
Abstract
In plants, a particular class of small non-coding RNAs, short interfering RNAs, can serve as a signal to induce cytosine methylation at homologous genomic DNA regions in the nucleus. If the targeted DNA regions have promoter function, this RNA-directed DNA methylation (RdDM) can result in transcriptional gene silencing (TGS). RNA-directed transcriptional gene silencing of transgenes provides a versatile system for the study of epigenetic gene regulation in plants. In our experimental setup in Arabidopsis thaliana, transcription of a promoter-inverted repeat provides a RNA signal that triggers de novo cytosine methylation and TGS of a homologous nopaline synthase promoter (proNOS) in trans. Utilising this two component transgene system in a forward-genetic screen for “suppressor of silencing” mutations, we were able to identify new candidates for factors involved in RdDM of transgenic as well as endogenous target regions.
Similar content being viewed by others

1 Introduction
DNA methylation in general and the mechanism of RNA-directed DNA methylation (RdDM) as well as related transcriptional gene silencing (TGS) in particular have been extensively studied in the model plant Arabidopsis thaliana (Matzke et al. 2009; Law and Jacobsen 2010). As in other plants, DNA methylation in A. thaliana can occur in symmetric CG and CNG as well as in asymmetric CHH (where H is A, C or T) context. In total, about 7 % of cytosines are methylated but methylation is not distributed randomly. 24 % of cytosines in CG, but only 6.7 % of cytosines in CNG and 1.7 % of cytosines in CHH context are methylated (Cokus et al. 2008).
In each round of the cell cycle, cytosine methylation patterns need to be re-established in genomic DNA, as only unmethylated cytosines are incorporated in DNA replication and thus no methylation is present on the de novo synthesized DNA strand. Cytosine methylation in CG context is maintained by METHYLTRANSFERASE 1 (MET1), a plant member of the Dnmt1 class of methyltransferases, while methylation in CNG context is maintained by the CHROMOMETHYLASE3 (CMT3). Cytosine methylation in CHH context persists by constitutive de novo DNA methylation involving the DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2), a plant member of the Dnmt3 class of methyltransferases.
The target specificity of de novo DNA methylation by DRM2 is achieved by a class of small non-coding RNAs, the short interfering (si)RNAs. The generation of these 24 nucleotides (nt) long RNAs is initiated by a plant-specific DNA-dependent RNA-polymerase, Pol IV, that transcribes regions undergoing RdDM. Two Pol IV subunits, NUCLEAR RNA POLYMERASE D1 and D2 (NRPD1 and NRPD2) were identified as essential for RdDM of endogenous target sequences (Huang et al. 2009; He et al. 2009; Lahmy et al. 2009; Bies-Etheve et al. 2009). The single stranded “aberrant” transcripts made by Pol IV are thought to be recognized by RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) and used as substrate to generate double stranded (ds)RNA. This dsRNA is cleaved by DICER-LIKE 3 (DCL3) into 24 nt fragments. One strand of the DCL3 cleavage products is then bound by ARGONAUT 4 (AGO4) and transferred to another plant-specific DNA-dependent RNA-polymerase, Pol V (Henderson and Jacobsen 2007; Matzke et al. 2009). Here, AGO4 interacts with the subunit NUCLEAR RNA POLYMERASE E1 (NRPE1). By interacting either with a Pol V-derived transcript or with the DNA itself, the siRNA bound by AGO4 then provides the sequence specificity to the de novo methylation of DNA by DRM2.
Beside NRPE1, the Pol V complex includes NUCLEAR RNA POLYMERASE E2 (NRPE2), the elongation factor SUPPRESSOR OF TY INSERTION 5-LIKE (SPT5L), the chromatin remodelling factor DEFECTIVE IN RNA-DIRECTED DNA METHYLATION 1 (DRD1) and the factor DEFECTIVE IN MERISTEM SILENCING 3 (DMS3). These factors are supposed to support the recruitment of the Pol V complex to the site of RdDM. The formation or recruitment of the complex is further aided by SET domain proteins SUPPRESSOR OF VARIEGATION 3–9 HOMOLOGUE SUVH2 and SUVH9.
