
Abstract
The diversity of genetically modified plants used for food and feed is increasing worldwide. For the detection and control of these products, efficient and reliable analytical tools are a prerequisite. This can be done by screening for specific DNA-elements and constructs characteristic of transgenic plants. In the past, numerous methods have already been published. However, several genetically modified plants are not covered by common screening methods. Here, a new qualitative triplex real-time polymerase chain reaction (PCR) method is presented, detecting two transgene flanking sequences and the transition between the Cassava Vein Mosaic Virus Promotor (P-CsVMV) and the phosphinothricin-N-acetyltransferase (pat) gene. These sequences are present in several transgenic plants and therefore, the described triplex method can be used as a screening tool to guide further analysis and increase the efficiency of the analysis strategy for GMO detection. The method is characterized by high specificity, sensitivity and robustness and is provided as a ring-trial validated method in the Official Collection of Methods according to the German Food and Feed Act.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
New genetically modified (GM) plants continue to be developed and commercialized. In many countries, especially within the European Union, strict authorization and specific labeling requirements must be met before the marketing of GM plants and their derived products is permitted. Therefore, the need for efficient and reliable analytical methods is paramount for control and monitoring purposes. For nearly 30 years, screening methods based on polymerase chain reaction (PCR) have been widely employed for detecting the presence of genetic modifications in routine analyses of food, feed, and seeds (European Network of GMO Laboratories 2011; Grohmann et al. 2009; 2015; Holst-Jensen et al. 2003; Waiblinger et al. 2010). These screening methods target DNA sequences that are frequently inserted into GM plants, such as specific promoters, terminators, or genes conferring tolerance to herbicides or protection against certain insects (European Network of GMO Laboratories 2011; Holst-Jensen et al. 2003). Due to the lack of specificity in identifying particular GM events, positive screening results need further confirmation using construct- and/or event-specific methods, if available (ISO 21569:2005).
The most commonly targeted sequences include the 35S promoter (P-35S) derived from the cauliflower mosaic virus (CaMV) or its derivatives, as well as the terminator sequence from the nopaline synthase (T-nos) gene found in Agrobacterium tumefaciens (ISO 21569:2005; Waiblinger et al. 2008).
Until a decade ago, the majority of GM plants listed in databases such as EUGINIUS: European GMO Initiative for a Unified Database System (2023) and ISAAA’s GM Approval Database could be detected in an initial screening with no more than 5 screening target sequences (Bahrdt et al. 2010; Waiblinger et al. 2010). However, over the last 10 years, the increasing diversity of genetic modifications has necessitated the identification of additional screening targets.
To economically and efficiently screen GM plants, multiplex PCR methods for simultaneously detecting various screening targets in a single PCR reaction have become increasingly important (BVL L 00.00-125:2008-12; BVL L 00.00-124:2008-12; BVL L 00.00-148:2014-02; Bahrdt et al. 2010; Waiblinger et al. 2008). These methods minimize analysis costs and time, providing a convenient way to develop analysis strategies using GMO screening elements (EUGINIUS: European GMO Initiative for a Unified Database System 2023; Federal Office of Consumer Protection and Food Safety 2020b; Waiblinger et al. 2010).
In this study, the development and validation of a novel triplex real-time PCR method that targets 3 distinct DNA sequences found in various GM plants is presented. Ti plasmids are utilized as vectors in Agrobacterium tumefaciens-mediated plant transformations. Such transformations result in the integration of the genetic construct, often flanked by short Ti plasmid-derived border sequence motifs located at the left and/or right ends of the transgenic construct. The presence of these border sequence motifs in a plant indicates that the plant has undergone genetic modification.
To identify these border sequence motifs, we conducted a sequence alignment using publicly available information related to the transformation method. We compared sequence information from various GM plants known to have been transformed by Agrobacterium using the NCBI BLASTN tool, leading to the identification of two border sequence motifs known as AgroBorder I and AgroBorder II.
The selection of 3 target sequences was based on enhancing the toolbox for routine GMO screening, allowing for the simultaneous detection of a significant number of GM plants in a single PCR run. Furthermore, this additional screening may significantly reduce the need for candidate event-specific tests for GM plant identification when employing common screening matrices (EUGINIUS: European GMO Initiative for a Unified Database System 2023; Waiblinger et al. 2010).
