
Abstract
Remote ischaemic conditioning (RIC) is a promising method of cardioprotection, with numerous clinical studies having demonstrated its ability to reduce myocardial infarct size and improve prognosis. On the other hand, there are several clinical trials, in particular those conducted in the setting of elective cardiac surgery, that have failed to show any benefit of RIC. These contradictory data indicate that there is insufficient understanding of the mechanisms underlying RIC. RIC is now known to signal indiscriminately, protecting not only the heart, but also other organs. In particular, experimental studies have demonstrated that it is able to reduce infarct size in an acute ischaemic stroke model. However, the mechanisms underlying RIC-induced neuroprotection are even less well understood than for cardioprotection. The existence of bidirectional feedback interactions between the heart and the brain suggests that the mechanisms of RIC-induced neuroprotection and cardioprotection should be studied as a whole. This review, therefore, addresses the topic of the neural component of the RIC mechanism.
Similar content being viewed by others

Introduction
It is almost 25 years since the phenomenon of remote ischaemic preconditioning (RIPre) was discovered by Przyklenk et al. [106]. They demonstrated in a canine model that a series of brief, alternating episodes of ischaemia and reperfusion, applied to one area of the left ventricle (LV) myocardium, protected a remote myocardial territory of the same heart from a subsequent prolonged ischaemic insult followed by reperfusion, limiting infarct size by almost 70%. In the same year, McClanahan et al. [94] achieved a similar infarct-limiting effect in rabbits by applying a single 10-min episode of ischaemia to a kidney prior to 30-min occlusion of the left anterior-descending coronary artery. However, since the RIPre stimulus must, by definition, be applied before the onset of ischaemia, it is clear that the only possible clinical niche for the RIPre phenomenon in cardioprotection could be in the setting of either elective cardiac surgery or elective percutaneous coronary intervention (PCI)—scenarios when the onset and the duration of ischaemia are carefully controlled (Fig. 1).

Schematic time lines representing the concepts of remote ischaemic pre-, per-, post- and delayed postconditioning, and the clinical scenarios in which they may be relevant. STEMI ST-elevated myocardial infarction. Asterisk—the evidence for the existence of the delayed remote ischaemic postconditioning phenomenon is currently limited having only been demonstrated in one experimental study
Twelve years after the discovery of RIPre, it was shown in a rat model that remote ischaemic conditioning (RIC) could be applied to a limb during myocardial ischaemia, and reduce infarct size by 50% [77]. This phenomenon became known as remote ischaemic preconditioning (RIPer). The next step in the study of RIC came with the demonstration by Andreka et al., that RIC could be successfully administered simultaneously with the onset of myocardial reperfusion—a phenomenon named as remote ischaemic postconditioning (RIPost) [1]. Taking this one step further, Basalay et al. demonstrated in a rat model of acute myocardial infarction that RIPost applied after a 10-min delay following the onset of reperfusion was still able to reduce infarct size similarly to RIPre [5]. Notably, the results included in this study were obtained in three research centres, using identical protocols [5]. The success of this procedure, termed ‘delayed RIPost’, was unexpected, in view of the prevailing hypothesis that the majority of myocardial reperfusion cell death is due to the opening of mitochondrial permeability transition pores (mPTP), thought to occur during the first few minutes of reperfusion [29, 46]. However, a series of studies demonstrated implication of non-mPTP/necrosis mechanisms in myocardial death [59] including pyroptosis [147], necroptosis [12, 127], and, more controversially, apoptosis [67]. We appreciate that the Basalay study is the only one to-date reporting the benefit of delayed RIPost [5], and is, therefore, still awaiting independent confirmation. Nonetheless, the clinical potential of this cardioprotective phenomenon can be significant, as it potentially broadens the time frame during which myocardial injury can be prevented. Moreover, the possibility to protect myocardium beyond the first few minutes of reperfusion has also been supported by studies from the groups of Ovize, Marbán and others [4, 27, 38, 71, 116], which have demonstrated cardioprotection with interventions applied up to the 20th min (rats, isolated rat hearts) [27, 38], 30th min (minipigs, mice) [71, 116] or 45th min (rats) [4] of myocardial reperfusion.
Though RIPre first emerged as a laboratory phenomenon, the cardioprotective potential of RIC strategies has subsequently drawn the attention of numerous clinical research groups [15, 28, 31, 36, 40, 82, 96, 105, 133, 135, 141]. In contrast to the repeated failures to translate cardioprotection from animal studies to clinical practice [42, 51, 59], RIC still appears to be a promising candidate for clinical use based both on the large number of successful experimental studies [17] and the fact that it satisfied all the recommendations for preclinical trials on cardioprotection [53, 56, 59, 86, 87].
The majority of clinical studies to date have demonstrated the infarct-limiting effect of RIC [15, 21, 28, 36, 105, 141] and improved outcome [40, 125] as a result of this procedure in patients with STEMI. The one exception is a recent study with neutral results, however, no long-term outcome data are currently available from this study [135]. In the setting of elective cardiac surgery, a single-centre trial involving 329 patients with isoflurane, used for the maintenance of anesthesia, suggested that RIPre provided perioperative myocardial protection [133] and improved the prognosis over 1.54 years [133] and 5 years, respectively [81]. Another small-scale clinical study on patients undergoing elective aortic valve replacement, demonstrated that RIPre reduced myocardial injury if applied on a background of sevoflurane, but not propofol anesthesia [9, 10]. Two, large, multicentre clinical trials on cardiac surgery patients using propofol for anesthesia [31, 96], and one small clinical study with perioperative anesthesia restricted to sevoflurane and fentanyl [99] failed to show any benefits of using RIPre. Bearing in mind the success of the previous (proof of concept) clinical trials both in STEMI patients and in patients undergoing cardiac surgery, these neutral results indicate the gaps in the knowledge of the mechanisms underlying the RIC phenomena, as well as lack of the differences between the mechanisms of myocardial ischaemia/reperfusion injury in these two clinical settings. It is, therefore, imperative to investigate the mechanism of RIC to facilitate the translation of this simple, non-invasive, low-cost intervention into patient benefit. Two large-scale multicentre clinical trials on STEMI patients are ongoing (NCT02342522; NCT01857414). Their results, due in 2019, will shed more light on the effects and possible confounding factors of cardioprotection by RIC.
The mechanisms of cardioprotection by remote ischaemic conditioning with regard to translational aspects
After the discovery of RIPre, two main competing hypotheses evolved regarding the mechanism of signal transduction from the preconditioned limb to the protected heart, one called the ‘humoral hypothesis’ and the other—the ‘neural hypothesis’. The humoral hypothesis is based on the fact that RIC-induced cardioprotection can be transferred to a naïve animal via whole blood transfusion [32]. Conversely, Gho et al. demonstrated that the neural blocker, hexamethonium, abolished cardioprotection by brief mesenteric artery occlusion [43], suggesting the important role for the autonomic ganglia in RIPre. However, in a later study by Weinbrenner et al., hexamethonium failed to inhibit the protection afforded by RIPre, performed as infrarenal occlusion of the aorta [140]. This result, as well as the necessity for a reperfusion period interspaced between the RIPre and myocardial ischaemia, during which time the humoral factor could travel to the heart, were viewed as reasons to exclude the hypothesis of a neuronal signal transmission from the remote area to the heart [140].
In 2010, Gourine et al. proposed the existence of a ‘remote preconditioning reflex’—a neural pathway of cardioprotection which is recruited by RIPre and protects the heart against ischaemia and reperfusion injury [44] (Fig. 2). This idea was supported by the data indicating the presence of all the components necessary for the realization of a classical reflex in the pathway mediating cardioprotection by RIPer: sensory (afferent) C-fiber neurons, an integration centre in the central nervous system, and motor (efferent) vagal neurons. In this regard, it has been demonstrated in a rat model of myocardial ischaemia/reperfusion injury, that cardioprotection by RIPre is completely abolished by denervation of the limbs performed as either mechanical transection of nerves [5, 130] or blocking afferent C-fibers by neonatal capsaicin treatment [5]. Similarly, stimulation of C-fiber afferents by topical capsaicin application limits myocardial injury, mimicking the effect of RIPre [5, 112]. It has been shown that calcitonin gene-related peptide (CGRP)—an important mediator of sensory neurons—mediates cardioprotection by RIPre [142]. Abrogation of protection by RIC with spinal cord transection at T7–T10 level [34], or with intrathecal spinal opioid receptor blockade [143], confirmed the involvement of the neural afferent pathway. The effector arm of the RIPer reflex appears to involve parasympathetic innervation. In the studies by Basalay et al., bilateral vagotomy in a rat model abolished cardioprotection induced by either RIPre [5, 6] or RIPer [6]. Mastitskaya et al. went further and directly inhibited parasympathetic motor neurons which are known to be located in two distinct brainstem sites [93]. This resulted in complete abolishment of the infarct-limiting effect of RIPre [93]. Notably, this study was the first to provide direct evidence for the involvement of vagal efferents in RIPre-mediated cardioprotection. It is noteworthy that bilateral vagotomy in the Basalay et al. study did not attenuate the infarct-limiting effect of delayed RIPost [5], which suggested that the mechanisms underlying these two phenomena may be distinct. Surprisingly, in a recent study by Buchholz et al., vagal stimulation in mice reduced myocardial infarct size when applied either before ischaemia or at the onset of reperfusion [18]. However, this study also revealed the difference in cardioprotective mechanisms of these two time-periods of vagal stimulation: the first activated the Akt/GSK-3β muscarinic pathway, while the second activated α7nAChR and JAK-2, independently of the cholinergic anti-inflammatory pathway [18]. The results of these studies [5, 18] raise the possibility that additive benefit could be achieved by combining different types of RIC. A recent study by Kleinbongard et al. confirms the neuronal transfer of the protective signal during RIC [78]. In this study, RIC was demonstrated to attenuate ischaemia-induced ST-segment elevation on the ECG during ongoing coronary artery occlusion [78].

