1,25(OH)2D has a short half-life (hours) and fluctuates rapidly.
However, a high result may be discovered even at trough level.
-
1,25(OH)2D levels are regulated by PTH, calcium, phosphate.
This is not true if extra-renal production is prevalent [143].
-
1,25(OH)2D does not decrease until 25(OH)D is very low.
A low 25(OH)D may be a sign that 1,25(OH)2D is abnormally high [55].
-
1,25(OH)2D is only over-produced in hypercalcemic disease states such as sarcoidosis.
Studies show this is not true [42].
-
1,25(OH)2D may be elevated as a result of up-regulation of the CYP27B1 enzyme.
This begs the question, Why is this enzyme elevated [146]?
Measuring both 25(OH)D and 1,25(OH)2D (and PTH, calcium, phosphate when indicated) as clinical markers in chronic disease is more likely to provide a true picture of vitamin D status, than measuring 25(OH)D alone [155, 156] (Table 1). Measuring 1,25(OH)2D should be considered in patients with low 25(OH)D, abnormal laboratory results (especially inflammatory markers), a diagnosis of autoimmune disease or other chronic inflammatory illness, or signs of chronic systemic inflammation. For example, elevated 1,25(OH)2D may serve as a marker of Crohn’s disease [52]. The 1,25(OH)2D test is a delicate assay which is only done in specialized laboratories. False low results have been observed due to apparent sample mishandling; freezing for transport is advised to prevent sample degradation due to agitation. A high result is always accurate.
Restoration of VDR competence
The ability to mount an appropriate immune system response to intracellular infection is highly dependent on a competent VDR [159]. When it appears that 1,25(OH)2D is unable to up-regulate the VDR due to microbial activity, VDR competence may be restored with another VDR ligand which acts as an agonist; an agonist increases the signal transduction activity of a cell when bound to a receptor on that cell. Over 3000 synthetic VDR ligands have been identified, but most of these 1,25(OH)2D analogues have no clinical use because of their undue disruption to calcium regulation [160]. A number of non-vitamin D VDR ligands have also been identified: curcumin, omega-6 fatty acids (e.g., arachidonic acid, linoleic acid), and lithocolic acid (LCA) but are not being used for this purpose [161, 162].
Angiotensin receptor blockers (ARBs) have been shown, via in silico molecular modeling, to modulate VDR activation [163]. The most promising ARB, olmesartan medoxomil (brand name Benicar®) was estimated to have a Ki value in the low nanomolar range, similar to the Ki values of the natural VDR ligands [163]. Olmesartan has been noted to cause a significant reduction in elevated 1,25(OH)2D within weeks of initiation, which provides further evidence of its ability to up-regulate the VDR [164]. Olmesartan is believed to decrease elevated 1,25(OH)2D by several VDR-mediated effects (Fig. 6). The up-regulated VDR:

Proposed hypothesis for restoring renal control of 1,25(OH)2D with olmesartan. Olmesartan is believed to decrease elevated 1,25(OH)2D by several mechanisms
-
transcribes CYP24A1 and CYP3A4 (enzymes which reduce 1,25(OH)2D production) [147].
-
represses CYP27B1 (the enzyme that hydroxylates 25(OH)D to 1,25(OH)2D) so less 1,25(OH)2D is produced [146].
Consequently, renal control of 1,25(OH)2D production is restored and extra-renal production of 1,25(OH)2D is reduced. A decrease in elevated 1,25(OH)2D means less systemic inflammation, as these studies of olmesartan indicate:
-
Improvement of glycemic control and insulin resistance was only observed in the olmesartan group and these effects of olmesartan might be mediated by an anti-inflammatory action [165].
-
Olmesartan treatment significantly reduced serum levels of inflammatory markers; h-CRP, h-TNFa, IL-6, MCP-1 after 6 weeks of therapy [166].
-
Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity [167].
-
Blocking angiotensin II receptor increases bone mass [168, 169].
Olmesartan acts in a manner similar to 1,25(OH)2D to reduce inflammation and, by inference, improve immune system function. VDR and RAS receptors are distributed in almost the same tissues. The endogenous VDR ligand 1,25(OH)2D down-regulates the RAS by repressing renin gene expression to reduce inflammation via the nuclear factor-kappa B pathway [170]. Olmesartan has a similar effect in that it reduces angiotensin II (a peptide that is implicated in the inflammatory process) [171]. Inappropriate stimulation of the RAS has been associated with the pathogenesis of hypertension, heart attack and stroke [151]. Ferder et al. [151] state, “Changes in RAS activity and activation of the VDR seem to be inversely related, making it possible to speculate that both systems could have a feedback relationship. The combination of RAS blockade and VDR stimulation appears to be more effective than each one used individually.” Blocking angiotensin II and stimulating the VDR is what olmesartan appears to accomplish and this function is consistent with a theory of VDR incompetence (Fig. 7).