The presented method facilitates the simple, parallel, sensitive, and robust qualitative detection of up to 3 DNA sequences found in transgenic plants. These sequences include the nos promoter sequence (AgroBorder I) from Agrobacterium tumefaciens (Ti plasmid type nopaline), a sequence (AgroBorder II) from Agrobacterium tumefaciens (Ti plasmid type octopin, often flanking various transgenic plants), and a genetic construct comprising the cassava vein mosaic virus promoter (P-CsVMV) and the phosphinothricin-N-acetyltransferase gene (pat) sequences from Streptomyces viridochromogenes (P-CsVMV-pat). The method has been validated in an international ring trial conducted within the § 64 working group for GMO at the Federal Office of Consumer Protection and Food Safety (BVL). It has also been included in the Official Collection of Test Methods in accordance with the German Food and Feed Act (BVL L 00.00-176:2022-12).
2 Materials and methods
2.1 Plant material and DNA-samples
Certified plant reference materials were obtained from the Joint Research Centre (JRC) of the European Commission or from the American Oil Chemists’ Society (AOCS) as leaf DNA or ground seed material. Materials from 11 different GM cotton events, 4 GM potato events, 24 GM maize events, 10 GM canola events, 1 GM rice event, 18 GM soybean events and 1 GM sugar beet event were used (Table 1). Detailed descriptions of commercial reference materials (catalogue numbers, GM content, status of the materials) are given in a publicly available list (Federal Office of Consumer Protection and Food Safety 2020a).
Further non-GM plant materials for specificity testing were collected from the market.
2.2 DNA extraction
DNA was extracted from ground seed materials with either the DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany) starting with a CTAB extraction (ISO 21571:2005), the NucleoSpin Food Mini Kit (Macherey-Nagel AG, Oensingen, Switzerland) or the Quick-DNA Plant/Seed Miniprep Kit (Zymo Research Europe GmbH, Freiburg, Germany). The concentration of DNA was estimated fluorimetrically using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, Wilmington, DE, USA) according to the manufacturer’s instructions. Copy numbers were calculated on the basis of the genome sizes (Arumuganathan and Earle 1991). Zygosity of GM plant material (CRMs) was applied as stated on the respective analytical certificate. When a more precise specification of copy numbers was necessary (e.g. for determination of LOD), the target copy numbers of the DNA extracts of the GM plant materials were quantified by droplet digital PCR on the QX200 system (Bio-Rad Laboratories, Feldkirchen, Germany) using event specific methods (Bonfini et al. 2012). For further dilution of extracts, 0.2 x TE buffer was used as diluent.
2.3 Oligonucleotides
The primers and probes were developed in this study. In silico testing for complementarity or primer dimerization was performed using primer3 software (Untergasser et al. 2012). Each combination of the underlying primer sequences (Table 2) was tested. To theoretically test the specificity of the triplex detection method, a comparison was performed against genome sequences of 1,760 animal and plant species (Benson et al. 2013). A similar alignment was performed against DNA sequences from authorized and non-authorized GMO from the CCSIS (Central Core DNA Sequence Information System) database of the Joint Research Centre (JRC) of the European Commission (retrieved on 10/03/2020) using the e-PCR tool (Schuler 1997).
2.4 Real-time PCR analysis
DNA extracts (5 µl) were added to 20 µl of reaction mix containing SensiFast™ Probe No-ROX Kit (Bioline, Meridian Bioscience, Cincinnati, USA), primers and probes (for final concentration see Table 2). The DNA concentration used depended on the respective question and is described in more detail in the corresponding sections. Oligonucleotides were purchased from Microsynth AG (Balgach, Switzerland). During the development phase PCR was performed on the Rotor-Gene 6000 (QIAGEN, Hilden, Germany) according to the following cycling protocol: Initial step of 15 min at 95 °C; followed by 45 cycles of 60 s at 94 °C and 90 s at 60 °C. Fluorescence data were collected during the annealing / elongation step at 60 °C. Further reaction master mixes and PCR devices were used as specified during the validation of the method (see 2.5.4).
2.5 Validation of the method
2.5.1 Specificity testing
For experimental testing of the specificity, i.e., the presence of the individual screening target in the material from the GM events, 10 copies of target DNA and at least 2,500 copies of non-target DNA, respectively, were used according to Table 1. Five replicates were tested for confirmation of the presence and duplicates for the confirmation of the absence of target sequences, respectively.