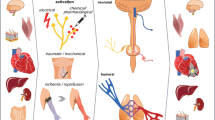
Neural mechanisms of remote ischaemic preconditioning. A diagram of connections (both known and controversial) between the neural and humoral mechanisms of remote ischaemic preconditioning of the heart and the brain. The core of this mechanism is the ‘remote preconditioning reflex’ comprising afferent somatic nerves, integration centre/centres in the central neural system, and efferent vagal nerves, innervating the heart and other organs, specifically, the intestine. However, it is not certain whether the increase in ACh concentration in the myocardium mediates the cardioprotective effects of remote preconditioning. It is also unclear which of the abdominal n. vagus fibres—afferent or efferent—are involved in this phenomenon. The known humoral factors that have the closest relationship with neural mechanisms are GLP-1 and nitrite. However, the source of their release in response to remote ischaemic stimulus is not clear. GLP-1 glucagon-like peptide-1, ACh acetylcholine
Yellon’s group were the first to demonstrate the involvement of both neural and humoral pathways in RIPre [89]. They found that in an RIPre model of femoral artery occlusion in mice, the absence of venous blood return from the preconditioned limb, or combined femoral and sciatic nerve resection, completely abolished the infarct-limiting effect. Moreover, resection of only one nerve—either femoral or sciatic—only partially abolished this effect [89]. In an elegant study, Jensen et al. confirmed the requirement for both the neural and humoral pathways in RIPre-mediated cardioprotection by testing whether human plasma dialysate obtained after RIC could reduce infarct size and improve hemodynamic recovery in isolated rabbit hearts [64]. They showed that the plasma dialysate was protective if obtained from healthy subjects or diabetic subjects without peripheral neuropathy, but when obtained from diabetic patients with neuropathy it was not protective. These findings further indicate that the mechanism of a humoral factor release involves neural pathways [64].
Because the initial studies investigating the role of the autonomic ganglia in RIPre-mediated cardioprotection had been controversial [43, 140], this question was re-visited by Yellon’s group in 2016 [102, 103]. They demonstrated that plasma dialysate obtained from RIPre-treated rats reduced infarct size in naïve isolated hearts subjected to ischaemia and reperfusion. However, the plasma was no longer cardioprotective if collected from vagotomised animals, or if the ganglionic blocker hexamethonium or muscarinic antagonist atropine were applied to isolated hearts [103]. This led to the conclusion that release of a protective factor following RIC is dependent on prior activation of the vagus nerve. In addition, the factor appears to induce cardioprotection via recruitment of intrinsic cardiac ganglia. Their subsequent study, demonstrating that coronary effluent from a preconditioned isolated heart could protect another isolated heart, but not isolated cardiomyocytes, also contributed to the hypothesis of the importance of intrinsic cardiac ganglia in the mechanisms of direct ischaemic preconditioning [102].
Since the first demonstration of the possibility to transfer ischaemic preconditioning cardioprotection with blood [32], the attention of several research groups has focused on identifying the humoral factor responsible for RIPre. Several candidate humoral factors of RIC have been proposed, including stromal cell-derived factor-1 [30], nitrite/nitric oxide [110], interleukin-1α [41], interleukin-10 [19], microRNA-144 [88], apolipoprotein A-I [57], alpha-ketoglutarate-dependent dioxygenase Egln1 [101] and glucagon-like peptide-1 (GLP-1) [7, 8]. However, for the purpose of this review, we focus on two of the proposed humoral mediators that are most relevant to the ‘neural hypothesis’ of RIC, namely nitrite/nitric oxide [110] and GLP-1 [7, 8].
Using an in vivo mouse model, Rassaf et al. demonstrated that shear stress-dependent stimulation of endothelial nitric oxide (NO) synthase within a femoral artery by RIPre yields a substantial release of NO. This NO is subsequently oxidized to nitrite and transferred humorally to the myocardium, where it reduces infarct size caused following 30-min myocardial ischaemia and 24-h reperfusion [110]. In addition, a series of experiments in which plasma from healthy volunteers subjected to RIPre was perfused through isolated mouse hearts, identified plasma nitrite as the cardioprotective agent [110]. Similarly, in a recent study by Hauerslev et al., scavengers of NO attenuated RIPre-induced protection in rat hearts [47]. Previously, it had been shown that either stimulation of cervical vagal nerves or perfusion with acetylcholine is associated with NO release in an isolated rabbit heart via the neuronal isoform of NO synthase [16]. Interestingly, in an earlier study, bilateral vagal nerve stimulation in rabbits was followed by an increase in nitrite formation at the level of the stomach and the colon [62]. In terms of planning future clinical studies on the effect of RIC, it is important to keep in mind that exogenous NO [98] and nitroglycerin [35, 50, 148, 155] have been shown to be able to reduce infarct size, and may therefore, potentially interfere with RIC.
The results obtained by Mastitskaya et al. [92] suggest that visceral organs, innervated by the posterior gastric branch of the vagus nerve, are the likely source of a humoral factor (or factors) of RIC cardioprotection. This finding is interesting as it provides the first potential mechanism uniting the ‘neural’ and ‘humoral’ hypotheses of RIC. Apart from nitric oxide [62], GLP-1 appears to be the most likely candidate for this role. Its release from the L-cells of the intestine is modulated by vagal efferent (motor) activity [58, 115] and there is also evidence that GLP-1 may interact with vagal sensory fibers innervating the viscera [58]. The molecular weight of GLP-1 is 3.3 kDa thereby satisfying the key criteria of the humoral preconditioning factor (including molecular weight of less than 8 kDa), suggested by Lang and colleagues on the basis of the proteomic analysis of blood samples obtained from experimental animals receiving the RIPre stimulus [85]. Studies conducted in animal models by the Yellon group [13, 14], and subsequently by others [111], demonstrated potent cardioprotection by GLP-1 receptor (GLP-1R) activation. The efficacy of GLP-1R agonists in limiting infarct size has also been shown in clinical trials on STEMI patients [90, 144]. Later, Basalay et al. showed that GLP-1 mediates cardioprotection by RIC, and demonstrated that cardioprotection induced by GLP-1R activation is mediated by a mechanism involving M3 muscarinic receptors [7, 8]. The location of these M3 muscarinic receptors, mediating the infarct-limiting effect of RIPre, is not yet established.
The main extracellular signaling molecules that have been shown to be involved in cardioprotection by RIPre include opioids, bradykinin and adenosine [79]. Opioid peptides can be secreted from cardiac nerves or produced in the cardiomyocytes themselves [107]. Shimizu et al. demonstrated that the protective effect of dialysate from a donor limb subjected to RIC, on isolated cardiomyocytes is blocked by pre-treatment of the cardiomyocytes with the opiate receptor blocker naloxone [122]. These results suggest that opioid receptors involved in RIC are located in cardiomyocytes and not connected with neural activation. On the other hand, in another study, hexamethonium abolished the protection provided by intramesenteric bradykinin infusion [119]. Regarding bradykinin, it is worth mentioning that it is degraded rapidly by angiotensin-converting enzyme (ACE) [136], while ACE inhibitors and angiotensin-1 receptor antagonists [63] mimic its cardioprotective effects. The role of adenosine in RIC cardioprotection is still controversial, being confirmed in a mouse model [120], but not a porcine model [48].
Analysis of the concentrations of metabolites in plasma samples of patients subjected to RIC revealed an increase in glycine concentration following RIC [23]. This is relevant to the current review, as glycine is known to be an inhibitory neurotransmitter in the central nervous system, and specifically, in parasympathetic nuclei [22, 131, 132]. In the above-mentioned study, the injection of glycine mimicked the protective effects of RIC in rats [23]. García-Dorado’s group demonstrated that glycine exerts cross-species cardioprotection against infarction through glycine receptor activation [66]. The potential involvement of glycine signalling in RIC-induced cardioprotection should be borne in mind when planning clinical trials, as certain drugs, such as Ketamine [137], can inhibit glycine neurotransmission to cardiac vagal neurons.
The importance of understanding the mechanism of RIC cardioprotection is clearly demonstrated by the neutral results of two recent multicentre trials—ERICCA and RIPHeart, in which the majority of the patients were given propofol as part of the anesthesia protocol [31, 96]. Importantly, it had previously been shown that propofol inhibits neurotransmission to cardiac vagal neurons in the nucleus ambiguous [138]. On the other hand, propofol had been shown to cause the enhanced nitrite production in cultured myocytes [146], whereas nitrite is known as one of the humoral mediators of RIPre [110]. The results of small-scale clinical trials completed in 2012 and 2014 confirmed the importance of the anesthetic regime, showing that RIPre during isoflurane but not during propofol anesthesia decreased myocardial damage in patients undergoing CABG surgery [82] or elective aortic valve replacement [9, 10]. A recent experimental study also clearly demonstrated that propofol, but not sevoflurane, abolished the infarct-limiting effect of RIPre in rats in vivo [11]. In a more recent single-centre study with a neutral effect of RIC in CABG patients, propofol was purposefully avoided [99]. However, in this study, the anesthetic protocol included fentanyl, which has been shown to inhibit neurotransmission to cardiac vagal neurons in the nucleus ambiguous [45]. It could also be argued, however, that fentanyl or sufentanil did not abolish RIC-induced cardioprotection in other clinical studies [9, 133]. Although there are multiple possible reasons for the lack of protection in these studies, as has been extensively discussed [25, 39, 55], the suppressed vagal system is likely to have contributed.