Effect of treatment with olmesartan and antibiotics on inflammatory symptoms. Olmesartan up-regulates the vitamin D receptor to improve innate immune system function and reduce elevated 1,25(OH)2D. Avoidance of immunosuppressants and elevated 25(OH)D also improves immune system function. Inflammatory symptoms gradually resolve as intracellular bacteria are slowly eliminated with the help of select low-dose, pulsed antibiotics
In patients with autoimmune disorders and inflammatory symptoms, olmesartan is noted to provoke an increase in inflammatory symptoms indicative of a Jarisch–Herxheimer reaction (JHR). JHR is a cascade of reactions including inflammation, cytokine release, and endotoxin release as part of the immune response to the disintegration of infected cells [172]. This immunopathology suggests transcription of AMPs by an activated VDR, points to the presence of occult infection and provides additional evidence that olmesartan is a VDR agonist [167, 173, 174]. Theoretically, olmesartan restores VDR competence and, thus, phagocytosis leads to bacterial death; consequently, inflammation is temporarily increased by cytokine reaction to microbial endotoxins and cellular debris from dead host cells and bacteria [175].
Hajishengallis and Lambris [92] conclude that a blockade of hijacked receptors of the innate immune system may offer promising options to control infection and associated immunopathology. Although this use of olmesartan is off-label, its safety profile is well established [176]. The multiple, documented beneficial effects of olmesartan, including the ability to reduce cardiovascular and kidney disease, prevent migraines, and reduce oxidative stress, also suggest it could play a key role in the resolution of chronic systemic inflammation [177, 178].
Clinical use of olmesartan
Olmesartan is being used as a novel VDR ligand in the clinical setting [179]. Immunotherapy with olmesartan may also include pulsed administration of select MIC (minimum inhibitory concentration) oral antibiotics to weaken and help eradicate the intracellular pathogens. With each antibiotic dose, inflammatory symptoms (JHR) wax and wane, providing further evidence of persistent infection [180]. Changes in laboratory findings (e.g., BUN, creatinine, CRP, blood counts, liver enzymes) often point to areas of occult inflammation. A correlating treatment strategy is the avoidance of excessive sunlight exposure, foods high in vitamin D and vitamin D supplements to maintain serum 25(OH)D at a level (20–30 ng/ml) that is not likely to suppress the immune system and inhibit bacterial elimination [122, 135, 137].
Accumulating case reports now support the observation that a number of complex, chronic conditions can be improved by restoring VDR function using this type of immunotherapy [179, 181, 182]. It is becoming increasingly clear that microbes slow down immune reactivity by dysregulating the VDR, ultimately to increase their chance of survival. Immune modulatory therapies that enhance VDR expression and activity should, therefore, be considered in the clinical setting [183].
Discussion
Vitamin D is essential for many important biological processes and most people get an adequate supply from exposure to sunlight (Table 2). Long-term studies are needed to determine if low 25(OH)D in healthy individuals leads to disease. Evidence that vitamin D supplementation cures or prevents chronic disease is inconsistent. Despite increased supplementation chronic inflammatory diseases are on the rise. Attention to the alternate hypothesis—low 25(OH)D is a consequence of the chronic disease process, provoked by persistent intracellular infection—may be crucial to reversing this trend and needs further research. The prevailing dogma that the level of serum 25(OH)D provides an accurate assessment of vitamin D status needs closer examination. Circulating levels of 25(OH)D may not be an accurate reflection of vitamin D status. In those with an autoimmune disease or chronic inflammatory symptoms, 1,25(OH)2D may be elevated. This can lead to osteoporosis and cause inhibition of innate immunity, which is contraindicated in the presence of infection. The resulting immunosuppression may promote persistent infection which has been implicated in chronic inflammatory diseases.
Human cells live in harmony with many types of microbes but some microbes may become pathogenic under commonly experienced conditions. The innate immune system is designed to kill pathogens via 1,25(OH)2D-mediated VDR transcription of anti-microbial peptides but microbes may use strategies which down-regulate the VDR in order to live and reproduce within nucleated host cells. Studies using more advanced cell culture and molecular techniques are confirming the presence of previously undetected intracellular bacteria. Defense mechanisms that intracellular bacteria use to persist and proliferate need to be investigated. Pathogen-induced VDR dysfunction which causes the release of pro-inflammatory cytokines appears to be at the root of chronic disease and low 25(OH)D. Improving VDR activation may be the key to reducing inflammatory diseases. Treatments that up-regulate the VDR to restore normal immune function, reduce inflammation and eradicate persistent bacterial infections require further research. An immunotherapy which has demonstrated efficacy in reversing vitamin D metabolism dysfunction and reducing inflammatory symptoms is currently being used by clinicians and warrants formal study.
In summary, elevated 1,25(OH)2D, often accompanied by reduced 25(OH)D, is a clinical sign of dysregulated vitamin D metabolism and evidence that the immune system is competing with parasitic microbes for VDR dominance. Failure of the immune system to mount an effective anti-microbial response results in persistent intracellular infection. This induces relentless inflammation (immunopathology) which causes tissue damage and disease symptoms.
References
Cheng T, Chimeh C, Lu Z, et al. Factors that influence the cutaneous synthesis and dietary sources of vitamin D. Arch Bio Biophs. 2007;460(2):213–7.
Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008;88(2):582–6.
Holick M, Schnoes H, Deluca H, Suda T, Cousins R. Isolation and identification of 1,25-dihydroxycholecalciferol. A metabolite of vitamin D active in intestine. Biochem. 1971;10(14):2799–804.