To find new relevant factors and increase the insight in the mechanism of RdDM and related TGS, we made use of a A. thaliana line containing two transgenes, a target transgene (K) and an unlinked silencer transgene (H) (Fig. 1, Aufsatz et al. 2002). The employed target transgene K chr1–10 contains an intact single copy T-DNA insertion including a NEOMYCIN PHOSPHOTRANSFERASE II (NPTII) gene conferring resistance to kanamycin, followed by an octopine synthase polyadenylation signal (ocs 3′), under the control of a nopaline synthase promoter (proNOS) and a GUS gene followed by an ocs 3′ under the control of a cauliflower mosaic virus 35S promoter (pro35S) (Fischer et al. 2008). The silencer transgene (H) consists of two copies of the proNOS oriented as inverted repeat (IR) under the control of a constitutive pro35S promoter and a hygromycin resistance gene controlled by pro19S. Transcription of this silencer proNOS IR leads to formation of proNOS dsRNA that is processed to siRNAs of predominantly 21–24 nucleotides in length (Papp et al. 2003). Just as endogenous 24 nt siRNAs, these transgene-derived siRNAs guide de novo DNA methylation to the proNOS copies and lead to transcriptional silencing.

Transgene system for the analysis of RNA dependent DNA methylation and related transcriptional gene silencing in Arabidopsis thaliana. H silencer transgene contains two copies of the nopaline synthase promotor (proNOS) in inverted repeat (IR) orientation under the control of a cauliflower mosaic virus 35S promoter (pro35S). Transcription of the IR generates self-complementary RNA that folds back to form double stranded RNA (dsRNA). The dsRNA is cleaved to 21–24 nt long small interfering (si)RNAs, of which the 24 nt species serves as signal for the methylation (black dots) of proNOS in trans. K target transgene, contains a NEOMYCIN PHOSPHOTRANSFERASE II (NPTII) gene under the control of a proNOS. NPTII expression confers kanamycin resistance; methylation of the proNOS (black dots) will result in transcriptional gene silencing and sensitivity to kanamycin
2 Material and methods
2.1 Plant material and growth conditions
Target and silencer transgenes were introduced separately into A. thaliana accession Columbia (Col-0) by Agrobacterium tumefaciens mediated transformation and were combined by crossing plants homozygous for target or silencer, respectively (Fischer et al. 2008).
About 105 seeds from plants homozygous for target and silencer transgene were submitted for custom ethyl methanesulfonate (EMS) mutagenesis to Lehle Seeds (Round Rock, TX, USA). The seeds were divided into 32 batches (~3 × 103 seeds per batch), treated with EMS, and germinated on soil. For each batch, the resulting M1 plants (~1.5 × 103 assuming a survival rate of 50 %) were grown and allowed to selfpollinate to generate M2 seeds, which were harvested in bulk. About 2 × 104 M2 seeds per batch (thus, ~13 M2 seeds per M1 plant) were germinated on ½ Murashige and Skoog medium (Murashige and Skoog 1962) containing 200 mg/l kanamycin under long day conditions (16 h light, 8 h dark, 21 °C) to screen for reactivation of kanamycin resistance. After 3 weeks, the most vigorous plants (10 per batch) were transferred to soil and allowed to selfpollinate. Release of silencing of the NPTII gene was confirmed by testing the M3 generation for viability on kanamycin containing medium. Lines with a survival rate of at least 95 % among M3 seeds were considered as true candidates for “suppressor of silencing” mutants.
Plants for expression and DNA methylation analysis were grown on soil for ~6 weeks under short day conditions (8 h light, 16 h dark, 21 °C).
2.2 DNA isolation
Genomic DNA was extracted from leaves using DNeasy® Plant Mini and Maxi Kit (Qiagen) according to the manufacturer’s protocol.