The effect of an anesthetic protocol on RIC efficacy was revealed in a Bayesian network meta-analysis of 55 randomized trials, which included 6921 patients undergoing cardiac surgery. The use of volatile agents and the combination of volatile agents with remote preconditioning were found to be associated with a reduction in mortality at the longest follow-up time available, when compared to total intravenous anesthesia [151]. Furthermore, it was observed that the combination of RIC with volatile agents was associated with a reduction in mortality when compared to RIC with total intravenous anesthesia [151].
Unresolved questions and discrepancies on the mechanisms of cardioprotection by remote ischaemic conditioning
Although the degree of cardioprotection conferred by RIPre and delayed RIPost is similar, the underlying mechanisms seem to be distinct [5]. In the Basalay et al.’s study (2012), the infarct-limiting effect of delayed RIPost was not affected by either vagotomy or peripheral denervation, as opposed to the effect of RIPre [5]. As peripheral or especially parasympathetic denervation, can potentially represent real clinical situations with patients suffering from peripheral polyneuropathies of different etiologies, or exposed to drugs with vagolytic effects, these results allow us to predict a lack of cardioprotective benefit from RIPre in these cohorts of patients. On the other hand, delayed RIPost could potentially be more effective in the same patients, being less susceptible to innervation impairments, though no clinical trials demonstrating this have been completed so far.
The other missing part in our knowledge is the mechanism of vagally mediated RIPre cardioprotection. The feasibility of vagally mediated cardioprotection (i.e., RIPre) in a left ventricle, which is known to be only sparsely innervated, can be explained by the observation that there is a local acetylcholine (ACh) synthesis system in the myocardium [37, 69, 109]. This system is positively modulated by cholinergic stimuli [69]. Conversely, atropine reduces the basal ACh content [69]. It has been shown that either bilateral vagotomy [5] or direct inhibition of the parasympathetic motor neurons in the central nervous system [93], or atropine treatment [34] abolishes the infarct-limiting effect of RIPre. Yet, it is not clear whether it is the increase of acetylcholine concentration that mediates the cardioprotective effect of RIC. There are a number of experimental observations relevant to the assumption that it is indeed acetylcholine which mediates RIPre. First, in support of this hypothesis, the hearts from transgenic mice overexpressing choline acetyltransferase (ChAT)—the main ACh-synthesizing enzyme in cardiomyocytes—are more resistant to ischaemia–reperfusion injury than wild-type hearts [70]. And second, RIC increases both ChAT expression and ACh content in mice hearts [68, 100]. However, there are also experimental data that contradict this hypothesis. In this regard, it has been shown that acute myocardial ischaemia provoked an increase of ACh concentration in an ischaemic myocardium, and vagotomy did not affect this increase [76], while on the other hand, vagotomy abolished the infarct-limiting effect of RIPre [5]. This indicates that the loss of RIPre-mediated cardioprotection in vagotomised rats [5] might be caused by some other factors rather than a decrease in ACh concentration. Another indirect argument suggesting that ACh concentration may not be of critical importance for ischaemic preconditioning is that vagotomy abolishes the myocardial interstitial ACh release induced by brief myocardial ischaemia [75]. On the other hand, bilateral vagotomy does not attenuate the infarct-limiting effect of direct ischaemic preconditioning [5], which indicates that ACh concentration in the myocardium does not correlate with the degree of cardioprotection. Currently, there are no data revealing whether RIPre causes further increase of ACh concentration in the ischaemic myocardium in comparison to the myocardial ischaemia itself, and whether vagotomy prevents this additional increase. The answer to this question is even more difficult, as the degree of ACh increase during myocardial ischaemia varies from study to study, depending on the animal species, samples and techniques used, as well as on the duration of the ischaemic episode [74, 76, 102].
Despite the lack of experimental data that would unequivocally answer the question of myocardial ACh concentration in RIC-mediated cardioprotection, there are several observations that may shed some light on this aspect. In this respect, it was demonstrated that cardiomyocytes of adult, but not neonatal rats, are able to synthesize, transport and excrete ACh in the heart [109]. The expression level of ChAT and the amount of ACh excreted were also significantly downregulated in cardiomyocytes of old animals [109]. If ACh concentration in myocardium determines the potency of protection by RIC, we may expect that RIC would be less effective in children and aged people. Indeed, RIPre was actually shown to impair ventricular function and increase infarct size in an isolated neonatal rabbit heart [118]. An earlier, small-scale clinical study had demonstrated myocardial-protective effects of RIPre in children [26]. However, in a more recent clinical trial performed on 299 children (aged neonate to 17 years), RIPre was not associated with important improvements in clinical outcomes and physiological markers after cardiac surgery [95]. It should be noted, however, that there was no standardized protocol of anesthesia in this study, and more than half the patients were exposed to propofol [95]. The effect of RIC in elderly patients (> 70 years) was investigated in the CONDI trial, and found to be equally as protective as in younger patients [126]. Similarly, in the LIPSIA CONDITIONING trial, the combination of RIC and local ischaemic postconditioning was effective in both age groups—under and over 65 years old [36]. Similar results were obtained after the analysis of confounders of RIPre cardioprotection in patients undergoing coronary artery bypass grafting [80]. In a recent experimental study by Heinen et al., plasma of young male volunteers, subjected to RIC, reduced infarct size in isolated hearts from aged rats [49]. However, RIC plasma of aged male volunteers had no protective effect in young rat hearts [49]. This indicates that aging affects the RIC-induced release of a humoral factor, but not the susceptibility of myocardium to the protective effect of this phenomenon.
One of the main purposes for investigating the mechanisms of endogenous cardioprotective phenomena is the potential to develop pharmacological therapy or electronic devices that can fully mimic its positive effects. These could have the advantage of being able to be applied or administered significantly more quickly than the RIC procedure, which requires up to 40 min. As RIPre/RIPer cardioprotection is mediated by the activation of vagal efferents [5, 93], it is logical to assume that vagal nerve stimulation could potentially reproduce the effects of RIC. The obvious question arising here is whether monolateral vagal nerve stimulation can achieve this goal. In experimental studies, electrical stimulation of right [3, 18, 20, 72, 73], left [123, 124] or both vagal nerves [134] limited myocardial infarct size when started either before [18, 20, 72] or during [3, 73, 123, 124, 134] myocardial ischaemia, or at the onset of reperfusion [18]. In a clinical study by Yu et al., low-level transcutaneous stimulation of the vagal branch within the area of the right tragus, in patients presenting with STEMI, was followed by the reduction of the incidence of reperfusion-related ventricular arrhythmia, the area under curve for creatine kinase-MB and myoglobin over 72 h, and blood levels of inflammatory markers [52, 150]. In the study by Basalay et al., the activity of only one vagal nerve—either right or left—was sufficient and contributed equally to mediate cardioprotection established by RIPre. On the other hand, functional integrity of both nerves was required to establish cardioprotection when the remote conditioning stimulus was applied during myocardial ischaemia [6]. However, the differences in the mechanisms of RIPre and RIPer leading to this difference [6] have not been explained. Another arising question is—whether electrical vagal nerve stimulation can completely mimic all aspect of RIC, which include the indirect stimulation of the release of humoral factors [52].
While the importance of the activation of parasympathetic efferents in RIC-mediated cardioprotection is now unambiguous, the role of the sympathetic nervous system in this phenomenon has not been described clearly. However, it has been demonstrated that beta-blockers reduce infarct size in STEMI patients undergoing PCI [61], and this group of drugs is currently included in the guidelines for the management of these patients [60]. It has been shown that beta-adrenoreceptors are involved in the mechanisms of myocardial infarct-limiting effect of remote pre- and postconditioning of trauma [128, 129]. The existence of some common, inherent mechanisms, such as the activation of C sensory fibers and the K(ATP) channels for both remote preconditioning of trauma [65] and RIPre/RIPer [5, 83, 117], may be an argument to suggest that sympathetic beta-adrenergic activation is involved in the mechanisms of RIC cardioprotection. On the other hand, it has been demonstrated in rats that exercise training, which shares some molecular mechanisms with RIC [108, 121], augments the dynamic heart rate response to vagal but not sympathetic stimulation [97]. Moreover, it is known that sympathetic nervous response to ischaemia–reperfusion injury is altered with RIPre [84]. In the CONDI trial, the infarct-limiting effect of RIC was preserved in beta-blocker users [126]. Although there are no available data on the effect of beta-blockers on the efficacy of RIC in other clinical trials with STEMI patients, the fact that remote conditioning was able to reduce infarct size in the clinical study, where almost all the patients were taking beta-blockers [36], reinforced the CONDI results (Table 1). Similarly, medication with beta-blockers was not a significant confounder of RIPre cardioprotection in patients undergoing cardiac surgery [80]. In contrast, a meta-analysis of 15 clinical trials, including 1155 patients randomised to treatment with or without RIPre, showed an attenuated effect of this intervention in cardiosurgery patients on perioperative beta-blocker treatment [153].
Remote ischaemic conditioning of the brain
In addition to protecting the heart, RIC is potentially able to protect any other organ or tissue. It has been shown in a series of experimental studies that RIPre, RIPer, RIPost and delayed RIPost reduce brain infarct size in a rodent model of acute focal ischaemia/reperfusion brain injury [24]. Interestingly, the reperfusion ‘time-window’, during which delayed RIPost was effective, seems to be longer in an ischaemic stroke model [104, 114] than in a STEMI model [4, 5]. In this regard, in a rat model with 100-min focal brain ischaemia, RIPost was able to reduce infarct size when initiated within 30 min of reperfusion [104]. Impressively, when subject to 30-min brain ischaemia, RIPost was effective up to 3 h of reperfusion [114]. A meta-analysis of 13 randomized controlled trials, which included a total of 794 study participants who either suffered from, or were at risk from brain ischaemia and reperfusion injury, suggested that RIPost can offer cerebral protection for stroke patients [152]. Compared with controls, RIPost was shown to be able to reduce the recurrence of stroke or transient ischaemic attacks, levels of National Institutes of Health Stroke Scale score, modified Rankin Scale score and high-sensitivity C-reactive protein [152].
