
Abstract.
Introduction
Macrophages/microglial cells are considered as immune cells in the central nervous system. Interleukin (IL)-16 is a proinflammatory cytokine produced by activated monocytic cells.
Materials and Methods
Expression of IL-16 was analyzed by immunohistochemistry in human astrocytic brain tumors and the rat C6 glioblastoma tumor model. IL-16 was detected in both human astrocytic brain tumors and rat C6 glioma.
Results
Compared with human control brains, a significant increase in the percentages of parenchymal IL-16+ macrophages/microglia was observed already in grade II astrocytomas, indicating that IL-16+ immunostaining could be a descriptor of a macrophage/microglia subset in astrocytic brain tumors. A further increase was observed at the transition from grade II to III astrocytomas. This increase in IL-16 immunoreactivity correlated with WHO grades of human astrocytic brain tumors.
Conclusions
Therefore, IL-16 might be a so far unknown factor in the regulation of the local inflammatory milieu of human and experimental astrocytomas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Macrophages/microglial cells constitute the first line of cellular defense against a variety of stressors to the central nervous system (CNS), participating in the regulation of innate and adaptive immune responses in human and rat gliomas [1]. Under neuropathological conditions, microglia are activated, and activated microglia are characterized morphologically by a gradual transition from a quiescent stellate form to a macrophage-like morphology, which is accompanied by upregulation of surface antigens and the formation of cell clusters. Recent studies indicate that activated microglial cells occur not only within and around brain tumors, but also contribute significantly to the actual tumor mass, notably in astrocytic gliomas [16]. However, their role in the pathogenesis of malignant brain tumor progression remains unclear. A better understanding of microglia-tumor interaction might be helpful in increasing the therapeutic efficacy of future immune modulatory approaches. As we know, several regulatory factors are secreted by activated microglia. It needs to be established whether such activation-associated peptides can be used to investigate microglial activation in gliomas of different WHO grade.
Interleukin (IL)-16 is one of the candidates expressed by activated monocytic cells. IL-16, known as lymphocyte chemoattractant factor (LCF), serves as a natural soluble ligand for CD4 that contributes to the regulatory process of CD4+ cell recruitment and activation at sites of inflammation in association with asthm a and several autoimmune diseases. Thus it is considered a proinflammatory cytokine produced by a variety of cells, including microglia [4, 5, 7, 14, 19]. In the human brain, IL-16 is constitutively but minimally expressed by a subpopulation of microglial cells [20]. In the chronic inflammatory milieu of multiple sclerosis lesions, IL-16 has been associated with the attraction of CD4+ lymphocytes across the blood-brain barrier (BBB) [21]. We showed previously that IL-16 is expressed by inflammatory cells in non-malignant CNS pathologies [9, 12, 24] as well as in human glioma [8]. Here we further describe the distribution of IL-16+ cells in different human astrocytomas, including protoplasmatic astrocytomas (WHO grade II), fibrillary astrocytomas (WHO grade II), anaplastic astrocytomas (WHO grade III), glioblastoma multiforme (WHO grade IV), and four neuropathologically unaltered control brains. Rat C6 gliomas and normal control brains were investigated as well.
Materials and Methods
Patient data
Control brains without neuropathological alterations were obtained from an established normal tissue brain bank of the Institute of Brain Research of the University of Tuebingen as published previously [20]. Autopsy data of these cases are listed in Table 1. We investigated 50 primary brain tumor specimens, consisting of 3 protoplasmatic astrocytomas (WHO grade II), 21 fibrillary astrocytomas (WHO grade II), 7 anaplastic astrocytomas (WHO grade III), and 19 glioblastoma multiforme (WHO grade IV; Table 2). The ages of the patients (19 females and 31 males) ranged 20–77 years. All tumor specimens were obtained before either radiotherapy or chemotherapy. Histological grading of the human tumor specimens was performed according to the revised WHO classification guidelines [15]. Tumors were resected at the Department of Neurosurgery at Tuebingen.