2.3 Confirmation of the presence of target and silencer transgene by PCR
PCR indicative for the presence of transgenes was performed with the primers indicated in Table 1. Primer pair pNOS-for and pNOS-rev was used to demonstrate the presence of the target transgene, primer pairs p19s-for and Hyg-rev to demonstrate the presence of the silencer transgene. The heterozygous (signal present) versus homozygous (signal absent) state of the silencer transgene was tested by PCR with flanking primers HinS-for and HinS-rev.
2.4 NPTII ELISA
NPTII protein levels in leaves were determined using a PathoScreen® kit for neomycin phosphotransferase II from Agdia according to the manufacturer’s protocol. The data read out was performed on an infinite200 plate reader (Tecan) with supplied software (Tecan i-control).
2.5 DNA methylation analysis
Quantitative (q) PCR was performed after cleavage of genomic DNA with methylation-sensitive restriction enzymes. About 0.1 μg of genomic DNA was diluted in 500 μl H2O. Aliquots of 90 μl of this stock were diluted to a total volume of 100 μl of the appropriate reaction buffer and incubated with 30 units of restriction enzyme (Fermentas) at 37 °C over night. After incubation, restriction enzymes were inactivated by incubation at 65 °C for 5 min and the reaction mixture diluted 1:10 with H2O. 10 μl of the dilution were added to 12.5 μl SyBr green Supermix® (Bio-Rad cat. no. 170-8882) and 1.25 μl of each primer solution (of 10 pmol/μl) according manufacturer’s protocol to perform qPCR in a final volume of 25 μl on an iQ5 cycler (Bio-Rad). The primers used for amplification of the proNOS-fragment (460 bp) were pNOSfor (5′-GATAGTTGGCGAAATTTTCAAAGT-3′) and pNOSrev (5′-CAATCCATCTTGTTCAACCATGG-3′). The PCR program included an initial step of 5 min 95 °C followed by 40 cycles of 15 s 95 °C, 30 s 62 °C and 30 s 72 °C (detection during elongation). Calibration using a serial dilution of genomic DNA resulted in a correlation coefficient of 0.999 and a PCR efficiency of 99.7 % for the quantification.
Bisulfite mediated chemical conversion of DNA was done using an EpiTect Bisulfite Kit (Qiagen) following the manufacturer’s instructions. Primers used for the amplification of a particular AtSN1 copy from bisulfite converted genomic A. thaliana DNA were ATS150 (5′-ACCAACGTGCTGTTGGCCCAGTGGTAAATC-3′) and AtSN1-F4 (5′-AAAATAAGTGGTGGTTGTACAAGC-3′). The PCR program included an initial step of 5 min 95 °C followed by 40 cycles of 15 s 95 °C, 30 s 60 °C and 30 s 72 °C. PCR amplification products were cloned with a Strataclone™ PCR Cloning kit (Stratagene) and the resulting plasmid clones were sequenced using a Thermo Sequenase Cycle Sequencing kit (USB) and a LI-COR 4300 DNA analyser.
Southern analysis in combination with methylation-sensitive restriction enzymes to determine DNA methylation in the proNOS was performed as described by Fischer et al. (2008).
3 Results
3.1 Identification of “suppressor of silencing” mutants releasing RNA-directed transcriptional gene silencing
The presence of the silencer transgene H reduces the level of NPTII transcripts from target transgene K chr1–10 ~100-fold (Fig. 2a, Fischer et al. 2008). Consistent with this, plants are resistant to kanamycin if they contain the target transgene K chr1–10 , but become sensitive if they contain, additionally, the silencer transgene H (Fig. 2b). Analysis of the DNA methylation by Southern blot analysis with methylation-sensitive restriction enzymes (Fig. 2c) and bisulfite sequencing (Fig. 2d) revealed that ~80 % of the cytosines in the proNOS of the target transgene were methylated. The methylation was evenly distributed among cytosines independent of their sequence context, which is a hallmark of RdDM.

