
Abstract
Introduction:
Recent studies indicate that bone marrow stromal cells (BMSCs) derived from patients with multiple myeloma (MM) differ from those of healthy donors in their expression of extracellular matrix compounds and in cytokine production. It is not known whether these abnormalities are primary or are acquired by BMSCs on contact with MM cells.
Materials and Methods:
Interleukin (IL)-6, IL-11, IL-10, and tumor necrosis factor (TNF)-α production by CD166+ mesenchymal BMSCs and the CD38+/CD138+ RPMI8226 myeloma cell line cultivated in vitro in monocultures or co-cultivated under cell-to-cell contact or non-contact conditions in the presence of a tissue culture insert were measured. Intracellular cytokines were measured by flow cytometry analysis as the percentage of cytokine-producing cells or by mean fluorescence intensity as the level of cytokine expression in cells. Additionally, ELISA was used to measure IL-6, soluble IL-6 receptor (sIL-6R), IL-11, IL-10, TNF-α, B-cell-activating factor of the TNF family (BAFF), hepatocyte growth factor (HGF), and osteopontin (OPN) production in the supernatants of the cultures and co-cultures.
Results:
A higher ability of the BMSCs of MM patients than in controls was detected to produce IL-6, IL-10, TNF-α, OPN, and especially HGF and BAFF in response to the RPMI8226 cells. Moreover, the BMSCs of the MM patients significantly enhanced the production of sIL-6R by the RPMI8226 cells.
Discussion:
Cytokines over-expressed by BMSCs of MM patients can function as growth factors for myeloma cells (IL-6, IL-10, HGF), migration stimulatory factors for tumor plasma cells (TNF-α, HGF), adhesion stimulatory factors (HGF, BAFF and OPN), stimulators of osteoclastogenesis (IL-6, TNF-α), and angiogenic factors (TNF-α). The results of this experiment strongly suggest that the BMSCs from MM patients differed in spontaneous and myeloma cell-induced production of cytokines, especially of HGF and BAFF, and these abnormalities were both primary and acquired by the BMSCs on contact with the MM cells. This in turn suggests the presence of an undefined, autocrine stimulation pathway resulting in a prolonged production of cytokines even in long-term cultures in vitro and in vivo. These abnormalities might provide optimal conditions for the proliferation and differentiation of residual tumor cells or their precursors in the affected bone marrow.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM) is an uncontrolled growth of cells with the phenotype of terminally differentiated B cells (plasma cells). The disease usually occurs in the bone marrow (BM) and is associated with skeletal pathology, which in most patients appears as multiple lytic bone lesions and general osteoporosis [36].
The BM environment contains a complex reticulum of extracellular matrix and nonhematopoietic cells, mainly of mesenchymal origin, which provides support for the growth, proliferation, and differentiation of hematopoietic stem cells and their progeny. Bone marrow stromal cells (BMSCs) have been shown to provide efficient support for MM cell survival and proliferation by producing a high level of interleukin (IL)-6, a major MM cell growth factor [51]. Through interaction with BMSCs, MM cells also stimulate the production of cytokines which support osteoclastogenesis and angiogenesis [14]. On the other hand, the impaired osteoblastic function in MM may be a consequence of a direct action on osteoblasts, since MM cells enhance their apoptosis [40], and a result of their differentiation by DKK-1 produced by MM cells [45]. MM cells also stimulate the production of osteoclast-activating cytokines, such as IL-1, tumor necrosis factor (TNF)-α, and a TNF family member, the receptor activator of nuclear factor κB ligand (RANKL). In contrast, osteoprotegerin, a decoy receptor of RANKL is down-regulated. In MM, the high RANKL/osteoprotegerin ratio leads to an increased number of osteoclasts and enhanced bone resorption [34].
Recent studies indicate that BMSCs in MM patients differ from those of healthy donors as they express less CD106 (VCAM-1) and fibronectin and several times more IL-1β and TNF-α and have an abnormal synthesis of hyaluronan [4, 5, 13, 48, 49]. A gene expression profile analysis revealed that among 145 distinct genes differentially expressed in MM and normal BMSCs there were genes encoding IL-6 and DKK-1, already known to be implicated in the pathophysiological features of MM, as well as new ones, such as GDF-15, a divergent member of the TGF-β superfamily involved in angiogenesis, osteogenic differentiation, or tumor growth [7]. BMSCs from MM patients also exhibited a reduced efficacy to inhibit T-cell proliferation compared with normal cells. These properties existed in the absence of MM cells and were not associated with the chromosome aberration that features in tumor plasma cells [1].
What is not known is whether these abnormalities are primary or are acquired by BMSCs on contact with MM cells. To address this question we conducted studies on the cytokine production by BMSCs, mainly of mesenchymal origin, derived from MM patients and compared it with that of BMSCs derived from control subjects. Since interactions of MM cells with BMSCs are important in the disease development, as they stimulate the production of several cytokines, we also measured cytokine production by the BMSCs after contact with the myeloma cell line RPMI8226.
Materials and Methods
Patients
Bone marrow samples of 16 patients with newly diagnosed MM and 11 healthy control subjects were obtained with the patients’ and controls’ written informed consent. The study was approved by the local ethics committee. The characteristics of the subjects are shown in Table 1.
Cells
Bone marrow from the patients and the controls was obtained by sternal puncture. Anti-coagulated BM samples were diluted 1:2 with Iscove’s Modified Dulbecco’s medium (IMDM; Gibco, BRL, UK) containing 0.2% methylcellulose (Sigma, St. Louis, MO, USA). After sedimentation of erythrocytes (30–40 min) at room temperature, supernatants were collected, washed twice with IMDM supplemented with 2% fetal bovine serum (FBS; Gibco), and resuspended in IMDM supplemented with 10% FBS, 10% horse serum (Gibco), 1 µM hydrocortisone (Sigma) and 1% antibiotic-antimycotic solution (Gibco). Cells from the supernatants were seeded in T25 cell culture flasks (Nunc, Roskilde, Denmark) at a density of 2×106 cells/ml. The cultures were incubated for 4 weeks at 37°C in a humidified atmosphere containing 5% CO2. After one week of incubation, the cultures were semi-populated and nonadherent cells were respirated together with half of the medium volume. After 2 and 3 weeks of incubation all the non-adherent cells were removed together with the medium. After 4 weeks of incubation a continuous network of adherent cells occupying the entire bottom of each flask was obtained and adherent cells were treated with 0.25% trypsin (Sigma) and frozen in IMDM medium supplemented with 20% FBS and 10% DMSO at −80°C until use. After thawing, the cells were propagated in T75 flasks in IMDM medium supplemented with 10% FBS. After trypsinization the BMSCs were suspended in IMDM medium with 10% FBS and seeded at a density of 2×105 cells/ml into six-well plastic plates (Nunc; 3 ml/well) with a final density of 6×105 BMSCs/well. After 24 h of incubation the supernatants were removed and new IMDM medium supplemented with 2% FBS was added.