2.5.2 Crosstalk
Any potential bleed-through of fluorescence signals between detection channels during multiplex PCR was tested experimentally (ENGL 2021). Mixtures of test samples containing no target DNA of the respective PCR module were tested in the presence of 2,500 copies of target DNA for the other two PCR modules (n = 3 per test). Triplex PCR was performed using the QuantiTect Multiplex PCR No ROX Kit (QIAGEN) on a LightCycler®480 Instrument II (LC 480 II, Roche Life Science, Mannheim, Germany). For this purpose, DNA from the GM events maize MIR162 (AgroBorder I), soybean MON87769 (AgroBorder II) and soybean DAS44406-6 (P-CsVMV-pat) was used.
2.5.3 Sensitivity
The limit of detection (LOD) was first determined by means of a dilution series of the target DNA according to the guidance document for single laboratory validation (Grohmann et al. 2016). For this purpose, DNA from the GM events maize MIR162 (AgroBorder I), soybean MON87769 (AgroBorder II) and soybean DAS44406-6 (P-CsVMV-pat) was used. Dilution levels of 20, 10, 5, 2, 1 and 0.1 mean copies of each DNA target per PCR were analyzed under symmetric conditions in a background of 100 ng of non-target DNA (UltraPure™ Salmon Sperm DNA, Thermo Fisher Scientific) in 12 replicates each. An additional calibration series was included in the PCR run of 2,500, 500, 100, and 50 copies of each DNA target per PCR in triplicates each. Two different thermocyclers were used: QuantStudio™ 5 (Thermo Fisher Scientific, Wilmington, DE, USA) and Rotor-Gene Q (QIAGEN).
For a first estimation, the lowest dilution level (i.e. the lowest number of copies) for which all 12 replicates are positive can be considered as the LOD95%. In addition, the concentration of the target DNA at which an amplification product is detected with a probability of at least 0.95 (LOD95%) was determined by means of a probability of detection model (POD curve) using a software tool as described in (Grohmann et al. 2016; Uhlig et al. 2015).
The confirmation of the LOD under asymmetric conditions was performed in accordance with the ENGL guidance document for multiplex PCR (European Network of GMO Laboratories 2021). In a single laboratory, 20 copies of each target were analyzed in the presence of 20,000 copies per reaction of each of the other targets (1:1,000). For each combination, 3 PCR replicates were tested. Additionally, sensitivity under both symmetric and asymmetric conditions were tested in an interlaboratory trial with 7 participating laboratories (see Sect. 2.5.4).
Moreover, the possible influence of background DNA present in routine samples was tested at the level of 30 copies per reaction of each target DNA together with 100 ng background DNA extracted from non-modified maize, canola, and soybean, respectively, corresponding to 0.03 to 0.08% cp/cp, in duplicates (Grohmann et al. 2016).
2.5.4 Robustness
Robustness was first tested inhouse under symmetric conditions on the QuantStudio™ 5 system (Thermo Fisher Scientific) using the Bioline SensiFast™ Probe No-ROX Kit. Twenty copies of target DNA in a mixture of DNA from the GM events maize MIR162 (AgroBorder I), soybean MON87769 (AgroBorder II) and soybean DAS44406-6 (P-CsVMV-pat) were analyzed in a background of 100 ng of non-target DNA (UltraPure™ Salmon Sperm DNA, Thermo Fisher Scientific) per PCR in triplicates using a multifactorial experimental design (European Network of GMO Laboratories 2015; Grohmann et al. 2016). The following factors were changed compared to the protocol: Volume of PCR master mix (19 and 21 µl, 5 µl of sample DNA to be added), annealing temperature (59 and 61 °C), concentration of primers and probes (unchanged and − 30%) (Table S1).
Similarly, a robustness test under asymmetric conditions was performed inhouse using the QuantiTect Multiplex PCR No ROX Kit in triplicates with 20 copies of one target DNA in the presence of 20,000 copies per reaction of the other targets (European Network of GMO Laboratories 2021). All reactions were run on a LightCycler®480 Instrument II (Roche Life Science, Mannheim, Germany). The following factors were changed compared to the standard protocol: Volume of PCR master mix (19 and 21 µl, 5 µl of sample DNA to be added), annealing temperature (59 and 61 °C), concentration of primers and probes (-10% and + 10%, tested for each module indepedently) (Table S2).