Adams J, Hewison M. Extrarenal expression of the 25-hydroxyvitamin d-1-hydroxylase. Arch Biochem Biophys. 2012;523(1):95–102.
Haddad J, Vitamin D. Binding protein (Gc-globulin): multiple tasks. J Steroid Biochem Mol Biol. 1996;53(1–6):579–82.
Sutton A, MacDonald P. Vitamin D: more than a “bone-a-fide” hormone. Mol Endocrinol. 2003;17(5):777–91.
White JH. Vitamin D signaling, infectious diseases, and regulation of innate immunity. Infect Immun. 2008;76(9):3837–43.
Lai Y, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30(3):131–41.
Wang T, Nestel F, Bourdeau V, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004;173(5):2909–12.
Hayes C, Nashold F, Spach K, Pedersen L. The immunological functions of the vitamin D endocrine system. Cell Mol Biol (Noisy-le-grand). 2003;49(2):277–300.
Topilski I, Flaishon L, Naveh Y, Harmelin A, Levo Y, Shachar I. The anti-inflammatory effects of 1,25-dihydroxyvitamin D3 on Th2 cells in vivo are due in part to the control of integrin-mediated T lymphocyte homing. Eur J Immunol. 2004;34(4):1068–76.
Maxmen A. Nutrition advice: the vitamin D-lemma. Nature. 2011;475(7354):23–5.
Autier P, Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med. 2007;167(16):1730–7.
For health professionals: position statement on supplementation, blood levels and sun exposure Vitamin D Council. Vitamin D Council. Jan 12, 2010. Available at http://www.vitamindcouncil.org/further-topics/for-health-professionals-position-statement-on-supplementation-blood-levels-and-sun-exposure/. Accessed 28 Apr 2013.
Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Guidelines for preventing and treating vitamin D deficiency and insufficiency revisited. J Clin Endocrinol Metab. 2012;97(4):1153–8.
Ross AC, Taylor CL, Yaktine AL, Del Valle HB. Dietary reference intakes for calcium and vitamin D. Washington: National Academy of Sciences; 2010. 0-309-16394-3.
Beers M. Vitamin D Deficiency and Dependency. Merck (Document). Oct 15, 2006. Available at http://web.archive.org/web/20061015162329/www.merck.com/mrkshared/mmanual/section1/chapter3/3d.jsp. Accessed 21 Apr 2013.
Topiwala S. 25-hydroxy vitamin D test. MedlinePlus. Oct 7, 2012. Available at http://www.nlm.nih.gov/medlineplus/ency/article/003569.htm. Accessed 21 Apr 2013.
Vitamin D, 25-Hydroxy, LC/MS/MS. Quest diagnostics. 2013. Available at http://www.questdiagnostics.com/home/physicians/testing-services/by-test-name/vitamind.html. Accessed 28 Apr 2013.
Snellman G, Melhus H, Gedeborg R, et al. Seasonal genetic influence on serum 25-hydroxyvitamin D levels: a twin study. PLoS One. 2009;4(11):e7747.
Bogh MK, Schmedes AV, Philipsen PA, Thieden E, Wulf HC. Vitamin D production depends on ultraviolet-B dose but not on dose rate: a randomized controlled trial. Exp Dermatol. 2011;20(1):14–8.
Bogh MK, Schmedes AV, Philipsen PA, Theiden E, Wulf HC. Vitamin D production after UVB exposure depends on baseline vitamin D and total cholesterol but not on skin pigmentation. J Invest Dermatol. 2010;130(2):546–53.
Matsuoka LY, Worstman J, Haddad JG, Kolm P, Hollis BW. Racial pigmentation and the cutaneous synthesis of vitamin D. Arch Dermatol. 1991;127(4):536–8.
Matsuoka LY, Wortsman J, Chen TC, Holick MF. Compensation for the interracial variance in the cutaneous synthesis of vitamin D. J Lab Clin Med. 1995;126(5):452–7.
Villa M, Kelsey J, Chen J, Marcus R. Skin Pigmentation does not affect vitamin D status in community-dwelling Mexican–American women. J Bone Miner Res. 1994;9(Suppl 1):S418.
Matsuoka LY, Wortsman J, Dannenberg MJ, Holis BW, Lu Z, Holick MF. Clothing prevents ultraviolet-B radiation-dependent photosynthesis of vitamin D3. J Clin Endocrinol Metab. 1992;75(4):1099–103.
Dietary supplement fact sheet: vitamin D. National Institutes of Health. Jan 24, 2011. Available at http://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional/?print=1. Accessed 10 Mar 2013.
Matsuoka LY, Ide L, MacLaughlin JA, Holick MF. Sunscreens suppress cutaneous vitamin D3 synthesis. J Clin Endocrinol Metab. 1987;64(6):1165–8.
Diehl JW, Chiu MW. Effects of ambient sunlight and photoprotection on vitamin D status. Dermatol Ther. 2010;23(1):48–60.
Youl PH, Janda M, Kimlin M. Vitamin D and sun protection: the impact of mixed public health messages in Australia. Int J Cancer. 2009;124(8):1963–70.
Global Solar UV Index: A Practical Guide. Geneva, Switzerland: World Health Organization; 2002. ISBN 9241590076.