Cytokine induction
The cultures were divided into three batches. The first batch of cultures consisted of BMSCs seeded on six-well plastic plates and served as a control of spontaneous cytokine production by BMSCs. Into the second batch of BMSCs, myeloma RPMI8226 cells (ECACC 87012702) were added at a density of 1×106 cells/ml in 3 ml of IMDM medium supplemented with 2% FBS (direct cell-to-cell contact) at a final density of 3×106 cells/well. In the third batch of wells, with a monolayer of BMSCs, RPMI8226 cells were seeded into tissue culture inserts (Transwell; Costar, Cambridge, MA, USA) with a pore diameter of 0.4 µm. After 18 h of co-culture incubation, brefeldin (a protein transport inhibitor) at a concentration of 10 µg/ml was added and the co-cultures were incubated for an additional 4 h at 37°C. The supernatants of the cultures and co-cultures were collected and stored at −80°C until cytokine measurement by ELISA. The cells were washed with PBS (without Ca2+ and Mg2+), trypsinized, resuspended in PBS, and subjected to flow cytometric analysis. Another batch of six-well plastic plates with co-cultures of BMSCs with RPMI8226 cells were incubated for 72 h at 37°C and the cytokine concentrations in the supernatants were measured by ELISA.
Assay for cytokines
The concentrations of IL-10, IL-11, B-cell-activating factor of the TNF family (BAFF)/BlyS, osteopontin (OPN), and hepatocyte growth factor (HGF) were measured by ELISA using commercially available kits from R&D Systems, Minneapolis, MN, USA. The detection limits were 3.9 pg/ml for IL-10, 8 pg/ml for IL-11, 0.73-6.67 pg/ml for BAFF, 0.006–0.024 ng/ml for OPN, and 40 pg/ml for HGF. The concentrations of TNF-α, IL-6, and soluble IL-6 receptor (sIL-6R) were measured by ELISA using kits from Bender MedSystems, GmbH, Vienna, Austria. The detection limits were 3.83 pg/ml for TNF-α, 0.92 pg/ml for IL-6, and 0.01 ng/ml for sIL-6R.
Intracellular cytokine staining
Reagents. Monoclonal antibodies (moAb) anti-CD38 PE-Cy5 (clone: HIT2, isotype: IgG1) and CD138 FITC (clone: Mi15, isotype: IgG1) were obtained from BD PharMingen (San Diego, CA, USA). The CD14 TRI-Color® (clone: Tük4, isotype: IgG2a) moAb was obtained from Caltag Laboratories (Burlingame, CA, USA). Anti-CD166 FITC (clone: 3A6, isotype: IgG1) was obtained from Ancell (Bayport, MN, USA). PE-conjugated moAb anti-TNF (clone: MAb11, isotype: IgG1), anti-IL-6 (clone: MQ2-6A3, isotype: IgG2a), and anti-IL-10 (clone: JES3-19F1, isotype: IgG2a) were purchased from BD PharMingen. Anti-IL-11 PE (clone: 22626, isotype: IgG2a) was obtained from R&D Systems (Minneapolis, MN, USA). Mouse IgG1 and rat IgG2a isotype controls were obtained from BD PharMingen. IntraPrep™ was purchased from Immunotech (Marseille, France).
Sample staining. The cultured cells were washed twice in PBS, divided into tubes at a concentration of 5×105 cells per tube, and then stained with moAb against the cell-surface markers CD138, CD38, and CD166 (20 min at room temperature). Following membrane staining, the cells were fixed and permeabilized with the IntraPrep kit according to the manufacturer’s instructions. Then the cells were incubated with anti-TNF, anti-IL-6, anti-IL-10, and anti-IL-11 moAb or an appropriate isotype control for 15 min at room temperature. Finally, the cells were washed and analyzed by flow cytometry directly after preparation.
Flow cytometric analysis
The samples were analyzed by two- and three-color flow cytometry using a Becton Dickinson FACS-Calibur instrument. For the analysis of CD38+/CD138+ cells staining for intracellular cytokine expression, five data parameters were acquired and stored: linear forward and side scatter (FSC, SSC), green fluorescence (FL-1), orange-fluorescence (FL-2), and red-fluorescence (FL-3). For each analysis, 10,000 events were acquired and analyzed using CellQuest software. An acquisition gate was established based on FSC and SSC that excluded dead cells and debris (Fig. 1a). The R1-gated events were analyzed for CD38+/CD138+ staining (Fig. 1b) and positive cells were selected (region R2). Dot plots of CD38 PE-Cy5 vs. CD138 FITC were used. The final dot plots, i.e. CD38 PE-Cy5/isotype control (Fig. 1c) and CD38 PE-Cy5/anti-cytokine moAb PE (Fig. 1d), were established by combined gating of events using R1 and R2. An isotype-matched antibody was used to verify the staining specificity and as a guide for setting the markers to delineate positive and negative populations.

The dot plots show representative data from one patient illustrating the method for the identification of CD38+/CD138+ cells expressing IL-11 following three-color staining. The cells were gated (R1) using FSC and SSC plots (a). Then the R1-gated events were analyzed for CD38+/CD138+ staining and positive cells were selected (region R2). We used a dot plot of CD38 PE-Cy5 vs. CD138 FITC (b). The final dot plots: CD38 PE-Cy5/isotype control (c) and CD38 PE-Cy5/IL-11 PE (d), were established by combined gating of events using R1 and R2. The number in the upper right quadrant on dot plot d represents the percentage of CD38+/CD138+/IL-11+ cells. For the analysis shown, the RPMI8226 control was used.