The studies on the mechanisms of RIC-induced neuroprotection are quite scarce in comparison with the amount of studies on the mechanisms of RIC-induced cardioprotection. Similar to RIC-induced cardioprotection, there is evidence for the involvement of the neural pathway in the mechanism of RIC of the brain [91, 104, 114, 139, 149], including the possible involvement of endogenous opioids [113, 154] and CGRP [113]. On the other hand, in one study, local electrical stimulation enhanced the neuroprotective effect of RIPost in a rat stroke model [145], which might suggest that the mechanisms underlying the therapeutic effects of these interventions are different. Pignataro et al. demonstrated that the neuronal isoform of nitric oxide synthase takes part in the neuroprotective effect of RIPost [104]. Further investigation of the neuroprotective mechanism of RIC is important to establish the most effective and safe implementation of this phenomenon to clinical practice.
The activity of the cardiovascular system is continually modulated by the central nervous system, ensuring coordination and regulation of regional cardiac electrical, mechanical and metabolic indexes throughout each cardiac cycle [2]. This fine regulation is provided by the neuronal elements, which are distributed from the level of the insular cortex to the intrinsic cardiac nervous system, and are in constant communication with one another [2]. In this regard, both ischaemic and haemorrhagic strokes can cause abnormalities in autonomic nervous system activity, followed by functional imbalance within the cardiovascular system, and in certain situations—even by irreversible cardiomyocyte damage, determined as serum troponin elevation [33]. On the other hand, nociceptive sensory inputs arising from the ischaemic heart represent a stimulus that can evoke discord within and among different levels of the hierarchy of the neuronal elements connecting heart, brain and central nervous system [2, 54]. Bearing in mind the existence of these bidirectional feedback interactions between the heart and brain [2], and specifically the involvement of brain subcortical structures in ‘remote preconditioning reflex’ [44, 93], more detailed understanding of the mechanisms of RIC-induced neuroprotection would shed more light on the mechanisms RIC-induced cardioprotection and myocardial ischaemia/reperfusion injury.
Conclusions
Investigating the mechanisms of RIC is the essential step on the road to its translation to patient benefit, and specifically to the discovery of therapies or electronic devices, which would be able to mimic the beneficial effects of this phenomenon. Experimental and clinical data indicate that neural mechanisms, known as the ‘remote preconditioning reflex’, have an important contribution into RIC phenomena. However, there are still gaps in our understanding of the neural mechanism:
-
Though the mechanism of RIPre has been extensively investigated, the mechanism of delayed RIPost is less clear. According to experimental data so far, delayed RIPost could be effective in patients with neural impairments, in whom the anti-ischaemic effect of RIPre is not expected.
-
Although the involvement of vagal nerves in RIPre and RIPer is certain, an important question in terms of future drug discovery is whether these phenomena provide cardioprotection via the increase of ACh concentration in left ventricle myocardium.
-
In terms of the future possibility to mimic or increase the effects of RIC using direct vagal nerve stimulation, a gap in the current knowledge is: why the effect of monolateral vagotomy is different for infarct-limiting effect of RIPre and RIPer.
-
Sympathetic involvement in RIC is another gap in our understanding of these phenomena. The interaction of the sympathetic and parasympathetic nervous systems is fundamental and complex, so that, from a translational point of view, it would hardly be possible to modulate one of them without influencing the other. Notably, drugs modulating sympathetic activity are widely used in routine clinical practice—this fact is important to bear in mind when designing clinical studies.
-
Revealing the mechanisms of RIC-induced neuroprotection is an important goal for future research not only to be able to use this phenomenon most efficiently in clinical practice, but also as it could help better understand the mechanisms of cardioprotective effects of these phenomena.
References
Andreka G, Vertesaljai M, Szantho G, Font G, Piroth Z, Fontos G, Juhasz ED, Szekely L, Szelid Z, Turner MS, Ashrafian H, Frenneaux MP, Andreka P (2007) Remote ischaemic postconditioning protects the heart during acute myocardial infarction in pigs. Heart 93:749–752. https://doi.org/10.1136/hrt.2006.114504
Ardell JL, Armour JA (2016) Neurocardiology: structure-based function. Compr Physiol 6:1635–1653. https://doi.org/10.1002/cphy.c150046
Arimura T, Saku K, Kakino T, Nishikawa T, Tohyama T, Sakamoto T, Sakamoto K, Kishi T, Ide T, Sunagawa K (2017) Intravenous electrical vagal nerve stimulation prior to coronary reperfusion in a canine ischemia–reperfusion model markedly reduces infarct size and prevents subsequent heart failure. Int J Cardiol 227:704–710. https://doi.org/10.1016/j.ijcard.2016.10.074
Barsukevich V, Basalay M, Sanchez J, Mrochek A, Whittle J, Ackland GL, Gourine AV, Gourine A (2015) Distinct cardioprotective mechanisms of immediate, early and delayed ischaemic postconditioning. Basic Res Cardiol 110:452. https://doi.org/10.1007/s00395-014-0452-7
Basalay M, Barsukevich V, Mastitskaya S, Mrochek A, Pernow J, Sjöquist P-O, Ackland GL, Gourine AV, Gourine A (2012) Remote ischaemic pre- and delayed postconditioning—similar degree of cardioprotection but distinct mechanisms. Exp Physiol 97:908–917. https://doi.org/10.1113/expphysiol.2012.064923
Basalay M, Barsukevich V, Mrochek A, Gourine AV, Gourine A (2013) Right vs left vagus and cardioprotection conferred by remote ischaemic pre- and perconditioning. Eur Heart J 34:3697. https://doi.org/10.1093/eurheartj/eht309.3697
Basalay M, Mastitskaya S, Mrochek A, Ackland GL, del Arroyo AG, Sanchez J, Sjoquist P-O, Pernow J, Gourine AV, Gourine A (2017) Reply: Glucagon-like peptide-1 mediates cardioprotection by remote ischaemic conditioning. Cardiovasc Res 113:13–14. https://doi.org/10.1093/cvr/cvw238
Basalay MV, Mastitskaya S, Mrochek A, Ackland GL, del Arroyo AG, Sanchez J, Sjoquist P-O, Pernow J, Gourine AV, Gourine A (2016) Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc Res 112:669–676. https://doi.org/10.1093/cvr/cvw216
Bautin AE, Galagudza MM, Datsenko SV, Tashkhanov DM, Marichev AO, Bakanov AI, Malaia EI, Naĭmushin AV, Rubinchik VE, Gordeev ML (2014) Effects of remote ischemic preconditioning on perioperative period in elective aortic valve replacement. Anesteziol Reanimatol 3:11–17
Bautin AE, Galagudza MM, Tashkhanov DM, Datsenko SV, Marichev AO, Kostareva AA, Kravchuk EN, Bakanov AY, Gordeev ML (2015) Protein kinase C expression following remote ischemic preconditioning in cardiac surgery. Anesteziol Reanimatol 60:4–8
Behmenburg F, van Caster P, Bunte S, Brandenburger T, Heinen A, Hollmann MW, Huhn R (2018) Impact of anesthetic regimen on remote ischemic preconditioning in the rat heart in vivo. Anesth Analg 126:1377–1380. https://doi.org/10.1213/ANE.0000000000002563
Bell RM, Bøtker HE, Carr RD, Davidson SM, Downey JM, Dutka DP, Heusch G, Ibanez B, Macallister R, Stoppe C, Ovize M, Redington A, Walker JM, Yellon DM (2016) 9th Hatter Biannual Meeting: position document on ischaemia/reperfusion injury, conditioning and the ten commandments of cardioprotection. Basic Res Cardiol 111:41. https://doi.org/10.1007/s00395-016-0558-1
Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM (2005) Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 54:146–151. https://doi.org/10.2337/diabetes.54.1.146
Bose AK, Mocanu MM, Carr RD, Yellon DM (2005) Glucagon like peptide-1 is protective against myocardial ischemia/reperfusion injury when given either as a preconditioning mimetic or at reperfusion in an isolated rat heart model. Cardiovasc Drugs Ther 19:9–11. https://doi.org/10.1007/s10557-005-6892-4
Bøtker HE, Kharbanda R, Schmidt MR, Bøttcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sørensen HT, Redington AN, Nielsen TT (2010) Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375:727–734. https://doi.org/10.1016/S0140-6736(09)62001-8