MacLaughlin J, Holick MF. Aging decreases the capacity of human skin to produce vitamin D3. J Clin Invest. 1985;76(4):1536–8.
Chuck A, Todd J, Diffey B. Subliminal ultraviolet-B irradiation for the prevention of vitamin D deficiency in the elderly: a feasibility study. Photodermatol Photoimmunol Photomed. 2001;17(4):168–71.
Chel VG, Ooms ME, Popp-Snijders C, et al. Ultraviolet irradiation corrects vitamin D deficiency and suppresses secondary hyperparathyroidism in the elderly. J Bone Miner Res. 1998;13(8):1238–42.
Webb AR, Kline L, Holick MF. Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. Clin Endocrinol Metab. 1988;67(2):373–8.
Kimlin MG, Olds WJ, Moore MR. Location and vitamin D synthesis: is the hypothesis validated by geophysical data? J Photochem Photobiol B. 2007;86(3):234–9.
Lubin D, Jensen EH, Gies HP. Global surface ultraviolet radiation climatology from TOMS and ERBE data. J Geo Res. 1998;103(D20):26061–91.
Schoenmakers I, Goldberg GR, Prentice A. Abundant sunshine and vitamin D deficiency. Br J Nutr. 2008;99(6):1171–3.
Al-Othman A, Al-Musharaf S, Al-Daghri NM, et al. Tea and coffee consumption in relation to vitamin D and calcium levels in Saudi adolescents. Nutr J. 2012;11:56.
Willis KS, Smith DT, Broughton KS, Larson-Meyer DE. Vitamin D status and biomarkers of inflammation in runners. Open Access J Sports Med. 2012;2012(3):35–42.
Binkley N, Novotny R, Krueger D, et al. Low vitamin D status despite abundant sun exposure. J Clin Endocrinol Metab. 2007;92(6):2130–5.
Blaney GP, Albert PJ, Proal AD. Vitamin D metabolites as clinical markers in autoimmune and chronic disease. Ann NY Acad Sci. 2009;1173:384–90.
Brot C, Jorgensen NR, Sorensen OH. The influence of smoking on vitamin D status and calcium metabolism. Eur J Clin Nutr. 1999;53(12):920–6.
Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab. 2009;27(4):392–401.
Rickets. MedlinePlus. Aug 1, 2012. Available at http://www.nlm.nih.gov/medlineplus/ency/article/000344.htm. Accessed 6 May 2013.
Lerch C, Meissner T. Interventions for the prevention of nutritional rickets in term born children. Cochrane Database Syst Rev. 2007;4:CD006164.
Pettifor JM, Ross P, Wang J, Moodley G, Couper-Smith J. Rickets in children of rural origin in South Africa: is low dietary calcium a factor? J Pediatr. 1978;92(2):320–4.
Pettifor JM. Nutritional rickets: deficiency of vitamin D, calcium, or both? Am J Clin Nutr. 2004;80(6 Suppl):1725S–9S.
Reid IR, Bolland MJ, Grey A. Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet. 2013;383:146–55.
Boden SD, Kaplan FS. Calcium homeostasis. Orthop Clin North Am. 1990;21(1):31–42.
Sun J. Vitamin D and mucosal immune function. Curr Opin Gastroenterol. 2010;26(6):591–5.
Abreu MT, Kantorovich V, Vasiliauskas EA, et al. Measurement of vitamin D levels in inflammatory bowel disease patients reveals a subset of Crohn’s disease patients with elevated 1,25-dihydroxyvitamin D and low bone mineral density. Gut. 2004;53(8):1129–36.
Brot C, Jørgensen N, Madsen OR, Jensen LB, Sørensen OH. Relationships between bone mineral density, serum vitamin D metabolites and calcium:phosphorus intake in healthy perimenopausal women. J Intern Med. 1999;245(5):509–16.
Ishizuka S, Kurihara N, Miura D, et al. Vitamin D antagonist, TEI-9647, inhibits osteoclast formation induced by 1alpha,25-dihydroxyvitamin D3 from pagetic bone marrow cells. J Steroid Biochem Mol Biol. 2004;89–90(1–5):331–4.
Vanderschueren D, Pye SR, O’Neill TW, et al. Active vitamin D (1,25-dihydroxyvitamin D) and bone health in middle-aged and elderly men: the European Male Aging Study (EMAS). J Clin Endocrinol Metab. 2013;98(3):995–1005.
Chung M, Lee J, Terasawa T, Lau J, Trikalinos TA. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the US. Preventive Services Task Force. Ann Intern Med. 2011;155(12):827–38.
Whayne TF Jr. Vitamin D: popular cardiovascular supplement but benefit must be evaluated. Int J Angiol. 2011;20(2):63–72.
Shelby J. Needs great, evidence lacking for people with multiple chronic conditions. Scribd. Apr 2013. Accessed 7 May 2013.
Mattke S, Klautzer L, Mengistu T, Hu J, Wu H. Health and well-being in the home: a global analysis of needs, expectations, and priorities for home health care technology. Rand Corporation. 2010. http://www.rand.org/pubs/occasional_papers/OP323.html. Accessed 7 May 2013.
Tseng L. Controversies in vitamin D supplementation. eScholarship. 2003. http://www.escholarship.org/uc/item/4m84d4fn#page-1. Accessed 7 May 2013.