Prior to the ring trial validation of the method, robustness data were determined by an interlaboratory test with 7 participating laboratories. Dilution levels of 50, 20, 10, 5, 1 and 0.1 mean copies/PCR of each DNA target (MIR162/AgroBorder I, MON87769/AgroBorder II, DAS44406-6/P-CsVMV-pat) under symmetric conditions (n = 12) and additional calibration series of 2,500, 500, 100, and 50 copies/PCR of each DNA target (n = 3) in a background of 100 ng of non-target DNA (UltraPure™ Salmon Sperm DNA, Thermo Fisher Scientific) were analyzed. In addition, a mixture of 10 copies of each target DNA in presence of 2,500 copies of the other 2 target sequences (1:250 asymmetric conditions) were analyzed using the following different combinations of PCR master mixes and real-time PCR cyclers: SensiFast Probe No-ROX Kit on Rotor-Gene 6000 (QIAGEN), Rotor-Gene Q and QuantStudio 5; QuantiTect Multiplex Kit on QuantStudio 5 and LC 480 II; PCRrely Mplex Mix (Gold Standard Diagnostics) on CFX96™ (Bio-Rad), and PerfeCTa® MultiPlex qPCR ToughMix® (Quantabio, MA, USA) on CFX96 ™ and Mx3005P™ (Agilent, CA, USA).
3 Results and discussion
3.1 Specificity testing
The specificity of the multiplex PCR method was initially assessed in silico to anticipate potential artifacts and false positive results (European Network of GMO Laboratories 2021). Using Primer3, no increased probability of self-complementarity or primer dimerization was observed. Additionally, querying the Genbank and CCSIS databases did not reveal any unexpected similarities to other target sequences. Combining the primers in the triplex method did not predict the presence of any additional unintended amplicons.
Experimental testing of specificity involved analyzing DNA extracts from available reference materials of GM events. Table 1 provides a list of these events along with their qualitative signals obtained using the triplex method. All results aligned with the expectations based on the available theoretical sequence information. Notably, when DNA extracted from cotton event MON88701 was measured with 2,500 copies per PCR, a weak amplification with a Cq value exceeding approximately 35 was observed in the AgroBorder I - PCR. This result deviates from the theoretical absence of this sequence in this event and may be attributed to traces or contamination of material containing the AgroBorder I sequence in the certified reference material (AOCS 0113-A) used for DNA extraction. In a few events, one mismatch between a primer and the target sequence was theoretically predicted; however, PCR amplification was successful in all cases except for Topas19/2, which did not exhibit unambiguous detection below 25 copies per PCR (Table 1).
DNA extracted from non-GM materials, such as maize, soybean, canola, cotton, potato, sugar beet, and rice, did not yield any positive results.
3.2 Crosstalk
No significant crosstalk was observed in any of the 3 detection channels of the LightCycler®480 Instrument II (Fig. S1). As a result, the acceptance criteria outlined in the ENGL guidelines (European Network of GMO Laboratories 2021) pertaining to crosstalk were met. Moreover, there were no indications of crosstalk on other PCR devices.
3.3 Sensitivity
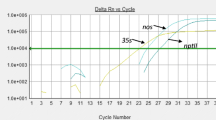
Amplification plots for the standard curves can be found in Fig. 1. The results of the in-house validation of the method’s sensitivity are summarized in both Table 3 and Table S3. Performance criteria for standard curves are delineated in several guidelines for GMO analysis (European Network of GMO Laboratories 2015; 2021; Grohmann et al. 2016). According to these guidelines, the Limit of Detection at 95% confidence (LOD95%) for a qualitative real-time PCR method should not exceed 20 to 25 copies per PCR.