Animal model
The rat C6 glioblastoma cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI 1640 medium with Glutamax II (Gibco, BRL, Paisely, UK) containing 10% fetal calf serum (Gibco BRL) and 1.2% penicillin/streptomycin (Fluka, Buchs, Switzerland) at 37°C and 5% CO2. C6 glioma cells were implanted as follows: 15 male Sprague-Dawley rats (220–280 g, Charles River, Sulzfeld, Germany) were anesthetized by intraperitoneal injection of Ketanest (Ketaminehydrochloride, Parke Davis, USA; 100 mg/kg) and Rompun (Xylazine-hydrochloride, Bayer, Germany; 10 mg/kg). In order to avoid xerophthalmia during anesthesia, the eyes were covered with Oculotect Gel (Retinolpalmitate, CIBA Vision, Novartis, Germany). After reaching an adequate depth of anesthesia, the skin of the head was shaved, cleaned, and disinfected with 75% ethanol and incised (midline incision) to expose the skull. After drilling a 2-mm hole 3 mm to the right and 3 mm posterior to bregma, the rat was fixed in a stereotactic apparatus (ASI Instruments, Germany). The needle of an Hamilton syringe (900 series; Hamilton Company, Reno, NV, USA) was inserted 4 mm from the pial surface vertically into the brain. Five μl of C6 glioblastoma cell suspension (2×105 cells/μl) were injected. Thereafter the needle was slowly withdrawn and the skin was restored with a suture (4/0, FS-2, Ethicon; Johnson & Johnson, Brussels, Belgium). After two weeks the rats were sacrificed and perfused intracardially with fixative (4% formalin in 0.1 M phosphatebuffered saline, pH 7.5). Four normal rats (without suffering sham injection) were used as controls. All rats were kept under controlled conditions of temperature and light, with food and water available ad libitum. The animals were cared for in accordance with published International Health Guidelines under a protocol approved by the local Institutional Animal Care and the Administration District Official Committee.
Immunohistochemistry
Tissue samples were fixed in buffered 4% formalin (pH 7.4) and paraffin-embedded by routine methods. After dewaxing, sections (3 μm) were immersed in 0.01 M citrate buffer and irradiated for 15 min in a microwave oven set at 600 W to enhance the fraction of accessible epitopes. Endogenous peroxidase was blocked with 1% H2O2 in methanol (15 min). Non-specific protein binding was inhibited by incubation with normal porcine serum (Biochrom, Berlin, Germany) for 15 min. Tissue sections were incubated with purified mouse anti-IL-16 monoclonal antibody (BMA, Augst, Switzerland; diluted at 1:100 in TBS-BSA) overnight at 4°C. Specific antibody binding was detected by incubating the sections with a secondary biotinylated rabbit anti-mouse IgG F(ab)2 antibody fragment (1:400; Dako, Hamburg, Germany) for 30 min, followed by incubation with a peroxidase-conjugated streptavidin-biotin-complex (Dako). Labeled antigens were visualized by application of 3,3′-diaminobenzidine (DAB; Fluka, Neu-Ulm, Germany) as a chromogen. All sections were counterstained with Mayer's Hemalaun.
Negative controls consisted of sections incubated in the absence of the primary antibody. The specificity of monoclonal IL-16 antibody was analyzed by isotype control and by preincubation of the monoclonal antibody with an irrelevant cytokine, EMAP II (endothelial-monocyte activation polypeptide II) [22]. As a positive control for IL-16 immunoreactivity we used rat and human spleen sections, as macrophages are abundantly found in this organ.