Bjelakovic G, Gluud LL, Nikolova D, et al. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst Rev. 2011;(7):CD007470.
Holick MF. Environmental factors that influence the cutaneous production of vitamin D. Am J Clin Nutr. 1995;61(3 Suppl):638S–45S.
Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69(5):842–56.
Noordam R, de Craen AJ, Pedram P, et al. Levels of 25-hydroxyvitamin D in familial longevity: the Leiden Longevity Study. CMAJ. 2012;184(18):E963–8.
Autier P, Boniol M, Pizot C, Mullie P. Vitamin D: chasing a myth? Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2013.
Bolland MJ, Grey A, Gamble GD, Reid IR. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014;4(2):307–20.
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the institute of medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53–8.
Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87:1080S–6S.
Albert PJ, Proal AD, Marshall TG. Vitamin D the alternative hypothesis. Autoimmun Rev. 2009;8(8):639–44.
Madoff S, editor. The Bacterial L-forms. 1st ed. USA: Marcel Dekker Inc; 1986.
Allan EJ, Hoischen C, Gumpert J. Bacterial L-forms. Adv Appl Microbiol. 2009;68:1–39.
Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal. 2010;17(5):766–74.
Domingue GJ, Woody HB. Bacterial persistence and expression of disease. Clin Microbiol Rev. 1997;10(2):320–44.
O’Connor SM, Taylor CE, Hughes JM. Emerging infectious determinants of chronic diseases. Emerg Infect Dis. 2006;12(7):1051–7.
Domingue GJ. Demystifying pleomorphic forms in persistence and expression of disease: are they bacteria, and is peptidoglycan the solution? Discov Med. 2010;10(52):234–46.
Rueter A. Microbes start immune response by sneaking inside cells. University of Michigan Health System. Apr 16, 2007. http://www2.med.umich.edu/prmc/media/newsroom/details.cfm?ID=577. Accessed 8 May 2013.
Rolhion N, Darfeuille-Michaud A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(10):1277–83.
Onwuamaegbu ME, Belcher RA, Soare C. Cell wall-deficient bacteria as a cause of infections: a review of the clinical significance. J Int Med Res. 2005;33(1):1–20.
Verway M, Behr MA, White JH. VitaminD NOD2, autophay and Crohn’s disease. Expert Rev Clin Immunol. 2010;6(4):505–8.
Pirofski L, Casadevall A. Q and A what is a pathogen? A question that begs the point. BMC Biol. 2012;12:6.
Hof H. Opportunistic intracellular bacteria and immunity. Antibiotic treatment of infections with intracellular bacteria. USA: Springer; 2002.
Fuller E, Elmer C, Nattress F, Horne G, Cook P, Fawcett T. Beta-lactam resistance in Staphylococcus aureus cells that do not require a cell wall for integrity. Antimicrob Agents Chemother. 2005;49(12):5075–80.
Wróblewska J, Janicka G, Gospodarek E, Szymankiewicz M. L-forms of Staphylococcus epidermidis induced by penicillin. Pol J Microbiol. 2006;55(3):243–4.
Garssen J, Vandebriel RJ, De Gruijl FR, et al. UVB exposure-induced systemic modulation of Th1- and Th2-mediated immune responses. Immunology. 1999;97(3):506–14.
Riminton DS, Hartung HP, Reddel SW. Managing the risks of immunosuppression. Curr Opin Neurol. 2011;24(3):217–23.
Wall S, Kunze ZM, Saboor S, et al. Identification of spheroplast-like agents isolated from tissues of patients with Crohn’s disease and control tissues by polymerase chain reaction. J Clin Microbiol. 1993;31(5):1241–5.
Negi M, Takemura T, Guzman J, et al. Localization of propionibacterium acnes in granulomas supports a possible etiologic link between sarcoidosis and the bacterium. Mod Pathol. 2012;25(9):1284–97.
Astrauskiene D, Bernotiene E. New insights into bacterial persistence in reactive arthritis. Clin Exp Rheumatol. 2007;25(3):470–9.
McDougal JS. Emerging infectious diseases. Centers for Disease Control and Prevention. Mar 2006. http://wwwnc.cdc.gov/eid/article/12/3/05-1409_article.htm#suggestedcitation. Accessed 8 May 2013.
Kozarov E. Bacterial invasion of vascular cell types: vascular infectology and atherogenesis. Future Cardiol. 2012;8(1):123–38.
Khan N, Gowthaman U, Pahari S, Agrewala JN. Manipulation of costimulatory molecules by intracellular pathogens: veni, vidi, vici! PLoS Pathog. 2012;8(6):e1002676.
Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol. 2011;11(33):187–200.
Dörr T, Vulić M, Lewis K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010;8(2):e1000317.
Fuks JM, Arrighi RB, Weidner JM, et al. GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog. 2012;8(12):e1003051.
Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480(7376):241–4.
Riley DR, Sieber KB, Robinson KM, et al. Bacteria–human somatic cell lateral gene transfer is enriched in cancer samples. PLOS Comput Biol. 2013;9(6):e1003107.
Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science. 2014;343(6167):204–208. Available at http://www.sciencemag.org. Accessed 21 Jan 2014.