For the analysis of CD166+ cells staining for intracellular cytokine expression, four data parameters were acquired and stored: FSC, SSC, FL-1, and FL-2. An acquisition gate was established based on FSC and SSC (region R1; Fig. 2a). Then the R1-gated events were analyzed for CD166 FITC staining and positive cells (CD166+) were gated (R2; Fig. 2b). The dot plots CD166 FITC versus an isotype control (Fig. 2c) and CD166 FITC versus an appropriate anti-cytokine moAb PE (Fig. 2d) were established by combined gating of events using R1 and R2.

The dot plots show representative data from one patient illustrating the method for the identification of CD166+ cells expressing IL-11 following two-color staining. The cells were gated (R1) using FSC and SSC plots (a). Then the R1-gated events were analyzed for CD166+ staining and positive cells were selected (region R2). We used a dot plot of CD166 FITC vs. SSC (b). The final dot plots: CD166 FITC/isotype control (c) and CD166 FITC/IL-11 PE (d), were established by combined gating of events using R1 and R2. The number in the upper right quadrant on dot plot d represents the percentage of CD166+/IL-11+ cells. For the analysis shown, a co-culture of CD166+ cells with RPMI8226 CD38+/CD138+ was used.
In the experiment, the mean percentage of cells with intracellular cytokine expression and the level of cytokine expression in cells indicated by mean fluorescence intensity (MFI) were analyzed. MFI is the average fluorescence intensity of all analyzed cells.
Statistics
Results are reported as the mean ±SD. The significance of differences was determined with the use of analysis of variance (STATISTICA computer package). A number of statistical tests were used, including the two-way ANOVA test with post-hoc Tukey’s test to measure the differences within a group and the Mann-Whitney U-test to measure the differences between groups. The concentrations of cytokines below the detection level were considered to be 0 for the purpose of the analysis. p values of 0.05 or less were considered significant.
Results
The BMSCs were selected by their adherence to plastic vessels. The cells were obtained from 16 MM patients and 11 control subjects. As BMSC proliferation is correlated with the age of donors, we had chosen control donors who were age matched with the MM patients. The MM and the control BMSCs did not differ significantly in the percentage of CD166+ (a marker for human BM stroma fibroblasts [42]) and CD14+ cells (Table 2). A FACS analysis did not detect CD38+/CD138+ cells in either case, thus demonstrating that the BMSC cultures were not contaminated by MM cells. Moreover, the production of tartrate-resistant acid phosphatase (TRAP) and alkaline phosphatase was also comparable between the BMSCs from the MM patients and the controls, indicating that there were no significant differences in TRAP-positive cells or osteoblast precursors between the BMSC cultures.
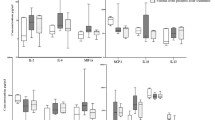
The aim of this study was to investigate the differences in cytokine production between BMSCs isolated from MM patients and those of control subjects. When the level of IL-6 was measured by ELISA (Fig. 3A), only BMSCs of MM patients cultivated under non-contact conditions with RPMI8226 cells produced more IL-6 than controls. To verify this finding, the level of IL-6 was additionally measured by flow cytometry in which the percentage of cells with the intracellular cytokine or the MFI level of cytokine expression was estimated in CD166+ (of both controls and MM patients) and CD38+/CD138+ myeloma RPMI8226 cells (Fig. 3C, D, E, F). When the percentage of cells expressing IL-6 was measured by flow cytometry, mesenchymal CD166+ cells of the MM patients produced significantly more IL-6 in response to RPMI8226 cells than controls, especially when the cells were separated by a porous membrane. Moreover, IL-6 was also produced by CD38+/CD138+ myeloma cells, especially when they were in direct contact with BMSCs. The BMSCs of the MM patients stimulated CD38+/CD138+ plasma cells to produce IL-6 in direct contact more efficiently than the BMSCs of controls. Moreover, MFI indicated a higher ability of the BMSCs from the MM patients to produce IL-6 spontaneously and after contact with RPMI8226 cells, though these differences were not statistically significant. However, when the sum of IL-6-producing CD166+ and CD38+/CD138+ cells was calculated, the sum was highest when the cells were in direct cell-to-cell contact. Such an unexpected difference between the results received from ELISA and cytometric analysis prompted us to measure the level of sIL-6R. We suspected that sIL-6R, which can bind IL-6, might have been the reason why IL-6 was not detected by the ELISA kit. The results presented in Fig. 3B indicated a very high level of sIL-6R, mainly produced by RPMI8226 in monocultures and in co-cultures with the BMSCs of controls and MM patients. The BMSCs in monocultures did not produce any sIL-6R. Moreover, sIL-6R expression was highest when the BMSCs and the RPMI8226 cells were cultivated together. Therefore it seems likely that the formation of a complex between sIL-6R and IL-6, not detected by the ELISA kit, was indeed the cause of the differences in the results obtained by the different methods.

Production of IL-6 in cultures and co-cultures of BMSCs of MM patients and control subjects with RPMI8226 myeloma cells. Production of IL-6 was measured by ELISA (A) and by cytometric analysis in monocultures of CD166+ cells (markers for mesenchymal stromal fibroblasts), monocultures of CD38+/CD138+ myeloma cells, and in co-cultures where both types of cells were in direct cell-to-cell contact (direct) or were separated by a transwell (non-contact). The level of IL-6 expression was measured by flow cytometry as the percentage of IL-6-expressing cells (C, D) or as the MFI (E, F), indicating the level of IL-6 expression in cells. Moreover, production of sIL-6R was measured by ELISA (B). *Significantly different from the control group, p<0.05; #significantly different from monoculture within the group, p<0.05.