Double-labeling experiments
Double-labeling experiments were performed on human tissues of GBM and rat C6 glioma. Antibodies used for GBM included monoclonal antibody against CD68 (clone KP-1, diluted 1:100 in TBS-BSA, Dako) for microglia/macrophage identification. Activated microglia/macrophages were detected with antibodies directed against HLA-DR, -DP, and -DQ (MHC class II, clone CR3/43, 1:50, Dako). Lymphocytic subsets were typed by monoclonal antibodies against CD4 (T-helper lymphocytes, clone 1F6, 1:10, Dako) and CD3 (pan-T-cell marker, clone PS1, 1:100, Novocastra, Dossenheim, Germany) and were applied overnight to tissue sections at 4°C. For rat C6 glioma, sections were incubated with monoclonal antibodies against ED1 (1:100; clone ED1, BMA, Augst, Switzerland) to demonstrate activated microglia/macrophages. Briefly, sections were dewaxed, irradiated in a microwave oven for antigen retrieval, and incubated with porcine serum as described above. Visualization was achieved by adding biotinylated secondary rabbit anti-mouse IgG diluted 1:400 for 30 min and alkaline phosphatase-conjugated ABC complex/AP (Dako, Kenmark) (1:400 in TBS-BSA) for 30 min. The sections were developed with Fast Blue BB salt (Fluka, Buchs, Switzerland), yielding a blue reaction product. To avoid cross-reactivity, the probes were irradiated again in a microwave oven. After administration of porcine serum for 15 min, the sections were labeled with IL-16 antibody as already described. Bound antibodies were visualized with DAB (brown).
Evaluation of human astrocytomas and rat C6 gliomas
IL-16+ cells with the typical morphology of activated microglial cells and with a clearly visible nucleus and all counterstained cells with a visible nucleus were counted in 10 high-power fields (×200 magnification with an eyepiece grid representing 0.25 mm2) in 10 independent areas of the tumor tissue for each section. The ratio of positively labeled and counterstained nuclei was calculated in each brain tumor specimen and the control brain section to obtain the percentage distribution of labeled TAMs. IL-16+ cells in perivascular spaces (cells nestled against the outer vessel wall and cells in perivascular spaces) were counted per 10 vessels and considered to be positive if a minimum of two IL-16+ cells were present.
Statistical analysis
In order to stabilize variances, percentages were subjected to an arcsine transformation before a one way analysis of variance (Anova) was performed. Differences of means were compared by a Tukey's honest significant difference post-hoc test. Data are presented as means and their 95% confidence intervals (95%CI) after back-transformation. The percentages of IL-16+ perivascular cells were calculated as means of labeled perivascular cells (MLPVC). Data of the density of IL-16+ parenchymal cells were calculated as means of labeled parenchymal cells (MLPC).
Results
Human control brains and astrocytomas
In four human control brains with no neuropathological alterations, IL-16 immunoreactivity was detected in single microglia/macrophages. Only a few IL-16+ cells were detected in perivascular spaces (Fig. 1A).

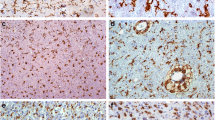
Single immunohistochemical staining of IL-16 in control human brain (A), WHO Grade II fibrillary astrocytoma (B), WHO Grade III anaplastic astrocytoma (C), and WHO Grade IV glioblastoma (D). Arrows indicate IL-16+ cells. In the glioma, double-labelling of IL-16 (brown) with CD68 (blue) confirms that macrophages are labelled with antibody against IL-16 (E). Double-labelling of IL-16 (brown) with MHC-II (blue) (F). Double-labelling of IL-16 (brown) with CD4 (blue) (G). Double-labelling of IL-16 (brown) with CD3 (blue) (H). A–D: ×200; E–H: ×400.
In human astrocytomas, the percentages of IL-16+ perivascular cells increased by a gradient from fibrillary (MLPVC: 11.3%, 95%CI: 4.9–19.8%, WHO grade II) to anaplastic astrocytomas (MLPVC: 77.0%, 95%CI: 57.9–91.5%, WHO grade III). In control brains (MLPVC: 9.4%, 95%CI: 0.2–30.4%), protoplasmatic astrocytomas (MLPVC: 13.0%, 95%CI: 0.3–39.8%), and fibrillary astrocytomas (mentioned above; Fig. 2A), no significant differences were observed between each entity. There was also no significant difference between anaplastic astrocytomas and glioblastomas multiforme (MLPVC: 84.3%, 95%CI: 74.3–92.2%).

Statistical evaluation of the distributions of IL-16+ cells in parenchyma sections (A) and in perivascular areas (B) in astrocytomas of different malignancies. C — control brain; PRO — protoplasmatic astrocytoma (WHO grade II); FA — fibrillary astrocytoma (WHO grade II); ANA — anaplastic astrocytoma (WHO grade III); GBM — glioblastoma multiforme (WHO grade IV).