Eishi Y. Etiologic aspect of sarcoidosis as an allergic endogenous infection caused by Propionibacterium acnes. Biomed Res Int. 2013;2013:935289.
Wang KX, Chen L. Helicobacter pylori L-form and patients with chronic gastritis. World J Gastroenterol. 2004;10(9):1306–9.
Chen Y, Liu W, Sun T, et al. 1,25-Dihydroxyvitamin D promotes negative feedback regulation of TLR signaling via targeting MicroRNA-155-SOCS1 in macrophages. J Immunol. 2013;190(7):3687–95.
Nauciel C. Immune defenses against intracellular bacterial infection. In: Paradise LJ, Friedman H, Bendinelli M, editors. Opportunistic intracellular bacteria and immunity. New York: Kluwer Academic; 2002.
Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311(5768):1770–3.
Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 2001;7(3):167–202.
McGeachy MJ, McSorley SJ. Microbial-induced Th17: superhero or supervillain? J Immunol. 2012;189(7):3285–91.
Oswald-Richter KA, Beachboard DC, Seeley EH, et al. Dual analysis for mycobacteria and propionibacteria in sarcoidosis BAL. J Clin Immunol. 2012;32(5):1129–40.
Labro M. Interference of antibacterial agents with phagocyte functions: immunomodulation or “immuno-fairy tales”? Clin Microbiol Rev. 2000;13(4):615–50.
Giongo A, Gano KA, Crabb DB, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5(1):82–91.
Anderson G, Horvath J. The growing burden of chronic disease in America. Public Health Rep. 2004;119(3):263–70.
Smith G. Angiotensin and systems thinking: wrapping your mind around the big picture. Ochsner J Spring. 2013;13(1):11–25.
Woolard MD, Frelinger JA. Outsmarting the host: bacteria modulating the immune response. Immunol Res. 2008;41(3):188–202.
Dermine JF, Desjardins M. Survival of intracellular pathogens within macrophages. Protoplasma. 1999;210(1–2):11–24.
Xu Y, Xie J, Li Y, et al. Using a cDNA microarray to study cellular gene expression altered by Mycobacterium tuberculosis. Chin Med J (Engl). 2003;116(7):1070–3.
Liu PT, Wheelwright M, Teles R, et al. MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat Med. 2012;18(2):267–73.
Coughlan CA, Chotirmall SH, Renwick J, et al. The effect of Aspergillus fumigatus infection on vitamin D receptor expression in cystic fibrosis. Am J Respir Crit Care Med. 2012;186(10):999–1007.
Yenamandra SP, Lundin A, Arulampalam V, et al. Expression profile of nuclear receptors upon Epstein—Barr virus induced B cell transformation. Exp Oncol. 2009;31(2):92–6.
Haug CJ, Aukrust P, Haug E, Mørkrid L, Müller F, Frøland SS. Severe deficiency of 1,25-dihydroxyvitamin D3 in human immunodeficiency virus infection: association with immunological hyperactivity and only minor changes in calcium homeostasis. J Clin Endocrinol Metab. 1998;83(11):3832–8.
Yoshizawa T, Handa Y, Uematsu Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16(4):391–6.
Sadeghi K, Wessner B, Laggner U, et al. Vitamin D3 down-regulates monocyte TLR expression and triggers. Eur J Immunol. 2006;36(2):361–70.
Barna BP, Culver DA, Kanchwala A, et al. Alveolar macrophage cathelicidin deficiency in severe sarcoidosis. J Innate Immun. 2012;4(5–6):569–78.
Mawer EB, Hayes ME, Still PE, et al. Evidence for nonrenal synthesis of 1,25-dihydroxyvitamin D in patients with inflammatory arthritis. J Bone Miner Res. 1991;6(7):733–9.
Wang TT, Dabbas B, Laperriere EG, et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J Biol Chem. 2010;285(4):2227–31.
Lemire J. 1,25-Dihydroxyvitamin D3—a hormone with immunomodulatory properties. Z Rheumatol. 2000;59(Suppl 1):24–7.
Deb DK, Chen Y, Zhang Z, et al. 1,25-Dihydroxyvitamin D3 suppresses high glucose-induced angiotensinogen expression in kidney cells by blocking the NF-{kappa}B pathway. 296(5):F1212–8.
Mouy R, Fischer A, Vilmer E, Seger R, Griscelli C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr. 1989;114(1):555–60.
Wu S, Sun J. Vitamin D receptor, and macroautophagy in inflammation and infection. Discov Med. 2011;11(59):325–35.
Domingue G, Turner B, Schlegel JU. Cell-wall deficient bacterial variants in kidney tissue. Detection by immunofluorescence. Urology. 1974;3(3):288–92.
Pleister A, Eckels DD. Cryptic infection and autoimmunity. Autoimmun Rev. 2003;2(3):126–32.
Chan TD, Wood K, Hermes JR, et al. Elimination of germinal-center-derived self-reactive B cells is governed by the location and concentration of self-antigen. Immunity. 2012;37(5):893–904.
Blander JM, Torchinsky MB, Campisi L. Revisiting the old link between infection and autoimmune disease with commensals and T helper 17 cells. Immunol Res. 2012;54(1–3):50–68.