Brack KE, Patel VH, Mantravardi R, Coote JH, Ng GA (2009) Direct evidence of nitric oxide release from neuronal nitric oxide synthase activation in the left ventricle as a result of cervical vagus nerve stimulation. J Physiol 587:3045–3054. https://doi.org/10.1113/jphysiol.2009.169417
Bromage DI, Pickard JMJ, Rossello X, Ziff OJ, Burke N, Yellon DM, Davidson SM (2017) Remote ischaemic conditioning reduces infarct size in animal in vivo models of ischaemia–reperfusion injury: a systematic review and meta-analysis. Cardiovasc Res 113:288–297. https://doi.org/10.1093/cvr/cvw219
Buchholz B, Kelly J, Muñoz MC, Bernatené EA, Méndez Diodati N, González Maglio DH, Dominici FP, Gelpi RJ (2018) Vagal stimulation mimics preconditioning and postconditioning of ischemic myocardium in mice by activating different protection mechanisms. Am J Physiol Circ Physiol 97:908–917. https://doi.org/10.1152/ajpheart.00286.2017
Cai ZP, Parajuli N, Zheng X, Becker L (2012) Remote ischemic preconditioning confers late protection against myocardial ischemia–reperfusion injury in mice by upregulating interleukin-10. Basic Res Cardiol 107:277. https://doi.org/10.1007/s00395-012-0277-1
Calvillo L, Vanoli E, Andreoli E, Besana A, Omodeo E, Gnecchi M, Zerbi P, Vago G, Busca G, Schwartz PJ (2011) Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion. J Cardiovasc Pharmacol 58:500–507. https://doi.org/10.1097/FJC.0b013e31822b7204
Cao B, Wang H, Zhang C, Xia M, Yang X (2018) Remote ischemic postconditioning (RIPC) of the upper arm results in protection from cardiac ischemia–reperfusion injury following primary percutaneous coronary intervention (PCI) for acute ST-segment elevation myocardial infarction (STEMI). Med Sci Monit 24:1017–1026. https://doi.org/10.12659/MSM.908247
Cassell MD, Yi H, Talman WT (2000) Glycine receptor (gephyrin) immunoreactivity is present on cholinergic neurons in the dorsal vagal complex. Neuroscience 95:489–497. https://doi.org/10.1016/S0306-4522(99)00454-6
Chao de la Barca JM, Bakhta O, Kalakech H, Simard G, Tamareille S, Catros V, Callebert J, Gadras C, Tessier L, Reynier P, Prunier F, Mirebeau-Prunier D (2016) Metabolic signature of remote ischemic preconditioning involving a cocktail of amino acids and biogenic amines. J Am Heart Assoc 5:e003891. https://doi.org/10.1161/JAHA.116.003891
Chen G, Thakkar M, Robinson C, Doré S (2018) Limb remote ischemic conditioning: mechanisms, anesthetics, and the potential for expanding therapeutic options. Front Neurol 9:40. https://doi.org/10.3389/fneur.2018.00040
Cheung CX, Healy DA, Walsh SR (2016) Remote preconditioning and cardiac surgery: regrouping after Remote Ischemic Preconditioning for Heart Surgery (RIPHeart) and Effect of Remote Ischemic Preconditioning on Clinical Outcomes in Patients Undergoing Coronary Artery Bypass Surgery (ERICCA). J Thorac Dis 8:E197–E199. https://doi.org/10.21037/jtd.2016.01.81
Cheung MM, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, Holtby HM, Cox PN, Smallhorn JF, Van Arsdell GS, Redington AN (2006) Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery. J Am Coll Cardiol 47:2277–2282. https://doi.org/10.1016/j.jacc.2006.01.066
de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, Arditi M, Marbán E (2015) Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Investig 125:3147–3162. https://doi.org/10.1172/JCI81321
Crimi G, Pica S, Raineri C, Bramucci E, De Ferrari GM, Klersy C, Ferlini M, Marinoni B, Repetto A, Romeo M, Rosti V, Massa M, Raisaro A, Leonardi S, Rubartelli P, Oltrona Visconti L, Ferrario M (2013) Remote ischemic post-conditioning of the lower limb during primary percutaneous coronary intervention safely reduces enzymatic infarct size in anterior myocardial infarction. JACC Cardiovasc Interv 6:1055–1063. https://doi.org/10.1016/j.jcin.2013.05.011
Crompton M, Costi A (1990) A heart mitochondrial Ca2(+)-dependent pore of possible relevance to re-perfusion-induced injury. Evidence that ADP facilitates pore interconversion between the closed and open states. Biochem J 266:33–39. https://doi.org/10.1042/bj2660033
Davidson SM, Selvaraj P, He D, Boi-Doku C, Yellon RL, Vicencio JM, Yellon DM (2013) Remote ischaemic preconditioning involves signalling through the SDF-1α/CXCR4 signalling axis. Basic Res Cardiol 108:377. https://doi.org/10.1007/s00395-013-0377-6
Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, Knight R, Kunst G, Laing C, Nicholas JM, Pepper J, Robertson S, Xenou M, Clayton T, Yellon DM (2016) Effect of Remote Ischaemic preconditioning on Clinical outcomes in patients undergoing Coronary Artery bypass graft surgery (ERICCA study): a multicentre double-blind randomised controlled clinical trial. Effic Mech Eval. https://doi.org/10.3310/eme03040
Dickson EW, Reinhardt CP, Renzi FP, Becker RC, Porcaro WA, Heard SO (1999) Ischemic preconditioning may be transferable via whole blood transfusion: preliminary evidence. J Thromb Thrombolysis 8:123–129. https://doi.org/10.1023/A:1008911101951
Doehner W, Ural D, Haeusler KG, Čelutkienė J, Bestetti R, Cavusoglu Y, Peña-Duque MA, Glavas D, Iacoviello M, Laufs U, Alvear RM, Mbakwem A, Piepoli MF, Rosen SD, Tsivgoulis G, Vitale C, Yilmaz MB, Anker SD, Filippatos G, Seferovic P, Coats AJS, Ruschitzka F (2018) Heart and brain interaction in patients with heart failure: overview and proposal for a taxonomy. A position paper from the Study Group on Heart and Brain Interaction of the Heart Failure Association. Eur J Heart Fail 20:199–215. https://doi.org/10.1002/ejhf.1100
Donato M, Buchholz B, Rodríguez M, Pérez V, Inserte J, García-Dorado D, Gelpi RJ (2013) Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp Physiol 98:425–434. https://doi.org/10.1113/expphysiol.2012.066217
Du YH, Peng J, Huang ZZ, Jiang DJ, Deng HW, Li YJ (2004) Delayed cardioprotection afforded by nitroglycerin is mediated by alpha-CGRP via activation of inducible nitric oxide synthase. Int J Cardiol 93:49–54. https://doi.org/10.1016/S0167-5273(03)00123-2
Eitel I, Stiermaier T, Rommel KP, Fuernau G, Sandri M, Mangner N, Linke A, Erbs S, Lurz P, Boudriot E, Mende M, Desch S, Schuler G, Thiele H (2015) Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: the randomized LIPSIA CONDITIONING trial. Eur Heart J 36:3049–3057. https://doi.org/10.1093/eurheartj/ehv463
Ekström J (1978) Fall in choline acetyltransferase activity in the ventricles of the rat heart after treatment with a ganglion blocking drug. Acta Physiol Scand 102:116–119. https://doi.org/10.1111/j.1748-1716.1978.tb06051.x
Ferrera R, Benhabbouche S, Da Silva CC, Alam MR, Ovize M (2015) Delayed low pressure at reperfusion: a new approach for cardioprotection. J Thorac Cardiovasc Surg 150(1641–1648):e2. https://doi.org/10.1016/j.jtcvs.2015.08.053
Garratt KN, Whittaker P, Przyklenk K (2016) Remote ischemic conditioning and the long road to clinical translation. Circ Res 118:1052–1054. https://doi.org/10.1161/CIRCRESAHA.115.308102
Gaspar A, Lourenço AP, Pereira MÁ, Azevedo P, Roncon-Albuquerque R, Marques J, Leite-Moreira AF (2018) Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Basic Res Cardiol 113:14. https://doi.org/10.1007/s00395-018-0672-3
Gedik N, Kottenberg E, Thielmann M, Frey UH, Jakob H, Peters J, Heusch G, Kleinbongard P (2017) Potential humoral mediators of remote ischemic preconditioning in patients undergoing surgical coronary revascularization. Sci Rep 7:12660. https://doi.org/10.1038/s41598-017-12833-2
Gerczuk PZ, Kloner RA (2012) An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol 59:969–978. https://doi.org/10.1016/j.jacc.2011.07.054
Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD (1996) Myocardial protection by brief ischemia in noncardiac tissue. Circulation 94:2193–2200. https://doi.org/10.1161/01.CIR.94.9.2193
Gourine A, Gourine AV, Mastitskaya S, Ackland G (2010) “Remote preconditioning reflex”—a neural pathway of cardioprotection during myocardial ischaemia and reperfusion induced by remote ischaemic preconditioning. Eur Hear J 31(Suppl 1):319
Griffioen KJ, Venkatesan P, Huang ZG, Wang X, Bouairi E, Evans C, Gold A, Mendelowitz D (2004) Fentanyl inhibits GABAergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Brain Res 1007:109–115. https://doi.org/10.1016/j.brainres.2004.02.010
Griffiths EJ, Halestrap AP (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307(Pt 1):93–98. https://doi.org/10.1042/bj3070093