Transcriptional gene silencing and RNA dependent DNA methylation of the target proNOS-NPTII reporter gene. a Remaining NPTII mRNA in presence of the silencer (light grey) relative to NPTII mRNA in absence of the silencer (dark grey) was detected by real time RT-PCR and is given in percent on a logarithmic scale. RNA was prepared from leaf tissue of individual hemizygous F1 (N = 5) and homozygous F3 (N = 5) plants (Fischer et al. 2008). Error bars show the total deviation. b Homozygous seedlings carrying the proNOS-NPTII target (K) in the absence (left) and the presence (right) of the silencer (H) on medium containing kanamycin. c Methylation analysis of the target proNOS in homozygous F3 plants in the absence (left) and the presence (right) of the silencer by Southern with methylation sensitive restriction enzymes. A basic fragment containing the proNOS was released by Bsu15I and PaeI cleavage (−). Methylation was tested by additional incubation with methylation-sensitive restriction enzymes EcoR47I (E, GGWCC), Psp1406I (P, AACGTT), NheI (Nh, GCTAGC), Alw26I (A, GTCTC, GAGAC) and NcoI (Nc, CCATGG). The underlined enzymes are recognition sites in the region covered by proNOS siRNAs derived from the silencer. d Methylation analysis of the target proNOS in homozygous F3 plants in the absence (left) and the presence (right) of the silencer by bisulfite sequencing analysis in percent of cytosines in total (white), CG context (light grey), CNG context (dark grey) and CHH context (black) (K/K: N = 11, K/K; H/H: N = 19)
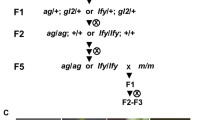
Ethyl methanesulfonate mutagenesis of seeds homozygous for target and silencer transgenes (K chr1–10/K chr1–10; H/H) in the Col-0 accession was performed by Lehle Seeds (Round Rock, TX, USA). From the obtained 32 batches of M2 seeds, (each batch representing the progeny from 1,500 M1 plants), 20,000 seeds per batch were germinated on medium containing kanamycin (200 mg/l) (Fig. 3). Resistant M2 plants were transferred to soil and allowed to set seeds by selfing. The suppression of TGS was confirmed by germinating resulting M3 seeds on kanamycin containing selective medium. True candidate lines for “suppressor of silencing” mutants showed root growth and development of primary leaves in all progeny. The lines passing these criteria were then tested by PCR for the presence of intact target and silencer transgenes (data not shown). The first round of screening resulted in six independent M3 lines from different seed batches which contained target and silencer transgene in the homozygous state and uniformly showed resistance to kanamycin, thus meeting our criteria for “suppressor of silencing” mutants.

Genetic screen for factors involved in RNA-directed transcriptional gene silencing. K: target transgene; H: silencer transgene; f/f: functional/non-functional allel of factor involved in RdTGS; M1: plants grown from seeds that were treated with EMS; M2, M3: generations obtained by selfing from M1; kanamycin-selection is performed on M2 seeds, derived M3 seeds are crossed to Landsberg erecta (Ler) to generate mapping populations
3.2 Target transgene expression and RNA-directed DNA methylation status in “suppressor of silencing” mutants
In order to test whether the kanamycin resistance in the mutant lines was due to reactivation of NPTII expression, NPTII protein levels in leaves were determined by a NPTII-specific ELISA (Fig. 4a). Four of the six analysed mutant lines (mut2-5, mut8-6, mut26-5 and mut29-8) showed a significant increase in NPTII protein compared to non-mutagenised plants containing the target transgene in the presence of the silencer, while two of the plant lines (mut19-7 and mut30-2) showed no detectable NPTII protein expression.