When IL-11 production was examined by the ELISA method, no statistically significant differences between the controls and the MM group was observed (Fig. 4A). The RPMI8226 cells produced a low but detectable level of IL-11. Cytometric analysis revealed that the RPMI8226 cells were the main producer of IL-11, especially after direct contact with the BMSCs of the MM patients (Fig. 4C). There were no significant differences in IL-11 production between the BMSCs derived from the BM of controls and MM patients.

Production of IL-11 in cultures and co-cultures of BMSCs of MM patients and control subjects with RPMI8226 myeloma cells was measured by ELISA (A) or by cytometric analysis in monocultures of CD166+ cells (markers for mesenchymal stromal fibroblasts), monocultures of CD38+/CD138+ myeloma cells, and in co-cultures where both types of cells were in direct cell-to-cell contact (direct) or were separated by a transwell (non-contact). The level of IL-11 expression in cells was measured by flow cytometry as the percentage of IL-11-expressing cells (B, C) or as the MFI (D, E), indicating the level of IL-11 expression in cells. #Significantly different from monoculture within the group, p<0.05.
In ELISA, both the spontaneous production of IL-10 by the BMSCs of the MM patients and, especially, its production after cell-to-cell contact with the RPMI8226 cells were significantly higher in the MM group than in the controls (Fig. 5). The percentage of IL-10-producing cells in the BMSCs estimated by cytometric analysis was highest when the BMSCs of the MM patients were separated from the RPMI8226 cells by a porous membrane. The RPMI8226 cells also expressed IL-10 when they were in cell-to-cell contact with the BMSCs of the myeloma patients. The sum of the percentages of the IL-10-producing BMSCs of the MM patients and RPMI8226 cells was highest when the cells were cultivated in cell-to-cell contact conditions. MFI analysis revealed that the BMSCs of the MM patients expressed more IL-10 spontaneously, without contact with the RPMI8226 cells, than the control BMSCs, confirming the results obtained by ELISA.

Production of IL-10 in cultures and co-cultures of BMSCs of MM patients and controls with RPMI8226 myeloma cells. Production of IL-10 was measured by ELISA (A) or by cytometric analysis in monocultures of CD166+ cells (markers for mesenchymal stromal fibroblasts), monocultures of CD38+/CD138+ myeloma cells, and in co-cultures where both types of cells were in direct cell-to-cell contact (direct) or were separated by a transwell (non-contact). The level of IL-10 expression was measured by flow cytometry as the percentage of IL-10-expressing cells (B, C) or as the MFI (D, E), indicating the level of IL-10 expression in cells. *Significantly different from control group, p<0.05; #significantly different from monoculture within the group, p<0.05.
The ELISA of TNF-α revealed that the BMSCs of the MM patients produced more TNF-α after direct contact with the RPMI8226 cells than did the control BMSCs. It should be noted that there were great differences in the ability of the BMSCs of MM patients to produce TNF-α, resulting in very high standard deviations. The percentage of TNF-α-producing cells measured by flow cytometry was significantly higher in the BMSCs derived from the MM patients compared with controls when they were separated from the myeloma RPMI8226 cells by a transwell (Fig. 6). The RPMI8226 cells also produced more TNF-α when they were in cell-to-cell contact with the BMSCs of the MM patients. MFI analysis revealed that the expression of TNF-α on cells was higher in the BMSCs of the MM patients than in those of the controls, but these differences were not statistically significant because of the high standard deviation values.

Production of TNF-α in cultures and co-cultures of BMSCs of MM patients and controls with RPMI8226 myeloma cells. Production of TNF-α was measured by ELISA (A) or by cytometric analysis in monocultures of CD166+ cells (markers for mesenchymal stromal fibroblasts), monocultures of CD38+/CD138+ myeloma cells, and in co-cultures, where both types of cells were in direct cell-to-cell contact (direct) or were separated by a transwell (non-contact). The level of TNF-α expression was measured by flow cytometry as the percentage of TNF-α-expressing cells (B, C) or as the MFI (D, E), indicating the level of TNF-α expression in cells. The level of TNF-α was also measured by ELISA (E). *Significantly different from the control group, p<0.05; #significantly different from monoculture within the group, p<0.05.
When the production of BAFF was measured by ELISA in the supernatants of the BMSC and RPMI8226 cultures and co-cultures, it was found that the BMSCs of the MM patients produced significantly more BAFF than control BMSCs (Fig. 7A). The amounts of BAFF produced by the BMSCs of the MM patients varied significantly, which resulted in a very high standard deviation; however, no correlations with the stage of the disease or the intensity of bone lesions were observed. Direct cell-to-cell contact of the BMSCs with the RPMI8226 cells did not result in increased BAFF production. The RPMI8226 cells cultivated alone did not release BAFF into the culture medium.

Production of BAFF (A), HGF (B), and OPN (C) in cultures of BMSCs of MM patients and control subjects and in co-cultures of both types of cells cultivated in cell-to-cell contact or in non-contact conditions, where the cells were separated by a transwell. The cytokine level was measured by ELISA in supernatants from the monocultures and co-cultures. *Significantly different from the control group, p<0.05; #significantly different from monoculture within the group, p<0.05.
The production of HGF was measured by ELISA (Fig. 7B). There were significant differences in the amounts of HGF released into the culture medium from the BMSCs of the MM patients and those of the controls. Especially after 72 h of incubation, significantly higher amounts of HGF were present in the media of the BMSCs of the MM patients than in those of the controls. A very slight, not statistically significant, enhancement of HGF production after co-culture of the RPMI8226 cells with the BMSCs was observed. The RPMI8226 cells cultivated alone produced very low levels of HGF.
ELISA measurements of OPN released into the culture medium revealed that the BMSCs of the MM patients and of the controls produced very low amounts of OPN (Fig. 7C). In contrast, the RPMI8226 cells produced high amounts of OPN. Additionally, the level of OPN increased after co-culture of the BMSCs with the RPMI8226 cells both in direct cell-to-cell contact and in the presence of a culture insert. Moreover, co-cultures of the BMSCs of the MM patients with the RPMI8226 cells produced more OPN than those of the control BMSCs.