Changes in the percentage of IL-16+ cells occurring in the brain parenchyma paralleled increased accumulation of IL-16+ cells in perivascular spaces. In detail, IL-16+ cell numbers increased significantly in the brain parenchyma ofcontrol brains (Fig. 1A, MLPC: 2.1%, 95%CI: 0.1–6.6%) to fibrillary astrocytomas (Fig. 1B, MLPC: 7.4%, 95%CI: 5.1–10.3%) and further to anaplastic astrocytomas (Fig. 1C, MLPC: 20.9%, 95%CI: 14.4–28.3%). In protoplasmatic astrocytomas (MLPC: 8.9%, 95%CI: 2.9–17.7%) and glioblastomas (Fig. 2D, MLPC: 24.4%, 95%CI: 20.0–29.1%), large numbers of IL-16+ cells were observed (Fig. 2B). The necrotic areas mainly contained cell debris and only very rare IL-16+ macrophages/microglial cells. Abundant labeling by IL-16 monoclonal antibody was observed in tumor cells and TAMs adjacent to infiltration areas (Fig. 1C and D). In the tumor parenchyma, the proportion of stained cells varied considerably between different areas of the tumor, especially in glioblastomas.
Double-labeling experiments revealed that the major contributors of IL-16 expression were CD68+/IL-16+ cells predominantly located at areas of infiltrative tumor growth and in perivascular spaces (Fig. 1E). Co-localization of IL-16 and MHC-II molecules was frequently seen (Fig. 1F). At the same time, only very few cells in glioblastoma were characterized as CD4+/IL-16+ and/or CD3+/IL-16+ T lymphocytes (Fig. 1G and H, respectively).
IL-16 expression in normal rat brain and C6 gliomas
In contrast to human control brains, the four normal rodent control brains revealed no perivascular (MLPVC: 0.0%, 95%CI: 0–0.8%) and parenchymal IL-16+ cells (MLPC: 0.0%, 95%CI: 0–0.04%; Fig. 3A).

Single-labelling of IL-16 in rat brain sections. In normal rat brain, IL-16 staining was hardly seen (A). In rats with implanted C6 glioma, IL-16+ cells were strikingly located in areas of tumor mass (B); in the tumor infiltration areas, IL-16+ cells were especially concentrated in perivascular areas (C). Double-staining of IL-16+ cells (brown) with ED1 (blue) in the tumor infiltration of C6 glioma revealed these cells as microglia/macrophages (arrows). A–C: ×200; D: ×400
In rat C6 glioblastomas, massive IL-16 staining was seen in areas of both compact tumor and tumor infiltration (Fig. 3B). Activated IL-16+ macrophages/microglial phenotypes exhibiting retracted elongations/processes were confined to perivascular spaces and regions of infiltrative tumor growth (Fig. 3B and C, respectively). In perivascular regions of rat C6 transplanted tumors, IL-16+ cells accumulated in perivascular spaces (MLPVC: 98.6%, 95%CI: 97.3–99.5%). In peritumoral regions, the majority of IL-16+ cells were identified as activated macrophages/microglial cells with long thin protrusions/elongations, which was confirmed by ED1+/IL-16+ staining (Fig. 3D). In control, in normal rat brains (MLPC: 0.0%, 95%CI: 0–0.04%), opposing hemispheres (MLPC: 0.0%, 95%CI: 0–0.01%), or necrotic tumor lesion sites (MLPC: 0.0%, 95%CI: 0–0.01%), only single IL-16-labeled cells were detectable, whereas the numbers of IL-16+ cells increased significantly in peritumoral (MLPC: 21.0%, 95%CI: 20.2–21.8%) and infiltration areas (MLPC: 28.9%, 95%CI: 28.0–29.8%).
Discussion
Microglia/macrophages actively participate in the immune response in human and rodent gliomas [1, 10]. The mechanisms of the activation of TAMs are still under investigation. Here we report that IL-16 was expressed on macrophages/microglial cells in human and rat astrocytic brain tumors of distinct tumor grades. In the present study we found a positive correlation between the numbers of IL-16+ cells and glioma WHO grades.