Hauerslev M, Mørk SR, Pryds K, Contractor H, Hansen J, Jespersen NR, Johnsen J, Heusch G, Kleinbongard P, Kharbanda R, Bøtker HE, Schmidt MR (2018) Influence of long-term treatment with glyceryl trinitrate on remote ischemic conditioning. Am J Physiol Circ Physiol. https://doi.org/10.1152/ajpheart.00114.2018
Hausenloy DJ, Iliodromitis EK, Andreadou I, Papalois A, Gritsopoulos G, Anastasiou-Nana M, Kremastinos DT, Yellon DM (2012) Investigating the signal transduction pathways underlying remote ischemic conditioning in the porcine heart. Cardiovasc Drugs Ther 26:87–93. https://doi.org/10.1007/s10557-011-6364-y
Heinen A, Behmenburg F, Aytulun A, Dierkes M, Zerbin L, Kaisers W, Schaefer M, Meyer-Treschan T, Feit S, Bauer I, Hollmann MW, Huhn R (2018) The release of cardioprotective humoral factors after remote ischemic preconditioning in humans is age- and sex-dependent. J Transl Med 16:112. https://doi.org/10.1186/s12967-018-1480-0
Heusch G (2001) Nitroglycerin and delayed preconditioning in humans: yet another new mechanism for an old drug? Circulation 103:2876–2878. https://doi.org/10.1161/01.CIR.103.24.2876
Heusch G (2017) Critical issues for the translation of cardioprotection. Circ Res 120:1477–1486. https://doi.org/10.1161/CIRCRESAHA.117.310820
Heusch G (2017) Vagal cardioprotection in reperfused acute myocardial infarction. JACC Cardiovasc Interv 10:1521–1522. https://doi.org/10.1016/j.jcin.2017.05.063
Heusch G, Bøtker HE, Przyklenk K, Redington A, Yellon D (2015) Remote ischemic conditioning. J Am Coll Cardiol 65:177–195. https://doi.org/10.1016/j.jacc.2014.10.031
Heusch G, Deussen A, Thämer V (1985) Cardiac sympathetic nerve activity and progressive vasoconstriction distal to coronary stenoses: feed-back aggravation of myocardial ischemia. J Auton Nerv Syst 13:311–326. https://doi.org/10.1016/0165-1838(85)90020-7
Heusch G, Gersh BJ (2016) ERICCA and RIPHeart: two nails in the coffin for cardioprotection by remote ischemic conditioning? Probably not! Eur Heart J 37:200–202. https://doi.org/10.1093/eurheartj/ehv606
Heusch G, Rassaf T (2016) Time to give up on cardioprotection? a critical appraisal of clinical studies on ischemic pre-, post-, and remote conditioning. Circ Res 119:676–695. https://doi.org/10.1161/CIRCRESAHA.116.308736
Hibert P, Prunier-Mirebeau D, Beseme O, Chwastyniak M, Tamareille S, Lamon D, Furber A, Pinet F, Prunier F (2013) Apolipoprotein a-I is a potential mediator of remote ischemic preconditioning. PLoS One 8:e77211. https://doi.org/10.1371/journal.pone.0077211
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87:1409–1439. https://doi.org/10.1152/physrev.00034.2006
Ibáñez B, Heusch G, Ovize M, Van de Werf F (2015) Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 65:1454–1471. https://doi.org/10.1016/j.jacc.2015.02.032
Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, Caforio ALP, Crea F, Goudevenos JA, Halvorsen S, Hindricks G, Kastrati A, Lenzen MJ, Prescott E, Roffi M, Valgimigli M, Varenhorst C, Vranckx P, Widimský P, ESC Scientific Document Group (2018) 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 39:119–177. https://doi.org/10.1093/eurheartj/ehx393
Ibanez B, Macaya C, Sanchez-Brunete V, Pizarro G, Fernandez-Friera L, Mateos A, Fernandez-Ortiz A, Garcia-Ruiz JM, Garcia-Alvarez A, Iniguez A, Jimenez-Borreguero J, Lopez-Romero P, Fernandez-Jimenez R, Goicolea J, Ruiz-Mateos B, Bastante T, Arias M, Iglesias-Vazquez JA, Rodriguez MD, Escalera N, Acebal C, Cabrera JA, Valenciano J, Perez de Prado A, Fernandez-Campos MJ, Casado I, Garcia-Rubira JC, Garcia-Prieto J, Sanz-Rosa D, Cuellas C, Hernandez-Antolin R, Albarran A, Fernandez-Vazquez F, de la Torre-Hernandez JM, Pocock S, Sanz G, Fuster V (2013) Effect of Early Metoprolol on Infarct Size in ST-Segment-Elevation Myocardial Infarction Patients Undergoing Primary Percutaneous Coronary Intervention: the Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) Trial. Circulation 128:1495–1503. https://doi.org/10.1161/CIRCULATIONAHA.113.003653
Iversen HH, Celsing F, Leone AM, Gustafsson LE, Wiklund NP (1997) Nerve-induced release of nitric oxide in the rabbit gastrointestinal tract as measured by in vivo microdialysis. Br J Pharmacol 120:702–706. https://doi.org/10.1038/sj.bjp.0700967
Jalowy A, Schulz R, Heusch G (1999) AT1 receptor blockade in experimental myocardial ischemia/reperfusion. J Am Soc Nephrol 10(Suppl 11):S129–S136. https://doi.org/10.1007/s003950050226
Jensen RV, Støttrup NB, Kristiansen SB, Bøtker HE (2012) Release of a humoral circulating cardioprotective factor by remote ischemic preconditioning is dependent on preserved neural pathways in diabetic patients. Basic Res Cardiol 107:285. https://doi.org/10.1007/s00395-012-0285-1
Jones WK, Fan GC, Liao S, Zhang JM, Wang Y, Weintraub NL, Kranias EG, Schultz JE, Lorenz J, Ren X (2009) Peripheral nociception associated with surgical incision elicits remote nonischemic cardioprotection via neurogenic activation of protein kinase C signaling. Circulation 120:S1–S9. https://doi.org/10.1161/CIRCULATIONAHA.108.843938
José Alburquerque-Béjar J, Barba I, Valls-Lacalle L, Ruiz-Meana M, Pecoraro M, Rodríguez-Sinovas A, García-Dorado D (2017) Remote ischemic conditioning provides humoural cross-species cardioprotection through glycine receptor activation. Cardiovasc Res 113:52–60. https://doi.org/10.1093/cvr/cvw242
Jose Corbalan J, Vatner DE, Vatner SF (2016) Myocardial apoptosis in heart disease: does the emperor have clothes? Basic Res Cardiol 111:31. https://doi.org/10.1007/s00395-016-0549-2
Kakinuma Y, Akiyama T, Okazaki K, Arikawa M, Noguchi T, Sato T (2012) A non-neuronal cardiac cholinergic system plays a protective role in myocardium salvage during ischemic insults. PLoS One 7:e50761. https://doi.org/10.1371/journal.pone.0050761
Kakinuma Y, Akiyama T, Sato T (2009) Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J 276:5111–5125. https://doi.org/10.1111/j.1742-4658.2009.07208.x
Kakinuma Y, Tsuda M, Okazaki K, Akiyama T, Arikawa M, Noguchi T, Sato T (2013) Heart-specific overexpression of choline acetyltransferase gene protects murine heart against ischemia through hypoxia-inducible factor-1α-related defense mechanisms. J Am Heart Assoc 2:e004887. https://doi.org/10.1161/JAHA.112.004887
Kanazawa H, Tseliou E, Malliaras K, Yee K, Dawkins JF, De Couto G, Smith RR, Kreke M, Seinfeld J, Middleton RC, Gallet R, Cheng K, Luthringer D, Valle I, Chowdhury S, Fukuda K, Makkar RR, Marbán L, Marbán E (2015) Cellular postconditioning: allogeneic cardiosphere-derived cells reduce infarct size and attenuate microvascular obstruction when administered after reperfusion in pigs with acute myocardial infarction. Circ Heart Fail 8:322–332. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001484
Katare RG, Ando M, Kakinuma Y, Arikawa M, Handa T, Yamasaki F, Sato T (2009) Vagal nerve stimulation prevents reperfusion injury through inhibition of opening of mitochondrial permeability transition pore independent of the bradycardiac effect. J Thorac Cardiovasc Surg 137:223–231. https://doi.org/10.1016/j.jtcvs.2008.08.020
Katare RG, Ando M, Kakinuma Y, Arikawa M, Yamasaki F, Sato T (2010) Differential regulation of TNF receptors by vagal nerve stimulation protects heart against acute ischemic injury. J Mol Cell Cardiol 49:234–244. https://doi.org/10.1016/j.yjmcc.2010.03.007
Kawada T, Akiyama T, Shimizu S, Kamiya A, Uemura K, Li M, Shirai M, Sugimachi M (2009) Detection of endogenous acetylcholine release during brief ischemia in the rabbit ventricle: a possible trigger for ischemic preconditioning. Life Sci 85:597–601. https://doi.org/10.1016/j.lfs.2009.08.015
Kawada T, Yamazaki T, Akiyama T, Mori H, Inagaki M, Shishido T, Takaki H, Sugimachi M, Sunagawa K (2002) Effects of brief ischaemia on myocardial acetylcholine and noradrenaline levels in anaesthetized cats. Auton Neurosci 95:37–42. https://doi.org/10.1016/S1566-0702(01)00352-6
Kawada T, Yamazaki T, Akiyama T, Sato T, Shishido T, Inagaki M, Takaki H, Sugimachi M, Sunagawa K (2000) Differential acetylcholine release mechanisms in the ischemic and non-ischemic myocardium. J Mol Cell Cardiol 32:405–414. https://doi.org/10.1006/jmcc.1999.1087
Kerendi F, Kin H, Halkos ME, Jiang R, Zatta AJ, Zhao ZQ, Guyton RA, Vinten-Johansen J (2005) Remote postconditioning: brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Basic Res Cardiol 100:404–412. https://doi.org/10.1007/s00395-005-0539-2