Table 3 lists the correlations between the cytokines expressed in CD166+ cells and CD38+/CD138+ (RPMI8226) cells. It can be seen that the production of IL-6 correlated positively with several cytokines produced both in the BMSCs of the MM patients and in the myeloma cell line.
Discussion
There is growing evidence for the existence of a network of cytokines operating in the growth, progression, and dissemination of MM [25]. Among the numerous growth and survival factors for myeloma cells, IL-6 is considered the most important and is the best characterized.
The induction of IL-6 in BMSCs from myeloma patients and normal donors by myeloma tumor cell lines has already been described by other authors [28]. It has been found that BMSCs of MM patients produce more IL-6 than those of healthy controls. While some authors stressed that direct contact between the two types of cells was necessary for this induction, others [2] found that a significant increase in IL-6 concentration occurred when the myeloma cells (isolated from patients or myeloma-derived cell lines) were co-cultured with BMSCs of MM patients in a non-contact transwell system. The latter authors suggested that this was caused by bFGF expressed by myeloma cells which bound with bFGF receptors on BMSCs and thus stimulated IL-6 production. These data demonstrate that paracrine interactions between myeloma cells and BMSCs trigger the stimulation of IL-6. In our study we also established that direct binding of plasma cells to BMSCs is not prerequisite for the induction of IL-6. When the RPMI8226 cells were separated from the BMSCs by a tissue culture insert which prevented direct cellular contact but allowed an exchange of soluble factors, IL-6 was also produced by both the BMSCs and the myeloma cells. These observations are in agreement with the results of other authors [12] who found that IL-6 could be produced in response to soluble factors, produced mainly by myeloma cells, such as RANKL, IL-1β, TNF-α, and TGF-β and cytokines produced by BMSCs, such as VEGF and HGF [8, 26]. These results suggest that in vivo not only myeloma cells attached to stroma, but also unattached cells may autocrinely produce IL-6. In our study we also discovered, for the first time in the literature, that direct contact of BMSCs of MM patients with RPMI8226 myeloma cells induces overproduction of sIL-6R. As sIL-6R is known to form a complex with IL-6, and such complexes can bind to gp130 even on cells which lack membrane-bound IL-6R and trigger gp130 cellular signaling [39], it seems likely that the effect of IL-6 overproduction by BMSCs of MM patients can have more pronounced consequences than that expected from IL-6 overproduction itself.
IL-6 is a member of the IL-6 family of cytokines, which includes, among others, IL-11, that commonly use gp130 as a signal transducing molecule. IL-6 and IL-11 have both been reported to stimulate myeloma cell growth [19]. Both are also known to promote osteoclastogenesis and bone resorption in BM cultures [11, 24]. It has already been found that in a co-culture system of BMSCs of MM patients with a human myeloma cell line, plasma cells upregulate IL-11 secretion by BMSCs through cell-to-cell contact [12]. In our experiments, a cytometric analysis confirmed that direct cell-to-cell contact significantly enhanced IL-11 production, although more efficiently in the RPMI8226 cells than in the BMSCs. It also showed that in co-cultures, the RPMI8226 cells were the main source of IL-11. In our experiments, however, there were no significant differences between the BMSCs from myeloma patients and controls in the ability to produce IL-11.
IL-10 is the most potent B cell differentiation factor. It can also stimulate the proliferation of myeloma cells in IL-6-deprived cultures and thus can be regarded as an IL-6 unrelated growth factor for malignant plasma cells [32, 33]. It is produced by the myeloma cells of about half of patients and is detected in the blood of MM patients. The myeloma cell growth activity of IL-10 is mediated through a gp130 cytokine, oncostatin M (OSM), which is frequently produced by myeloma cells. IL-10 induces receptors for OSM on myeloma cells [23, 33]. Moreover, IL-10 inhibits osteogenic activity in mouse BM, and may thus also be involved in bone lytic lesions in MM [47]. The high expression of IL-10 which we detected in unstimulated BMSCs of MM patients confirms our previous observations [50] and strongly suggests that the overproduction of IL-10 by stromal cells may function as a microenvironmental factor in BM promoting residual myeloma cell growth even in patients in remission. It should be noted that IL-10 overexpression induced by direct cell-to-cell contact of BMSCs with RPMI8226 cells was described by us for the first time in the literature.
In the BM, TNF-α is produced not only by the cells of the BM stroma, but also by myeloma cells themselves [37]. TNF-α has been shown to act as a survival factor for myeloma cells [21] and, in combination with IL-4, to be implicated in myeloma precursor cell differentiation [38]. Moreover, TNF-α is able to trigger the secretion of IL-6 from BMSCs [15, 44] and displays proangiogenic properties [10]. It has been observed that BM cells explanted from MM patients produce higher amounts of TNF-α than those of comparable controls. The extent of production correlated with the stage of disease, especially with the presence of extensive osteolytic bone disease [27]. Furthermore, TNF-α enhanced the transendothelial migration of myeloma cells [20]. In our experiment, TNF-α was produced mainly by the BMSCs of MM patients under non-contact and cell-to-cell contact conditions in the presence of RPMI8226 cells, but the differences between the BMSCs from MM patients and controls were not striking. However, the RPMI8226 cells also produced TNF-α, especially after direct cell-to-cell contact with the BMSCs of MM patients.