Inflammation occurs in most precancerous and cancerous tissues with pronounced infiltration of innate immune cells like microglia and the production of specific inflammatory signaling molecules, including various cytokines and chemokines [2, 3, 6, 18]. It has been reported that in inflammatory conditions, IL-16 triggers 1) the generation of inositol trisphosphate, 2) translocation of protein kinase C, 3) the expression of proinflammatory cytokines (e.g. IL-6 and TNF-α) and growth factors (e.g. TGF-β) [7, 21], and 4) an increase in intracellular Ca2+ levels, which can elicit the activation of macrophages and control various microglial cell functions [13, 17]. The role of IL-16 in brain tumors is unclear so far. In the present study we observed the expression of IL-16 on TAMs in both human and rat astrocytic brain tumors. It has been reported that IL-16 plays a chemoattractant and immunomodulatory role in CNS diseases [7]. In the present study we observed that IL-16+ microglia/macrophages accumulated in perivascular spaces of rat C6 tumors [26]. This suggests that this cytokine might have the ability to promote the infiltration of large numbers of monocytes and lower number of lymphocytes into the tumor, which could be in line with the CD4+/IL-16+ and/or CD3+/IL-16+ T cells observed in the present study. Therefore, IL-16+ TAMs might contribute to the local inflammatory response and the release of various factors supports tumor growth and progression [6].
In the current study the correlation of IL-16+ macrophages/microglia with glioma WHO grading of human astrocytic brain tumors was striking. Activation of TAMs was suggested by coexpression of IL-16 and CD-68 or MHC II antigen, which are up-regulated by activated microglia present in CNS lesions. Conceding a heterogeneous population of macrophages/microglial cells, IL-16 in the present study might define a specific subpopulation of activated microglial cell in brain tumor pathology. Furthermore, IL-16 is a potent inducer of TNF-α and IL-1β expression, both of which can increase vascular permeability. Perivascular spaces are of particular neuroimmunological interest as they are prime portals of monocyte transmigration and lymphocyte drainage [11]. Expression of IL-16 by TAMs suggests that IL-16 might have autocrine functions on microglial cells. A paracrine mechanism of macrophage recruitment based on IL-16 and CD4, which are weakly expressed by monocytes, appears feasible. While TAMs are capable of causing tumor cytotoxicity [10], paradoxically they can stimulate tumor growth by the secretion of growth-inducing factors and angiogenic mediators [23, 25]. The mechanism of malignancy-associated macrophage recruitment is of further interest, as the migratory pathways across the BBB might be exploited in the development of novel tumor-homing therapeutic agents.
In conclusion, IL-16 immunoreactivity is a putative marker of a monocytic subset in human and rat gliomas in vivo. Furthermore, upregulation of IL-16 expression by this cell subset correlates with malignancy of tumors. Therefore, IL-16 might be a novel factor in the regulation of the local inflammatory milieu and tumor progression in both clinical and experimental astrocytoma/glioma.
References
Badie B. and Schartner J. (2001): Role of microglia in glioma biology. Microsc. Res. Tech., 54, 106–113.
Balkwill F., Charles K. A. and Mantovani A. (2005): Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell, 7, 211–217.
Balkwill F. and Mantovani A. (2001): Inflammation and cancer: back to Virchow? Lancet, 357, 539–545.
Bazan J. F. and Schall T. J. (1996): Interleukin-16 or not? Nature, 381, 29–30.
Center D. M. and Cruikshank W. W. (1982): Modulation of lymphocyte migration by human lymphokines. Identification and characterization of chemoattractant activity for lymphocytes from mitogen-stimulated mononuclear cells. J. Immunol., 128, 2563–2568.
Coussens L. M. and Werb Z. (2002): Inflammation and cancer. Nature, 420, 860–867.
Cruikshank W. W., Kornfeld H. and Center D. M. (1998): Signaling and functional properties of interleukin-16. Int. Rev. Immunol., 16, 523–540.
Deininger M. H., Pater S., Strik H. and Meyermann R. (2001): Macrophage/microglial cell subpopulations in glioblastoma multiforme relapses are differentially altered by radiochemotherapy. J. Neurooncol., 55, 141–147.