PCR amplification of the standard curves. Sample mixtures containing DNA from maize MIR162 (containing AgroBorder I), soybean MON87769 (containing AgroBorder II) and soybean DAS44406-6 (containing P-CsVMV-pat) were measured on a QuantStudio 5 in triplicates under symmetric conditions with copy numbers as indicated. (a) AgroBorder I channel; (b) AgroBorder II channel; (c) P-CsVMV-pat channel. NTC, non-template control
Furthermore, the slope of the standard curve, which is obligatory only for quantitative methods, should fall within the range of -3.1 to -3.6, with a coefficient of determination (R2) equal to or greater than 0.98. The triplex method successfully met all of these requirements. The LOD95% was approximately 7 copies/PCR for AgroBorder I, 8 copies/PCR for AgroBorder II, and 9 copies/PCR for P-CsVMV-pat, respectively (Table 3). These results were also confirmed with similar outcomes on another PCR device (Table S3). The coefficients of determination (R2) and the slopes of the calibration curves underscore the method’s quantitative potential.
Additionally, in accordance with the guidelines for GMO analysis (European Network of GMO Laboratories 2015; 2021; Grohmann et al. 2016), the sensitivity of a multiplex method should be tested individually for each target in the presence of high amounts of the other targets to assess potential competitive effects.
To confirm the method’s sensitivity under asymmetric conditions, we conducted an analysis involving 20 copies of each target in the presence of 20,000 copies per reaction of the respective other targets (Fig. S2). Furthermore, sufficient sensitivity under asymmetric conditions was validated in an interlaboratory trial (as discussed in sect. 3.4). Moreover, the presence of background DNA from maize, soybean, or canola (100 ng DNA per PCR), as commonly encountered in routine samples of official control, did not affect the sensitivity of the method (data not shown).
3.4 Robustness
According to guidelines (European Network of GMO Laboratories 2015, 2021; Grohmann et al. 2016), robustness should be assessed to simulate the practical implementation of the method, ensuring that sensitivity remains sufficient even in the presence of minor deviations from the experimental conditions.
First, in-house robustness testing was conducted under symmetric conditions by analyzing 20 copies of each target DNA per reaction using a multifactorial experimental design (Grohmann et al. 2016) with the SensiFast Probe No-ROX Kit on a QuantStudio 5. Various factors, including changes in PCR master mix volume, annealing temperature, and the concentration of primers and probes, were investigated. It was found that these variations did not compromise the detectability of the targets (as shown in Table S4).
Furthermore, we assessed robustness in-house under asymmetric conditions by analyzing 20 copies of each target DNA in the presence of 20,000 copies of the other targets per reaction (at a 1:1,000 ratio) using a multifactorial experimental design (Grohmann et al. 2016) with the QIAGEN QuantiTect Multiplex Kit on a LightCycler 480 Instrument II. In all combinations of experimental factors tested, all PCR replicates consistently produced positive detection signals for the 3 targets (Table S5), demonstrating the method’s satisfactory robustness.
Robustness was also verified through an interlaboratory ring trial involving 7 different laboratories. Mixtures containing 10 copies of each target DNA in the presence of 2,500 copies of the other 2 target sequences (asymmetric conditions at a 1:250 ratio) were unambiguously detected by all participating laboratories, despite the use of various combinations of PCR master mixes and real-time PCR devices (Table 4). This trial reaffirmed the high sensitivity of the triplex method, with an average Limit of Detection at 95% confidence (LOD95%) of approximately 5 copies/PCR for AgroBorder I, 3 copies/PCR for AgroBorder II, and 5 copies/PCR for P-CsVMV-pat, respectively. Once again, the mean standard curve performance parameters were consistent with the guidelines for GMO analysis (European Network of GMO Laboratories 2015, 2021; Grohmann et al. 2016).
4 Conclusions
In this study, we present the development and validation of a novel triplex real-time PCR method designed for the efficient screening of food, feed, and seed products for GM traits encoding transgene flanking sequences known as ‘AgroBorder’ and the Cassava Vein Mosaic Virus Promoter-pat construct. Our results confirm the theoretically expected presence and absence of the 3 target sequences in 37 GM plants, demonstrating the method’s adequate specificity. Additionally, we have demonstrated sufficient sensitivity and robustness, even under challenging asymmetric conditions. Importantly, the multiplex amplification did not exhibit any interference between the individual PCR reactions. This method is already being successfully used for GMO screening in the routine analysis of numerous food and feed samples, as well as in proficiency tests conducted by various laboratories. The screening results obtained using this method have been consistently confirmed by subsequent identification of the respective GM plant events (data not shown).