Christen U, Hintermann E, Holdener M, von Herrath MG. Viral triggers for autoimmunity: is the ‘glass of molecular mimicry’ half full or half empty? J Autoimmun. 2010;34(1):38–44.
Proal AD, Albert PJ, Marshall TG. Autoimmune disease and the human metagenome. In: Nelson KE, editor. Metagenomics of the human body. 1st ed. USA: Springer; 2011.
Berlin T, Zandman-Goddard G, Blank M, et al. Autoantibodies in nonautoimmune individuals during infections. Ann NY Acad Sci. 2007;1108:584–93.
Molina V, Shoenfeld Y. Infection, vaccines and other environmental triggers of autoimmunity. Autoimmunity. 2005;38(3):235–45.
Griffin MD, Xing N, Kumar R. Vitamin D and its analogs as regulators of immune activation and antigen presentation. Annu Rev Nutr. 2003;23:117–45.
Zhang Y, Leung DY, Richers BN, et al. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol. 2012;188(5):2127–35.
Böhm M, Luger TA, Schneider M, Schwarz T, Kuhn A. New insight into immunosuppression and treatment of autoimmune diseases. Clin Exp Rheumatol. 2006;24(1 Suppl 40):S67–71.
Arnson Y, Amital H, Shoenfeld Y. Vitamin D and autoimmunity: new aetiological and therapeutic considerations. Ann Rheum Dis. 2007;66(9):1137–42.
Kim HM, Chung MJ, Chung JB. Remission and relapse of autoimmune pancreatitis: focusing on corticosteroid treatment. Pancreas. 2010;39(5):555–60.
Collins FS. Reengineering translational science: the time is right. Sci Transl Med. 2011;3(90):9017.
Norman A. From vitamin D to hormone D: fundamentals of the vitamin D endocrine system essential for good health. Am J Clin Nutr. 2008;88(2):491S–9S.
Rasmussen SB, Reinert LS, Paludan SR. Innate recognition of intracellular pathogens: detection and activation of the first line of defense. APMIS. 2009;117(5–6):323–37.
Dusso AS, Kamimura S, Gallieni M, et al. gamma-Interferon-induced resistance to 1,25-(OH)2D3 in human monocytes and macrophages: a mechanism for the hypercalcemia of various granulomatoses. J Clin Endocrinol Metab. 1997;82(7):2222–32.
Edfeldt K, Liu PT, Chun R, et al. T-cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Natl Acad Sci USA. 2010;107(52):22593–8.
Lambert PW, Stern PH, Avioli RC, et al. Evidence for extrarenal production of 1 alpha,25-dihydroxyvitamin D in man. J Clin Invest. 1982;69(3):722–5.
Reichrath J. Vitamin D and the skin: an ancient friend, revisited. Exp Dermatol. 2007;16(7):618–25.
Bikle DD. Vitamin D and immune function: understanding common pathways. Curr Osteoporos Rep. 2009;7(2):58–63.
Jones G, Prosser DE, Kaufman M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch Biochem Biophys. 2012;523(1):9–18.
Xu Y, Hashizume T, Shuhart MC, et al. Intestinal and hepatic CYP3A4 catalyze hydroxylation of 1alpha,25-dihydroxyvitamin D(3): implications for drug-induced osteomalacia. Mol Pharmacol. 2006;69(1):56–65.
Bell NH, Shaw S, Turner RT. Evidence that 1,25-dihydroxyvitamin D3 inhibits the hepatic production of 25-hydroxyvitamin D in man. J Clin Invest. 1984;74(4):1540–4.
Waldron JL, Ashby HL, Cornes MP, et al. Vitamin D: a negative acute phase reactant. J Clin Pathol. 2013 (Epub ahead of print).
Ferder M, Inserra F, Manucha W, Ferder L. The world pandemic of Vitamin D deficit could possibly be explained by cellular inflammatory response activity induced by the renin angiotensin system. Am J Physiol Cell Physiol. 2013 (Epub ahead of print).
Akbaraly TN, Hamer M, Ferrie JE, et al. Chronic inflammation as a determinant of future aging phenotypes. CMAJ. 2013 (Epub ahead of print).
Phillips CM, Perry IJ. Does inflammation determine metabolic health status in obese and nonobese adults? J Clin Endocrinol Metab. 2013;98(10):E1610–9.
Strathmann FG, Laha TJ, Hoofnagle AN. Quantification of 1α,25 dihydroxy vitamin D by immunoextraction and liquid chromatography–tandem mass spectrometry. Clin Chem. 2011;57(9):1279–85.
Hollis B. Detection of vitamin D and its major metabolites. In: Feldman D, Pike JW, Glorieux F, editors. vitamin D. San Diego: Elsevier Academic; 2005.
Perez T. Bacteria induced vitamin D receptor dysfunction in autoimmune disease; theoretical and practical implication for interpretation of serum vitamin D metabolite levels. Paper presented at: 6th International Congress on Autoimmunity, 2006; Porto, Portugal.
Vitamin D Assay Testing. Medicare Limited Coverage Tests—Covered Diagnosis Codes. Nov 14, 2011. Available at http://www.sonoraquest.com/uploads/docs/Vitamin_D_Assay_Testing.pdf?AspxAutoDetectCookieSupport=1. Accessed 31 Jan 2014.