Characterisation of candidate “suppressor of silencing” mutant lines. a NPTII protein levels in ng per 100 mg fresh weight determined by ELISA: Col-0 (non-transformed); target unsilenced (homozygous for target and an inactivated silencer variant); target silenced (homozygous for target and silencer); M3 mutant plants from lines mut2-5; mut8-6; mut19-7; mut26-5; mut29-8; mut30-2; measurements were done for two independent plants. b Target proNOS methylation determined by quantitative PCR after cleavage of genomic DNA with methylation-sensitive restriction enzymes NheI (black asymmetric context: GCTAGC); Psp1406I (grey symmetric context: AACGTT); Alw26I (black lines asymmetric context: GTCTC, GAGAC) and NcoI (white control: CCATGG): target unsilenced (homozygous for target); target silenced (homozygous for target and silencer); M3 mutant plants from lines mut2-5; mut8-6; mut19-7; mut26-5; mut29-8; mut30-2; measurements were done for two independent plants, except for mut30-2. c AtSN1 DNA methylation region determined by bisulfite sequencing in percent of cytosines in total (white), CG context (light grey), CNG context (dark grey) and CHH context (black): Col-0 (non-mutagenised, N = 12); M3 mutant plants from lines mut2-5 (N = 12); mut8-6 (N = 10); mut19-7 (N = 12); mut26-5 (N = 22); mut29-8 (N = 12); mut30-2 (N = 9) (N number of sequences analysed)
To see whether RdDM was affected in the mutant lines, DNA methylation of the proNOS in the target transgene construct was tested by a DNA methylation assay based on cytosine methylation-sensitive restriction enzymes and quantitative PCR (Fig. 4b). In agreement to prior analysis (Fischer et al. 2008), in all plants analysed no methylation was detected at the NcoI site outside of the region targeted by proNOS siRNAs derived from the silencer. In all plants that contained the silencer transgene, substantial symmetric DNA methylation was detected (Psp1406I site). In the four mutant lines with elevated NPTII protein levels (mut2-5, mut8-6, mut26-5 and mut29-8), we detected a reduction of asymmetric DNA methylation (NheI and Alw26I sites), while the amount of symmetric DNA methylation (Psp1406I) was comparable to non-mutagenised control plants containing target and silencer transgenes. The two mutant lines without detectable NPTII protein (mut19-7 and mut30-2) showed mostly unaltered DNA methylation.
To test for general hypomethylation in the identified mutant lines, we analysed the level of DNA methylation at AtSN1, an endogenous target of RdDM, by bisulfite sequencing (Fig. 4c). This confirmed the reduced level of DNA methylation at cytosines in asymmetric CHH context in lines mut2-5, mut8-6, mut26-5 and mut29-8. The AtSN1 methylation in CHH context in mutant line mut19-7 was partially reduced, while no change was observed for mutant line mut30-2.
4 Discussion
Using a forward genetic approach based on a two component transgene system, we were able to identify six independent mutant lines displaying kanamycin resistance. In at least four (mut2-5, mut8-6, mut26-5 and mut29-8) of the six mutant lines, kanamycin resistance of the plants is due to a release of silencing of the NPTII reporter gene in the target transgene. The reactivation of NPTII expression was correlated with DNA hypomethylation at the reporter gene promoter. Methylation loss was most prominent for cytosines in an asymmetric context, while methylation in CG context was hardly affected. In the same four mutant lines, bisulfite sequencing of the AtSN1 locus revealed a hypomethylation for cytosines in asymmetric context also for this endogenous target of RdDM. Thus, the mutations in these four lines most likely generate “loss of function” alleles of factors essential to RdDM. An interesting aspect is that these mutants show partial reactivation of reporter gene expression. Thus, although symmetric-context DNA methylation is persistent at the proNOS, it is solely not sufficient to grant full silencing. The inactivation of reporter gene expression seems to be an additive effect of different types of DNA methylation.
The line mut19-7 shows only a very weak reduction of reporter gene promoter methylation, but nevertheless a reduction by half of asymmetric context cytosine methylation in AtSN1. The mutation in mut19-7 might generate a “weak” allele of a factor with still some remaining gene function. Depending on the particular RdDM target, this remaining activity might be sufficient or not to sustain normal de novo methylation levels. Mutant line mut30-2 does neither show reactivation of reporter gene expression nor reduction of RdDM. It might contain a mutation conferring kanamycin resistance independent of transgene-based NPTII expression, e.g. by affecting a gene required for kanamycin uptake, possibly similar to a chloroplast transporter that has been described by Aufsatz et al. (2009).