Kleinbongard P, Amanakis G, Skyschally A, Heusch G (2018) Reflection of cardioprotection by remote ischemic perconditioning in attenuated ST-segment elevation during ongoing coronary occlusion in pigs: evidence for cardioprotection from ischemic injury. Circ Res 122:1102–1108. https://doi.org/10.1161/CIRCRESAHA.118.312784
Kleinbongard P, Heusch G (2015) Extracellular signalling molecules in the ischaemic/reperfused heart—druggable and translatable for cardioprotection? Br J Pharmacol 172:2010–2025. https://doi.org/10.1111/bph.12902
Kleinbongard P, Neuhäuser M, Thielmann M, Kottenberg E, Peters J, Jakob H, Heusch G (2016) Confounders of cardioprotection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. Cardiology 133:128–133. https://doi.org/10.1159/000441216
Kleinbongard P, Peters J, Jakob H, Heusch G, Thielmann M (2018) Persistent survival benefit from remote ischemic pre-conditioning in patients undergoing coronary artery bypass surgery. J Am Coll Cardiol 71:252–254. https://doi.org/10.1016/j.jacc.2017.10.083
Kottenberg E, Thielmann M, Bergmann L, Heine T, Jakob H, Heusch GPJ (2012) Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol—a clinical trial. Acta Anaesthesiol Scand 56:30–38. https://doi.org/10.1111/j.1399-6576.2011.02585.x
Kristiansen SB, Henning O, Kharbanda RK, Nielsen-Kudsk JE, Schmidt MR, Redington AN, Nielsen TT, Bøtker HE (2005) Remote preconditioning reduces ischemic injury in the explanted heart by a KATP channel-dependent mechanism. Am J Physiol Circ Physiol 288:H1252–H1256. https://doi.org/10.1152/ajpheart.00207.2004
Lambert EA, Thomas CJ, Hemmes R, Eikelis N, Pathak A, Schlaich MP, Lambert GW (2016) Sympathetic nervous response to ischemia–reperfusion injury in humans is altered with remote ischemic preconditioning. Am J Physiol Circ Physiol 311:H364–H370. https://doi.org/10.1152/ajpheart.00369.2016
Lang SC, Elsässer A, Scheler C, Vetter S, Tiefenbacher CP, Kübler W, Katus HA, Vogt AM (2006) Myocardial preconditioning and remote renal preconditioning–identifying a protective factor using proteomic methods? Basic Res Cardiol 101:149–158. https://doi.org/10.1007/s00395-005-0565-0
Lecour S, Bøtker HE, Condorelli G, Davidson SM, Garcia-Dorado D, Engel FB, Ferdinandy P, Heusch G, Madonna R, Ovize M, Ruiz-Meana M, Schulz R, Sluijter JPG, Van Laake LW, Yellon DM, Hausenloy DJ (2014) ESC working group cellular biology of the heart: position paper: improving the preclinical assessment of novel cardioprotective therapies. Cardiovasc Res 104:399–411. https://doi.org/10.1093/cvr/cvu225
Lefer DJ, Bolli R (2011) Development of an NIH consortium for preclinicAl AssESsment of CARdioprotective therapies (CAESAR): a paradigm shift in studies of infarct size limitation. J Cardiovasc Pharmacol Ther 16:332–339. https://doi.org/10.1177/1074248411414155
Li J, Rohailla S, Gelber N, Rutka J, Sabah N, Gladstone RA, Wei C, Hu P, Kharbanda RK, Redington AN (2014) MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol 109:423. https://doi.org/10.1007/s00395-014-0423-z
Lim SY, Yellon DM, Hausenloy DJ (2010) The neural and humoral pathways in remote limb ischemic preconditioning. Basic Res Cardiol 105:651–655. https://doi.org/10.1007/s00395-010-0099-y
Lønborg J, Kelbæk H, Vejlstrup N, Bøtker HE, Kim WY, Holmvang L, Jørgensen E, Helqvist S, Saunamäki K, Terkelsen CJ, Schoos MM, Køber L, Clemmensen P, Treiman M, Engstrøm T (2012) Exenatide reduces final infarct size in patients with ST-segment-elevation myocardial infarction and short-duration of ischemia. Circ Cardiovasc Interv 5:288–295. https://doi.org/10.1161/CIRCINTERVENTIONS.112.968388
Malhotra S, Naggar I, Stewart M, Rosenbaum DM (2011) Neurogenic pathway mediated remote preconditioning protects the brain from transient focal ischemic injury. Brain Res 1386:184–190. https://doi.org/10.1016/j.brainres.2011.02.032
Mastitskaya S, Basalay M, Hosford PS, Ramage AG, Gourine A, Gourine AV (2016) Identifying the source of a humoral factor of remote (pre)conditioning cardioprotection. PLoS One 11:e0150108. https://doi.org/10.1371/journal.pone.0150108
Mastitskaya S, Marina N, Gourine A, Gilbey MP, Spyer KM, Teschemacher AG, Kasparov S, Trapp S, Ackland GL, Gourine AV (2012) Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre-ganglionic neurones. Cardiovasc Res 95:487–494. https://doi.org/10.1093/cvr/cvs212
McClanahan TB, Nao BS, Wolke LJ, Martin BJ, Mertz TE, Gallagher KP (1993) Brief renal occlusion and reperfusion reduces myocardial infarct size in rabbits. FASEB J 7:A118
McCrindle BW, Clarizia NA, Khaikin S, Holtby HM, Manlhiot C, Schwartz SM, Caldarone CA, Coles JG, Van Arsdell GS, Scherer SW, Redington AN (2014) Remote ischemic preconditioning in children undergoing cardiac surgery with cardiopulmonary bypass: a single-center double-blinded randomized trial. J Am Heart Assoc 3:e000964–e000964. https://doi.org/10.1161/JAHA.114.000964
Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C, Coburn M, Schaelte G, Böning A, Niemann B, Roesner J, Kletzin F, Strouhal U, Reyher C, Laufenberg-Feldmann R, Ferner M, Brandes IF, Bauer M, Stehr SN, Kortgen A, Wittmann M, Baumgarten G, Meyer-Treschan T, Kienbaum P, Heringlake M, Schön J, Sander M, Treskatsch S, Smul T, Wolwender E, Schilling T, Fuernau G, Hasenclever D, Zacharowski K, RIPHeart Study Collaborators (2015) A multicenter trial of remote ischemic preconditioning for heart surgery. N Engl J Med 373:1397–1407. https://doi.org/10.1056/nejmoa1413579
Mizuno M, Kawada T, Kamiya A, Miyamoto T, Shimizu S, Shishido T, Smith SA, Sugimachi M (2011) Exercise training augments the dynamic heart rate response to vagal but not sympathetic stimulation in rats. Am J Physiol Integr Comp Physiol 300:R969–R977. https://doi.org/10.1152/ajpregu.00768.2010
Nakano A, Liu GS, Heusch G, Downey JM, Cohen MV (2000) Exogenous nitric oxide can trigger a preconditioned state through a free radical mechanism, but endogenous nitric oxide is not a trigger of classical ischemic preconditioning. J Mol Cell Cardiol 32:1159–1167. https://doi.org/10.1006/jmcc.2000.1152
Nederlof R, Weber NC, Juffermans NP, de Mol BAMJ, Hollmann MW, Preckel B, Zuurbier CJ (2017) A randomized trial of remote ischemic preconditioning and control treatment for cardioprotection in sevoflurane-anesthetized CABG patients. BMC Anesthesiol 17:51. https://doi.org/10.1186/s12871-017-0330-6
Oikawa S, Mano A, Takahashi R, Kakinuma Y (2015) Remote ischemic preconditioning with a specialized protocol activates the non-neuronal cardiac cholinergic system and increases ATP content in the heart. Int Immunopharmacol 29:181–184. https://doi.org/10.1016/j.intimp.2015.06.004
Olenchock BA, Moslehi J, Baik AH, Davidson SM, Williams J, Gibson WJ, Chakraborty AA, Pierce KA, Miller CM, Hanse EA, Kelekar A, Sullivan LB, Wagers AJ, Clish CB, Vander Heiden MG, Kaelin WG (2016) EGLN1 inhibition and rerouting of α-ketoglutarate suffice for remote ischemic protection. Cell 164:884–895. https://doi.org/10.1016/j.cell.2016.02.006
Pickard JMJ, Burke N, Davidson SM, Yellon DM (2017) Intrinsic cardiac ganglia and acetylcholine are important in the mechanism of ischaemic preconditioning. Basic Res Cardiol 112:11. https://doi.org/10.1007/s00395-017-0601-x
Pickard JMJ, Davidson SM, Hausenloy DJ, Yellon DM (2016) Co-dependence of the neural and humoral pathways in the mechanism of remote ischemic conditioning. Basic Res Cardiol 111:50. https://doi.org/10.1007/s00395-016-0568-z
Pignataro G, Esposito E, Sirabella R, Vinciguerra A, Cuomo O, Di Renzo G, Annunziato L (2013) nNOS and p-ERK involvement in the neuroprotection exerted by remote postconditioning in rats subjected to transient middle cerebral artery occlusion. Neurobiol Dis 54:105–114. https://doi.org/10.1016/j.nbd.2013.02.008
Prunier F, Angoulvant D, Saint Etienne C, Vermes E, Gilard M, Piot C, Roubille F, Elbaz M, Ovize M, Bière L, Jeanneteau J, Delépine S, Benard T, Abi-Khalil W, Furber A (2014) The RIPOST-MI study, assessing remote ischemic perconditioning alone or in combination with local ischemic postconditioning in ST-segment elevation myocardial infarction. Basic Res Cardiol 109:400. https://doi.org/10.1007/s00395-013-0400-y
Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P (1993) Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87:893–899. https://doi.org/10.1161/01.cir.87.3.893
Pugsley MK (2002) The diverse molecular mechanisms responsible for the actions of opioids on the cardiovascular system. Pharmacol Ther 93:51–75. https://doi.org/10.1016/S0163-7258(02)00165-1