Most recently, the BAFF of the TNF family produced by myeloid cells was identified as a critical factor in normal B-cell development and homeostasis. It is expressed predominantly by macrophages and dendritic cells in both membrane-bound and proteolytically cleaved soluble protein [29]. BAFF was detected in myeloma cells and in sera derived from MM patients, suggesting an autocrine loop of stimulation from the tumor cells. However, the biological role of BAFF in myeloma pathogenesis has not been fully characterized. It has been revealed that BAFF is present on BMSCs derived from MM patients and its secretion is higher in BMSCs than in myeloma cells. Moreover, tumor cell adhesion to BMSCs augments BAFF secretion [43]. In our experiment we detected using ELISA that RPMI8226 cells without contact with BMSCs did not produce BAFF, whereas the BMSCs of MM patients spontaneously produced high amounts of BAFF, which did not increase after co-culture with the myeloma cells. Our results are not in agreement with the results of other authors [43], mentioned above, which indicate that adhesion of myeloma cells to BMSCs augments BAFF production. These discrepancies may be due to the fact that different myeloma cells were used for the experiments. Tai et al. [43] used purified CD138+ myeloma cells isolated from patients which, as described by other authors [30], did not express BAFF-R, but expressed B-cell-maturating antigen (BCMA), another BAFF receptor. In contrast to that, RPMI8226 cells were described to express three BAFF receptors: BCMA, BAFF-R, and TACI (transmembrane activator and calcium modulator and cyclophilin ligand receptor). Therefore, in our experiment BAFF could have been bound to the surface receptors on RPMI8226 cells and removed from the culture media. Moreover, Moreaux et al. [30] reported that BMSCs, in contrast to osteoclasts, are poor producers of BAFF. In our BMSC cultures, CD14+ cells were detected, which can be precursors of osteoclasts. Therefore it cannot be excluded that CD14+ producing cells were the main source of BAFF. On the other hand there were no differences in the number of CD14+ cells between the controls and the MM group, so the question why the BMSCs of the MM patients produced more BAFF than those of the controls needs further experiments.
Recent studies have identified HGF as a potential key signal in the evolution of MM as HGF exerts a strong proliferative and antiapoptotic effect on myeloma cells by way of the RAS/MAPK and PI3K/PKB pathways. BMSCs producing HGF are a paracrine source of HGF, but myeloma cells are a source of HGF as well. Moreover, myeloma cells catalyze HGF activation by secreting an activator of HGF [46]. In our experiments, however, we showed that RPMI8226 myeloma cells cultivated alone produced a very low level of HGF. These results are not in agreement with the results reported by Borset et al. [3], who did detect production of HGF by RPMI8226 cells. We believe that this discrepancy is due to differences in the culture conditions, such as the time of incubation and the culture medium used, with 10% of FBS in Borset's experiment and 2% of FBS in our experiment. In contrast to the poor secretion of HGF by RPMI8226, we found that BMSCs of myeloma patients are a rich source of HGF. These results are in agreement with earlier publications indicating a high production of HGF by BM cells of MM patients [16, 22]. It was recently found that HGF enhances myeloma cell adhesion to the BM matrix protein fibronectin [17] and plays an important role in the proliferation, adhesion, and migration of myeloma cells [18]. The spontaneous overproduction of HGF by BMSCs of MM patients, even in the absence of myeloma cells, strongly suggests that the BM microenvironment of persons with MM may enhance the proliferation and dissemination of myeloma cells residual after chemotherapy.
OPN is a multifunctional protein that promotes cell adhesion and migration, inhibits hydroxyapatite formation, and binds Ca2+ [35]. OPN is strongly associated with tumorgenesis. In patients with MM, plasma levels of soluble OPN are associated with poor prognosis. Multiple myeloma cells adhere to OPN, which implies that elevated stromal expression of OPN may be one of the factors responsible for the retention of myeloma cells in the BM. Elevated plasma OPN levels in MM patients could be due both to the production of OPN by tumor cells and to tumor-induced production of OPN by non-tumor cells [41]. In our experiments we confirmed that OPN can be produced by myeloma cells. Moreover, we discovered that OPN production in co-cultures of the BMSCs from myeloma patients with RPMI8226 cells is significantly higher than in respective co-cultures of control BMSCs with myeloma cells. We do not know whether BMSCs or RPMI8226 cells are the producers of OPN, but these results strongly suggest that either the BMSCs of MM patients produce more OPN or they more effectively support OPN production by myeloma cells.
Summing up, we detected a high ability of BMSCs of MM patients, in comparison with controls, to produce IL-6, IL-10, TNF-α, and OPN in response to RPMI8226 cells. However, the differences in their production by the BMSCs of MM patients and of controls were not striking. In contrast, the production of HGF, BAFF, and sIL-6R was significantly higher. Cytokines over-expressed by the BMSCs of MM patients exhibit several biological properties and can function as growth factors for myeloma cells (IL-6, IL-10, HGF), migration stimulatory factors for tumor plasma cells (TNF-α, HGF), adhesion stimulatory factors (HGF, OPN), stimulators of osteoclastogenesis (IL-6, TNF-α), and proangiogenic factors (TNF-α). Therefore the results of our experiments strongly suggest that in spite of a lack of differences in morphology, surface antigen, and enzyme production between BMSCs from MM patients and controls, they differ in spontaneous and myeloma cell-induced production of cytokines. Interestingly a continued production of cytokines was observed in the BMSCs, independent of the long time of in vitro cultivation (several weeks). This suggests the presence of an undefined autocrine stimulation pathway resulting in prolonged production of cytokines in vivo. If these in vitro findings reflect in vivo behavior, the continued production of cytokines, even during disease remission, might provide optimal conditions for the proliferation and differentiation of residual tumor cells or their precursors in the affected BM.
References
Arnulf B., Lecourt S., Soulier J., Ternaux B., Lacassagne M. N., Crinquette A., Dessoly J., Sciaini A. K., Berbunan M., Chomienne C., Fermand J. P., Marolleau J. P., and Larghero J. (2007): Phenotypic and functional characterization of bone marrow mesenchymal stem cells derived from patients with multiple myeloma. Leukemia, 21, 158–163.
Bisping G., Leo R., Wenning D., Dankbar B., Padro T., Kropff M., Scheffold C., Kroger M., Mesters R. M., Berdel W. E. and Kienast J. (2003): Paracrine interactions of basic fibroblast growth factor and interleukin-6 in multiple myeloma. Blood, 101, 2775–2783.
Borset M., Lien E., Espevik T., Helseth E., Waage A. and Sudan A. (1996): Concomitant expression of hepatocyte growth factor/scatter factor and the receptor c-met in human myeloma cell lines. J. Biol. Chem., 271, 24655–24661.
Calabrio A., Oken M. M., Hascal V. C. and Masellis A. M. (2002): Characterization of hyaluronian synthetase expression and hyaluronian synthesis in bone marrow mesenchymal progenitor cells: predominant expression of HAS1 mRNA and up-regulated hyaluronian synthesis in bone marrow cells derived from multiple myeloma patients. Blood, 100, 2578–2585.