To further evaluate the performance of this method, an international ring trial validation study was conducted within the § 64 working group of the BVL, confirming the method’s high sensitivity, specificity, robustness, and practicability (as per BVL L 00.00-176:2022-12).
The triplex real-time PCR method described in this study serves as an efficient screening approach, reducing both analysis time and costs. It is fully suitable for official GMO control of food, feed, and seed products. Furthermore, the method will be incorporated into the BVL GMO screening matrix (Federal Office of Consumer Protection and Food Safety 2020b) and the EUginius database (EUGINIUS: European GMO Initiative for a Unified Database System (2023). Information regarding the presence or absence of the 3 target sequences in upcoming new GM plant events will be regularly updated in these databases.
References
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9:208–218. https://doi.org/10.1007/BF02672069
Bahrdt C, Krech AB, Wurz A, Wulff D (2010) Validation of a newly developed hexaplex real-time PCR assay for screening for presence of GMOs in food, feed and seed. Anal Bioanal Chem 396:2103–2112. https://doi.org/10.1007/s00216-009-3380-x
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2013) GenBank Nucleic Acids Res 41:D36–42. https://doi.org/10.1093/nar/gks1195
Bonfini L, van den Bulcke MH, Mazzara M, Ben E, Patak A (2012) GMOMETHODS: the European Union database of reference methods for GMO analysis. J AOAC Int 95:1713–1719. https://doi.org/10.5740/jaoacint.12-050
BVL L 00.00-124:2008-12 (2008) Untersuchung von Lebensmitteln - Nachweis einer bestimmten, häufig in gentechnisch veränderten Organismen (GVO) verwendeten DNA-Sequenz aus dem bar-Gen von Streptomyces hygroscopicus in Lebensmitteln - Screening-Verfahren. Amtliche Sammlung von Untersuchungsverfahren (Official Collection of Methods of Analysis)
BVL L 00.00-125:2008-12 (2008) Untersuchung Von Lebensmitteln - Nachweis Der CTP2-CP4-EPSPS-Gensequenz zum screening auf Bestandteile Aus Gentechnisch veränderten Organismen (GVO) in Lebensmitteln - Konstrukt-spezifisches Verfahren. Amtliche Sammlung von Untersuchungsverfahren (Official Collection of Methods of Analysis
BVL L 00.00-148:2014-02 (2014) Untersuchung von Lebensmitteln - Nachweis einer DNA-Sequenz des FMV-Promotors (pFMV) in Lebensmitteln mittels real-time PCR - Element-spezifisches Verfahren. Amtliche Sammlung von Untersuchungsverfahren (Official Collection of Methods of Analysis)
BVL L 00.00-176:2022-12 (2022) Untersuchung von Lebensmitteln - Nachweis von flankierenden Ti-Plasmid-(AgroBorder)-Sequenzen sowie einer Sequenz des Cassava Vein Mosaic Virus Promotor-pat-Konstruktes (P-CsVMV-pat) zum Screening auf Bestandteile aus gentechnisch veränderten Organismen (GVO) in Lebensmitteln mittels Multiplex real-time PCR (Konstrukt- und Element-spezifisches Verfahren). Amtliche Sammlung von Untersuchungsverfahren (Official Collection of Methods of Analysis)
EUGINIUS: European GMO Initiative for a Unified Database System (2023) https://www.euginius.eu/euginius/pages/home.jsf. Accessed 25 August 2023
European Network of GMO Laboratories (2011) Overview on the detection, interpretation and reporting on the presence of unauthorised genetically modified materials materials. JRC 67297. Publications Office of the European Union
European Network of GMO Laboratories (2015) Definition of Minimum Performance requirements for Analytical methods of GMO Testing. JRC95544. Publications Office of the European Union
European Network of GMO Laboratories (2021) Guidance document on multiplex real-time PCR methods. JRC125188. Publications Office of the European Union
Federal Office of Consumer Protection and Food Safety (2020b) Sceening table for the detection of genetically modified plants. https://www.bvl.bund.de/SharedDocs/Downloads/07_Untersuchungen/screening_tabelle_gvoNachweis.html. Accessed 25 August 2023