Engel S., Schluesener H.J., Mittelbronn M., Seid K., Adjodah D., Wehner H. D. and Meyermann R. (2000): Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathol., 100, 313–322.
Graeber M. B., Scheithauer B. W. and Kreutzberg G. W. (2002): Microglia in brain tumors. Glia, 40, 252–259.
Guillemin G. J. and Brew B. J. (2004): Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J. Leukoc. Biol., 75, 388–397.
Guo L. H., Mittelbronn M., Brabeck C., Mueller C. A. and Schluesener H. J. (2004): Expression of interleukin-16 by microglial cells in inflammatory autoimmune, and degenerative lesions of the rat brain. J. Neuroimmunol., 146, 39–45.
Hoffmann A., Kann O., Ohlemeyer C., Hanisch U. K. and Kettenmann H. (2003): Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signalling and control of release function. J. Neurosci., 23, 4410–4419.
Kazatchkine M. D. (1997): Terminology note: interleukin 16 (IL-16). Cytokine, 9, 72.
Kleihues P. and Cavenee W. K. (2000): Pathology and genetics of tumors of the nervous system. IRAC Press, Lyon.
Kurpad S. N., Wikstrand C. J. and Bigner D. D. (1994): Immunobiology of malignant astrocytomas. Semin. Oncol., 21, 149–161.
Li W. H., Llopis J., Whitney M., Zlokarnik G. and Tsien R. (1998): Cell-permanent caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature, 392, 936–940.
Mantovani A. (2005): Cancer: inflammation by remote control. Nature, 435, 752–753.
Mathy N. L., Bannert N., Norley S. G. and Kurth R. (2000): Cutting edge: CD4 is not required for the functional activity of IL-16. J. Immunol., 164, 4429–4432.
Mittelbronn M., Dietz K., Schluesener H. J. and Meyermann R. (2001): Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol., 101, 249–255.
Schluesener H. J., Seid K., Kretzschmar J. and Meyermann R. (1996): Leukocyte chemotactic factor, a natural ligand to CD4, is expressed by lymphocytes and microglial cells of the MS plaque. J. Neurosci. Res., 44, 606–611.
Schluesener H. J., Seid K., Zhao Y. and Meyermann R. (1997): Localization of Endothelial-monocyte activation polypeptide II (EMAP II) a novel proinflammatory cytokine, to lesions of experimental autoimmune encephalomyelitis, neuritis and uveitis: expression by monocytes and activated microglial cells. Glia, 20, 365–372.
Schoppmann S. F., Birner P., Stockl J., Kalt R., Ullrich R., Caucig C., Kriehuber E., Nagy K., Alitalo K. and Kerjaschki D. (2002): Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol., 161, 947–956.
Schwab J. M., Schluesener H. J., Seid K. and Meyermann R. (2001): IL-16 is differentially expressed in the developing human fetal brain by microglial cells in zones of neuropoiesis. Int. J. Dev. Neurosci., 19, 93–100.
Stoll G. and Jander S. (1999): The role of microglia and macrophages in the pathophysiology of the CNS. Prog. Neurobiol., 58, 233–247.
Streit W. J., Sammons N. W., Kuhns A. J. and Sparks D. L. (2004): Dystrophic microglia in the aging human brain. Glia, 45, 208–212.
Acknowledgment
This work has been supported by a grant of the Deutsche Forschungsgemeinschaft (GK 686-1) and the Wilhelm Sander-Foundation. Ms. L.-H. Guo is member of the Graduate College “Cellular mechanisms of immune-associated process (DFG: GK 794)”. Dr. J. M. Schwab received the Poste Rouge Fellowship of the Centre National de la Recherche Scientifique (CNRS), Paris, France, and is at present supported by a DFG research Fellowship #1164/1-1. We thank Sabine Conrad for excellent technical help and support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Markus Liebrich and Liang-Hao Guo: Contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Liebrich, M., Guo, LH., Schluesener, H.J. et al. Expression of interleukin-16 by tumor-associated macrophages/activated microglia in high-grade astrocytic brain tumors. Arch. Immunol. Ther. Exp. 55, 41–47 (2007). https://doi.org/10.1007/s00005-007-0003-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-007-0003-0