Caligaris-Cappio F., Bergui L., Gregoretti M. G., Gaidano G., Gaboli M., Schena M., Zallone A. Z. and Marchisio P. C. (1991): Role of bone marrow stromal cells in the growth of human multiple myeloma. Blood, 77, 2688–2693.
Coelho M. J. and Fernandez M. H. (2000): Human bone cell cultures in biocompatibility testing. Part II. Effect of ascorbic acid, beta-glycerophosphate and dexamethasone on osteoblastic differentiation. Biomaterials, 21, 1095–1102.
Corre J., Mahtouk K., Attal M., Gadelorge M., Huynh A., Fleury-Cappellesco S., Danho C., Laharague P., Klein B., Reme T. and Bourin P. (2007): Bone marrow mesenchymal cells are abnormal in multiple myeloma. Leukemia, 21, 1079–1088.
Dankbar B., Padro T., Leo R., Feldman B., Kropff M., Mesters R. M., Serve H., Berdel W. E. and Kienast J. (2000): Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood, 95, 2630–2636.
Durie B. G. and Salmon S. E. (1975): A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment and survival. Cancer, 36, 842–845.
Frater-Schroder M., Risau W., Hallmann R., Gautschi P. and Bohlen P. (1987): Tumor necrosis factor type-α, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proc. Natl. Acad. Sci. USA, 84, 5277–5281.
Fuller K., Wong B., Fox S., Choi Y. and Chambers T. J. (1998): TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J. Exp. Med., 188, 997–1002.
Giuliani N., Colla S., Morandi F. and Rizzoli V. (2004): The RANK/RANK ligand system is involved in interleukin-6 and IL-11 up-regulation by human myeloma cells in bone marrow microenvironment. Haematologica, 89, 1118–1123.
Gregoretti M. G., Gottardi D., Ghia P., Bergui L., Merico F. and Marchisio P. C. (1994): Characterization of bone marrow stromal cells from multiple myeloma. Leukemia Res., 18, 675–682.
Hayashi T., Hidishima T. and Anderson K. C. (2003): Novel therapies for multiple myeloma. Br. J. Haematol., 120, 10–17.
Hideshima T., Chauhan D., Schlossman R., Richardson P. and Anderson K. C. (2001): The role of tumor necrosis factor-α in pathophysiology of human multiple myeloma: therapeutic applications. Oncogene, 20, 4519–4527.
Hjertner O., Torgersen M. L., Seidel C., Hjorth-Hansen H., Waage A., Borset M. and Sudan A. (1999): Hepatocyte growth factor (HGF) induces interleukin-11 secretion from osteoblasts: a possible role for HGF in myeloma associated osteolytic bone disease. Blood, 94, 3883–3888.
Holt R. U., Baykov V., Ro T. B., Brabrand S., Waage A., Sundan A. and Borset M. (2005): Human myeloma cells adhere to fibronectin in response to hepatocyte growth factor. Haematologica, 90, 479–488.
Hov H., Holt R. U., Ro T. B., Fagerli U. M., Hjorth-Hansen H., Baykov V., Christensen J. G., Waage A., Sudan A. and Borset M. (2004): A selective c-met inhibitor blocks an autocrine hepatocyte growth factor growth loop in ANBL-6 cells and prevents migration and adhesion of myeloma cells. Clin. Cancer Res., 10, 6686–6694.
Ishikawa H., Tsuyama N., Obata M. and Kawano M. M. (2006): Mitogenic signals initiated via inteleukin-6 receptor complexes in corporation with other transmembrane molecules in myelomas. J. Clin. Exp. Hematopathol., 46, 55–66.
Jöhrer K., Janke K., Krugmann J., Fiegel M. and Greil R. (2004): Transendothelial migration of myeloma cells is increased by tumor necrosis factor (TNF)-α via TNF receptor 2 and autocrine up-regulation of MCP-1. Clin. Cancer Res., 10, 1901–1910
Jourdan M., Tarte K., Legouffe E., Brochier J., Rossi J. F. and Klein B. (1999): Tumor necrosis factor is survival and proliferation factor for human myeloma cells. Eur. Cytokine Netw., 10, 65–67.
Kara I. O., Sahin B., Gunesacar R. and Unsal C. (2006): Clinical significance of hepatocyte growth factor, platelet-derived growth factor AB, and transforming growth factor-alpha in bone marrow and peripheral blood of patients with multiple myeloma. Adv. Ther., 23, 635–645.
Klein B., Lu Z. Y., Gu Z. J., Costes V., Jourdan M. and Rossi J. F. (1999): Interleukin-10 and gp130 cytokines in multiple myeloma. Leuk. Lymphoma, 34, 63–70.
Kudo O., Sabokbar A., Pocock A., Itonaga I., Fujikawa Y. and Athanasou N. A. (2003): Inteleukin-6 and IL-11 support human osteoclast formation by RANKL-independent mechanism. Bone, 32, 1–7.
Lauta V. M. (2003): A review of cytokine network in multiple myeloma. Cancer, 97, 2440–2452.
Lauta V. M. (2001): Interleukin-6 and the network of several cytokines in multiple myeloma: an overview of clinical and experimental data. Cytokine, 16, 79–86.
Lichtenstein A., Berenson J., Norman D., Chang M. P. and Cartile A. (1989): Production of cytokines by bone marrow cells obtained from patients with multiple myeloma. Blood, 74, 1266–1273.
Horst H. M., Lamme T., de Smet M., Klein S., de Weger R. A., van Oers R. and Bloem A. C. (1994): Primary tumor cells of myeloma patients induce interleukin-6 secretion in long-term bone marrow cultures. Blood, 84, 2269–2277.
Moore P. A., Belvedere O., Orr A., Pieri K., LaFleur D. W., Feng P., Soppet D., Charters M., Gentz R., Parmelee D., Li Y., Galperina O., Giri J., Roschke V., Nardelli B., Carrell J., Sosnovtseva S., Greenfield W., Ruben S. M., Olsen H. S., Fikes J. and Hilbert D. M. (1999): BlyS: member of tumor necrosis factor family and B lymphocyte stimulator. Science, 285, 260–263.