Durup DJHCJSPHALB. A reverse J-shaped association of all-cause mortality with serum 25-hydroxyvitamin D in general practice: the CopD study. J Clin Endocrinol Metab. 2012;97(8):2644–52.
Liu PT, Schenk M, Walker VP, et al. Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS One. 2009;4(6):e5810.
Carlberg C, Molnar F. Detailed molecular understanding of agonistic and antagonistic vitamin D receptor ligands. Curr Top Med Chem. 2006;6(12):1243–53.
Haussler M, Haussler C, Bartik L, et al. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutr Rev. 2008;66(10 Suppl 2):98–112.
Ishizawa M, Matsunawa M, Adachi R, et al. Lithocholic acid derivatives act as selective vitamin D receptor modulators without inducing hypercalcemia. J Lipid Res. 2008;49(4):763–72.
Marshall TG, Lee RE, Marshall FE. Common angiotensin receptor blockers may directly modulate the immune system via VDR, PPAR and CCR2b. Theor Biol Med Model. 2006;3:1.
Waterhouse JC, Marshall TG, Fenter B, Mangin M, Blaney G. High levels of active 1,25-dihydroxyvitamin D despite low levels of the 25-hydroxyvitamin D precursor-implications of dysregulated vitamin D for diagnosis and treatment of chronic disease. In: Stolzt VD, editor. Vitamin D: New Research. New York: Nova Science Publishers; 2006.
Arao T, Okada Y, Mori H, Nishida K, Tanaka Y. Antihypertensive and metabolic effects of high-dose olmesartan and telmisartan in type 2 diabetes patients with hypertension. Endocr J. 2013; (Epub ahead of print).
Fliser D, Buchholz K, Haller H. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110(9):1103–7.
Platten M, Youssef S, Hur EM, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci USA. 2009;106(35):14948–53.
Izu Y, Mizoguchi F, Kawamata A, et al. Angiotensin II type 2 receptor blockade increases bone mass. J Biol Chem. 2009;284(8):4857–64.
Shimuru H, Nakagami H, Osako MK, et al. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008;22(7):2465–75.
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229–38.
Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35(6):881–900.
Herxheimer K, Martin H. So-called Herxheimer reaction. Arch Derm Syphilol. 1926;13(1):115–7.
Sonawane A, Santos JC, Mishra BB, et al. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell Microbiol. 2011;13(10):1601–17.
Nickel D, Busch M, Mayer D, Hagemann B, Knoll V, Stenger S. Hypoxia triggers the expression of human beta defensin 2 and antimicrobial activity against Mycobacterium tuberculosis in human macrophages. J Immunol. 2012;188(8):4001–7.
Hurley JC. Antibiotic-induced release of endotoxin. A therapeutic paradox. Drug Saf. Mar 1995;12(3):183–95.
Schwocho LR, Masonson HN. Pharmacokinetics of CS-866, a new angiotensin II receptor blocker, in healthy subjects. J Clin Pharmacol. 2001;41(5):515–27.
Ferrario C. Effect of angiotensin receptor blockade on endothelial function: focus on olmesartan medoxomil. Vasc Health Risk Manag. 2009;5(1):301–14.
Takizawa S, Dan T, Uesugi T, et al. A sartan derivative with a very low angiotensin II receptor affinity ameliorates ischemic cerebral damage. J Cereb Blood Flow Metab. 2009;29(10):1665–72.
Proal AD, Albert PJ, Marshall TG, Blaney GP, Lindseth LA. Immunostimulation in the treatment for chronic fatigue syndrome/myalgic encephalomyelitis. Immunol Res. 2013;56:398–412.
Hof H. Antibiotic treatment of infections with intracellular bacteria. In: Paradise LJ, Friedman H, Bendinelli M, editors. Opportunistic intracellular bacteria and immunity. New York: Kluwer Academic; 2002.
Waterhouse JC, Perez TH, Albert PJ. Reversing bacteria-induced vitamin D receptor dysfunction is key to autoimmune disease. Ann NY Acad Sci. 2009;1173:757–65.
Mangin M. Monitoring recovery from autoimmune disease with an interactive, internet-based clinical trial based on a molecular model of chronic disease. Paper presented at: 6th International Congress on Autoimmunity, 2008; Porto, Portugal.
Kongsbak M, Levring TB, Geisler C, von Essen MR. The vitamin D receptor and T cell function. Front Immunol. 2013;4:148.
Acknowledgments
The authors wish to gratefully acknowledge the reviewers who commented on the original manuscript and provided helpful, expert advice. We thank the editor of Inflammation Research, Dr. Mauro Teixeira, for his kind consideration of our work. Special thanks to Tapen Sinha for sharing with us his expertise regarding manuscript formatting and submission. We thank Tom Mangin, Belinda Fenter, Terry Fenter, Debbie Yeager and John Yeager for their support. And we acknowledge with gratitude the generous donors to Chronic Illness Recovery who have enabled us to continue our research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Mauro Teixeira.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Mangin, M., Sinha, R. & Fincher, K. Inflammation and vitamin D: the infection connection. Inflamm. Res. 63, 803–819 (2014). https://doi.org/10.1007/s00011-014-0755-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-014-0755-z