The map-based cloning of mutations will be focused on the ones clearly affecting RdDM and will be performed with the help of CAPS and InDel markers in classical Col-0 X Ler mapping populations (Fig. 3). Our approach has a high potential to lead us to new components and further insights into the complex mechanism of DNA methylation mediated by small non-coding RNAs.
References
Aufsatz W, Mette MF, van der Winden J, Matzke AJ, Matzke M (2002) RNA-directed DMA methylation in Arabidopsis. Proc Natl Acad Sci USA 99(Suppl l4):16499–16506
Aufsatz W, Nehlin L, Voronin V, Schmidt A, Matzke AJ, Matzke M (2009) A novel strategy for obtaining kanamycin resistance in Arabidopsis thaliana by silencing an endogenous gene encoding a putative chloroplast transporter. Biotechnol J 4:224–229
Bies-Etheve N, Pontier D, Lahmy S, Picart C, Vega D, Cooke R, Lagrange T (2009) RNA-directed DNA methylation requires an AGO4-interacting member of the SPT5 elongation factor family. EMBO Rep 10:649–654
Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE (2008) Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452:215–219
Fischer U, Kuhlmann M, Pecinka A, Schmidt R, Mette MF (2008) Local DNA features affect RNA-directed transcriptional gene silencing and DNA methylation. Plant J 53:1–10
He XJ, Hsu YF, Pontes O, Zhu J, Lu J, Bressan RA, Pikaard C, Wang CS, Zhu JK (2009) NRPD4, a protein related to the RPB4 subunit of RNA polymerase IV and V is required for RNA-directed DNA methylation. Genes Dev 23:318–330
Henderson IR, Jacobsen SE (2007) Epigenetic inheritance in plants. Nature 447:418–424
Huang L, Jones AM, Searle I, Patel K, Vogler H, Hubner NC, Baulcombe DC (2009) An atypical RNA polymerase involved in RNA silencing shares small subunits with RNA polymerase II. Nat Struct Mol Biol 16:91–93
Lahmy S, Pontier D, Cavel E, Vega D, El-Shami M, Kanno T, Lagrange T (2009) PolV (PolIVb) function in RNA-directed DNA methylation requires the conserved active site and an additional plant-specific subunit. Proc Natl Acad Sci USA 106:941–946
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220
Matzke M, Kanno T, Daxinger L, Huettel B, Matzke AJ (2009) RNA-mediated chromatin-based silencing in plants. Curr Opin Cell Biol 21:367–376
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Papp I, Mette MF, Aufsatz W, Daxinger L, Schauer SE, Ray A, van der Winden J, Matzke M, Matzke AJ (2003) Evidence for nuclear processing of plant micro RNA and short interfering RNA precursors. Plant Physiol 132:1382–1390
Acknowledgments
We thank Christa Fricke, Inge Glaser and Beate Kamm for excellent technical assistance. This work received support from IPK Gatersleben (to A.F.) and the German Research Foundation (DFG) collaborative research centre (SFB) 648 “Molecular Mechanisms of Information Processing in Plants” (to M.K.).
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article represents a lecture given by Doctor Markus Kuhlmann at the 6th Genetic Workshop on Small Non-coding RNAs organised by the Federal Office of Consumer Protection and Food Safety in co-operation with the Berliner Wissenschaftliche Gesellschaft and the Free University, 24 and 25 November 2010 in Berlin, Germany.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Finke, A., Mette, M.F. & Kuhlmann, M. Genetic analysis of RNA-mediated gene silencing in Arabidopsis thaliana . J. Verbr. Lebensm. 7, 27–33 (2012). https://doi.org/10.1007/s00003-011-0754-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00003-011-0754-8