Moreaux J., Cremer F.W., Reme T., Raab M., Mahtouk K., Kaukel P., Pantesco V., De Vos J., Jourdan E., Jauch A., Legouffe E., Moos M., Fiol G., Goldshmidt H., Rossi J. F., Hose D. and Klein B. (2005): The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood, 106, 1021–1030.
Nakasato Y. R., Janckila A. J., Halleen J. M., Vaananen H. K., Walton S. P. and Yam L. T. (1999): Clinical significance of immunoassay for type-5 tartrate-resistant acid phosphatase. Clin. Chem., 45, 2150–2157.
Otsuki T., Yamada O., Yata K., Sakaguchi H., Kurebayashi J., Yawata Y. and Ueki A. (2000): Expression and production of interleukin 10 in human myeloma cell lines. Br. J. Haematol., 111, 835–842.
Otsuki T., Yata K., Sakaguchi H., Uno M., Fujii T., Wada H., Sugihara T. and Ueki A. (2002): IL-10 in myeloma cells. Leuk. Lymphoma, 43, 969–974.
Pearse R. N., Sordillo E. M., Yaccoby S., Wong B. R., Liau D. F., Colman N., Michaeli J., Epstein J. and Choi Y. (2001): Multiple myeloma disrupts the TRANCE/osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc. Natl. Acad. Sci. USA, 98, 11581–11586.
Razzouk S., Brunn J. C., Qin C., Tye C. E., Goldberg H. A. and Butler W. T. (2002): Osteopontin posttranscriptional modifications, possibly phosphorylation, are required for the in vitro bone resorption but not osteoclast adhesion. Bone, 30, 40–47.
Roodman G. D. (2004): Pathogenesis of myeloma bone disease. Blood Cells Mol. Dis., 32, 290–292.
Sati H. I., Greaves M., Apperley J. F., Russell R. G. and Croucher P. I. (1999): Expression of IL-1β and tumor necrosis factor-α in plasma cells from patients with multiple myeloma. Br. J. Haematol., 1104, 350–357.
Sawamura M., Murakami H. and Tsychiya J. (1996): Tumor necrosis factor-α and interleukin-4 in myeloma cell precursor differentiation. Leuk. Lymphoma, 21, 31–36.
Scheller J. and Rose-John S. (2006): Interleukin-6 and its receptor: from bench to bedside. Med. Microbiol. Immunol., 195, 173–183.
Silvestris F., Caffario P., Calvani N. and Dammacco F. (2004): Impaired osteoblastogenesis in myeloma bone disease: role of upregulated apoptosis by cytokines and malignant plasma cells. Br. J. Haematol., 126, 475–486.
Standal T., Hjorth-Hansen H., Rasmussen T., Dahl I.M.S., Lenhoff S., Brenne A.T., Seidel C., Baykov V., Waage A., Borset M., Sudan A. and Hjertner O. (2004): Osteopontin is an adhesive factor for myeloma cells and is found in increasing levels in plasma of patients with multiple myeloma. Haematologica, 89, 174–182.
Stewart K., Monk P., Walsh S., Jefferiss C. M., Letchford J. and Beresford J. N. (2003): STRO-1, HOP-26 (CD63), CD49a and SB10 (CD166) as markers of primitive human marrow stromal cells and their more differentiated progeny: a comparative investigations in vitro. Cell Tissue Res., 313, 281–290.
Tai Y. T., Li X. F., Breitkreutz I., Song W., Neri P., Catley L., Podar K., Hideshima T., Chauhan D., Raje N., Schlossman R., Richardson P., Munshi N. C. and Anderson K. C. (2006): Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res., 66, 6675–6682.
Thomas X., Anglared B., Magaud J. P., Epstein J. and Archimbaud E. (1998): Independence between cytokines and cell adhesion molecules to induce interleukin-6 production by stromal cells in myeloma. Leuk. Lymphoma, 32, 107–119.
Tian E., Zhan F., Walker R., Rasmusen E., Ma Y., Barlogie B. and Shaugnessy J. D. (2003): The role of Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med., 349, 2483–2494.
Tjin E. P., Derksen P. W., Kataoka H., Spaargaren M. and Pals S. Y. (2004): Multiple myeloma cells catalyze hepatocyte growth factor (HGF) activation by secreting the serine protease HGF-activator. Blood, 104, 2172–2175.
Van Vlasselaer P., Borremans B., van Heuvel R., van Gorp U. and de Wall Malefyt R. (1993): Interleukin-10 inhibits osteogenic activity of mouse bone marrow. Blood, 82, 2361–2370.
Vincent T., Jourdan M., Sy M. S., Klein B. and Mechti N. (2001): Hyaluronic acid induces survival and proliferation of human myeloma cells through an interleukin-6-mediated pathway involving the phosphorylation of retinoblastoma protein. J. Biol. Chem., 276, 14728–14736.
Wallace S. R., Oken M. M., Lunetta K. L., Panoskaltsis-Mortari A. and Masellis A. M. (2001): Abnormalities of bone marrow mesenchymal cells in multiple myeloma patients. Cancer, 91, 1219–1230.
Zdzisińka B., Walter-Croneck A., Dmoszyńska A. and Kandefer-Szerszeń M. (2006): Matrix metalloproteinase and cytokine production by bone marrow adherent cells from multiple myeloma patients. Arch. Immunol. Ther. Exp., 54, 289–296.
Zhang X. G., Bataille R., Widjenes J. and Klein B. (1992): Interleukin-6 dependence of advanced malignant plasma cell dyscrasias. Cancer, 69, 1373–1376.
Acknowledgment
This work was supported by grant No. 2 P05A 095 28 from the Polish Ministry of Science and Higher Education.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zdzisińska, B., Bojarska-Junak, A., Dmoszyńska, A. et al. Abnormal cytokine production by bone marrow stromal cells of multiple myeloma patients in response to RPMI8226 myeloma cells. Arch. Immunol. Ther. Exp. 56, 207–221 (2008). https://doi.org/10.1007/s00005-008-0022-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-008-0022-5