Federal Office of Consumer Protection and Food Safety (2020a) Publicly Available Reference Materials for GMO Detection. https://www.bvl.bund.de/SharedDocs/Downloads/06_Gentechnik/nachweis_kontrollen/reference_materials.pdf?__blob=publicationFile&v=4. Accessed 25 August 2023
Grohmann L, Brünen-Nieweler C, Nemeth A, Waiblinger H-U (2009) Collaborative trial validation studies of real-time PCR-based GMO screening methods for detection of the bar gene and the ctp2-cp4epsps construct. J Agric Food Chem 57:8913–8920. https://doi.org/10.1021/jf901598r
Grohmann L, Reiting R, Mäde D, Uhlig S, Simon K, Frost K, Randhawa GJ, Zur K (2015) Collaborative trial validation of cry1Ab/Ac and pubi-cry TaqMan-based real-time PCR assays for detection of DNA derived from genetically modified Bt plant products. Accred Qual Assur 20:85–96. https://doi.org/10.1007/s00769-015-1108-5
Grohmann L, Broll H, Dagand H, Hildebrandt S, Hübert P, Kiesecker H, Lieske K, Mäde D, Mankertz J, Reiting R, Schulze M, Speck B, Uhlig S, Wahler D, Waiblinger H-U, Woll K, Zur K (2016) Guidelines for the single-laboratory validation of qualitative real-time PCR methods. https://www.bvl.bund.de/SharedDocs/Downloads/07_Untersuchungen/Guidelines%20for%20the%20single%20laboratory.html. Accessed 2023
Holst-Jensen A, Rønning SB, Løvseth A, Berdal KG (2003) PCR technology for screening and quantification of genetically modified organisms (GMOs). Anal Bioanal Chem 375:985–993. https://doi.org/10.1007/s00216-003-1767-7
ISAAA’s GM Approval Database. https://www.isaaa.org/gmapprovaldatabase/. Accessed 2023
ISO 21569: 2005 foodstuffs - methods of analysis for the detection of genetically modified organisms and derived products - qualitative nucleic acid based methods. International Organization of Standardization (ISO)
ISO 21571 (2005) Foodstuffs - methods of analysis for the detection of genetically modified organisms and derived products - nucleic acid extraction. International Organization of Standardization (ISO)
Schuler GD (1997) Sequence mapping by electronic PCR. Genome Res 7:541–550. https://doi.org/10.1101/gr.7.5.541
Uhlig S, Frost K, Colson B, Simon K, Mäde D, Reiting R, Gowik P, Grohmann L (2015) Validation of qualitative PCR methods on the basis of mathematical–statistical modelling of the probability of detection. Accred Qual Assur 20:75–83. https://doi.org/10.1007/s00769-015-1112-9
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3–new capabilities and interfaces. Nucleic Acids Res 40:e115. https://doi.org/10.1093/nar/gks596
Waiblinger H-U, Ernst B, Anderson A, Pietsch K (2008) Validation and collaborative study of a P35S and T-nos duplex real-time PCR screening method to detect genetically modified organisms in food products. Eur Food Res Technol 226:1221–1228. https://doi.org/10.1007/s00217-007-0748-z
Waiblinger H-U, Grohmann L, Mankertz J, Engelbert D, Pietsch K (2010) A practical approach to screen for authorised and unauthorised genetically modified plants. Anal Bioanal Chem 396:2065–2072. https://doi.org/10.1007/s00216-009-3173-2
Acknowledgements
The ring trial was organized by § 64 LFGB GMO working group, hosted at the Federal Office for Consumer Protection and Food Safety (BVL). We warmly thank all the participating laboratories to provide the resources for this work: Dominik Moor, Ingo Krujatz and Martin Weigel. Furthermore, we wish to thank Jennifer Kaiser, Miriam Schillinger and Marcel Schulze for their excellent technical assistance. We are grateful to Alexandre Angers and his colleagues (Joint Research Centre, European Commission) for the in-silico specificity testing. Furthermore, we would like to thank Jakob Frenzel for his support during the organization of ring trials and panel meetings of the § 64 LFGB GMO working group and for carefully reading the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weidner, C., Köppel, R., Freyer, R. et al. Development and validation of a multiplex real-time PCR method for screening genetically modified plants. J Consum Prot Food Saf 19, 165–174 (2024). https://doi.org/10.1007/s00003-024-01499-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00003-024-01499-4