Quindry JC (2017) Exercise-induced cardioprotection and the therapeutic potential of RIPC. J Cardiovasc Pharmacol Ther 22:397–403. https://doi.org/10.1177/1074248417715005
Rana OR, Schauerte P, Kluttig R, Schröder JW, Koenen RR, Weber C, Nolte KW, Weis J, Hoffmann R, Marx N, Saygili E (2010) Acetylcholine as an age-dependent non-neuronal source in the heart. Auton Neurosci 156:82–89. https://doi.org/10.1016/j.autneu.2010.04.011
Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M (2014) Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114:1601–1610. https://doi.org/10.1161/CIRCRESAHA.114.303822
Ravassa S, Zudaire A, Diez J (2012) GLP-1 and cardioprotection: from bench to bedside. Cardiovasc Res 94:316–323. https://doi.org/10.1093/cvr/cvs123
Redington KL, Disenhouse T, Strantzas SC, Gladstone R, Wei C, Tropak MB, Dai X, Manlhiot C, Li J, Redington AN (2012) Remote cardioprotection by direct peripheral nerve stimulation and topical capsaicin is mediated by circulating humoral factors. Basic Res Cardiol 107:241. https://doi.org/10.1007/s00395-011-0241-5
Rehni AK, Singh N, Jaggi AS (2007) Possible involvement of insulin, endogenous opioids and calcitonin gene-related peptide in remote ischaemic preconditioning of the brain. Yakugaku Zasshi 127:1013–1020. https://doi.org/10.1248/yakushi.127.1013
Ren C, Yan Z, Wei D, Gao X, Chen X, Zhao H (2009) Limb remote ischemic postconditioning protects against focal ischemia in rats. Brain Res 1288:88–94. https://doi.org/10.1016/j.brainres.2009.07.029
Rocca AS, Brubaker PL (1999) Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology 140:1687–1694. https://doi.org/10.1210/endo.140.4.6643
Roubille F, Franck-Miclo A, Covinhes A, Lafont C, Cransac F, Combes S, Vincent A, Fontanaud P, Sportouch-Dukhan C, Redt-Clouet C, Nargeot J, Piot C, Barrère-Lemaire S (2011) Delayed postconditioning in the mouse heart in vivo. Circulation 124:1330–1336. https://doi.org/10.1161/CIRCULATIONAHA.111.031864
Schmidt MR, Smerup M, Konstantinov IE, Shimizu M, Li J, Cheung M, White PA, Kristiansen SB, Sorensen K, Dzavik V, Redington AN, Kharbanda RK (2007) Intermittent peripheral tissue ischemia during coronary ischemia reduces myocardial infarction through a KATP-dependent mechanism: first demonstration of remote ischemic perconditioning. Am J Physiol Circ Physiol 292:H1883–H1890. https://doi.org/10.1152/ajpheart.00617.2006
Schmidt MR, Støttrup NB, Michelsen MM, Contractor H, Sørensen KE, Kharbanda RK, Redington AN, Bøtker HE (2014) Remote ischemic preconditioning impairs ventricular function and increases infarct size after prolonged ischemia in the isolated neonatal rabbit heart. J Thorac Cardiovasc Surg 147:1049–1055. https://doi.org/10.1016/j.jtcvs.2013.05.022
Schoemaker RG, van Heijningen CL (2000) Bradykinin mediates cardiac preconditioning at a distance. Am J Physiol Circ Physiol 278:H1571–H1576. https://doi.org/10.1152/ajpheart.2000.278.5.H1571
Schulte G, Sommerschild H, Yang J, Tokuno S, Goiny M, Lovdahl C, Johansson B, Fredholm BB, Valen G (2004) Adenosine A1 receptors are necessary for protection of the murine heart by remote, delayed adaptation to ischaemia. Acta Physiol Scand 182:133–143. https://doi.org/10.1111/j.1365-201X.2004.01350.x
Sharma V, Marsh R, Cunniffe B, Cardinale M, Yellon DM, Davidson SM (2015) From protecting the heart to improving athletic performance—the benefits of local and remote ischaemic preconditioning. Cardiovasc Drugs Ther 29:573–588. https://doi.org/10.1007/s10557-015-6621-6
Shimizu M, Tropak M, Diaz RJ, Suto F, Surendra H, Kuzmin E, Li J, Gross G, Wilson GJ, Callahan J, Redington AN (2009) Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection. Clin Sci (Lond) 117:191–200. https://doi.org/10.1042/CS20080523
Shinlapawittayatorn K, Chinda K, Palee S, Surinkaew S, Kumfu S, Kumphune S, Chattipakorn S, KenKnight BH, Chattipakorn N (2014) Vagus nerve stimulation initiated late during ischemia, but not reperfusion, exerts cardioprotection via amelioration of cardiac mitochondrial dysfunction. Hear Rhythm 11:2278–2287. https://doi.org/10.1016/j.hrthm.2014.08.001
Shinlapawittayatorn K, Chinda K, Palee S, Surinkaew S, Thunsiri K, Weerateerangkul P, Chattipakorn S, KenKnight BH, Chattipakorn N (2013) Low-amplitude, left vagus nerve stimulation significantly attenuates ventricular dysfunction and infarct size through prevention of mitochondrial dysfunction during acute ischemia–reperfusion injury. Hear Rhythm 10:1700–1707. https://doi.org/10.1016/j.hrthm.2013.08.009
Sloth AD, Schmidt MR, Munk K, Kharbanda RK, Redington AN, Schmidt M, Pedersen L, Sørensen HT, Bøtker HE, CONDI Investigators (2014) Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J 35:168–175. https://doi.org/10.1093/eurheartj/eht369
Sloth AD, Schmidt MR, Munk K, Schmidt M, Pedersen L, Toft Sorensen H, Botker HE, Bottcher M, Kaltoft A, Terkelsen C, Andersen N, Hansen T, Trautner S, Lassen J, Christiansen E, Krusell L, Kristensen S, Thuesen L, Nielsen S, Rehling M, Nielsen T (2015) Impact of cardiovascular risk factors and medication use on the efficacy of remote ischaemic conditioning: post hoc subgroup analysis of a randomised controlled trial. BMJ Open 5:e006923–e006923. https://doi.org/10.1136/bmjopen-2014-006923
Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM (2007) Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther 21:227–233. https://doi.org/10.1007/s10557-007-6035-1
Song Y, Shan JG, Xue Z, Wang SY, Xu H, Liu Y, Guo YS, Ren X (2016) Remote postconditioning induced by trauma protects the mouse heart against ischemia reperfusion injury. Involvement of the neural pathway and molecular mechanisms. Cardiovasc Drugs Ther 30:271–280. https://doi.org/10.1007/s10557-016-6661-6
Song Y, Ye YJ, Li PW, Zhao YL, Miao Q, Hou DY, Ren XP (2016) The cardioprotective effects of late-phase remote preconditioning of trauma depends on neurogenic pathways and the activation of PKC and NF-κB (But Not iNOS) in mice. J Cardiovasc Pharmacol Ther 21:310–319. https://doi.org/10.1177/1074248415609435
Steensrud T, Li J, Dai X, Manlhiot C, Kharbanda RK, Tropak M, Redington A (2010) Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine. Am J Physiol Heart Circ Physiol 299:H1598–H1603. https://doi.org/10.1152/ajpheart.00396.2010
Sved AF, Curtis JT (1993) Amino acid neurotransmitters in nucleus tractus solitarius: an in vivo microdialysis study. J Neurochem 61:2089–2098. https://doi.org/10.1111/j.1471-4159.1993.tb07446.x
Talman WT, Robertson SC, Cassell MD (1992) Mechanisms of cardiovascular responses to glycine injected into the dorsal vagal motor nucleus in rat. Hypertension (Dallas, Tex 1979) 19:187–192. https://doi.org/10.1161/01.hyp.19.2_suppl.ii18
Thielmann M, Kottenberg E, Kleinbongard P, Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhäuser M, Peters J, Jakob H, Heusch G (2013) Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-centre randomised, double-blind, controlled trial. Lancet 382:597–604. https://doi.org/10.1016/S0140-6736(13)61450-6
Uitterdijk A, Yetgin T, te Lintel Hekkert M, Sneep S, Krabbendam-Peters I, van Beusekom HMM, Fischer TM, Cornelussen RN, Manintveld OC, Merkus D, Duncker DJ (2015) Vagal nerve stimulation started just prior to reperfusion limits infarct size and no-reflow. Basic Res Cardiol 110:51. https://doi.org/10.1007/s00395-015-0508-3
Verouhis D, Sörensson P, Gourine A, Henareh L, Persson J, Saleh N, Settergren M, Sundqvist M, Tornvall P, Witt N, Böhm F, Pernow J (2016) Effect of remote ischemic conditioning on infarct size in patients with anterior ST-elevation myocardial infarction. Am Heart J 181:66–73. https://doi.org/10.1016/j.ahj.2016.08.004
Walter T, Helber U, Bail D, Heller W, Hoffmeister HM (2002) Influence of ACE inhibition on myocardial damage, the Kallikrein–Kinin system and hemostasis during cardiopulmonary bypass surgery. Thorac Cardiovasc Surg 50:150–154. https://doi.org/10.1055/s-2002-32409
Wang X, Huang ZG, Dergacheva O, Bouairi E, Gorini C, Stephens C, Andresen MC, Mendelowitz D (2005) Ketamine inhibits inspiratory-evoked gamma-aminobutyric acid and glycine neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Anesthesiology 103:353–359. https://doi.org/10.1097/00000542-200508000-00019
Wang X, Huang ZG, Gold A, Bouairi E, Evans C, Andresen MC, Mendelowitz D (2004) Propofol modulates gamma-aminobutyric acid-mediated inhibitory neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Anesthesiology 100:1198–1205. https://doi.org/10.1097/00000542-200405000-00023
Wei D, Ren C, Chen X, Zhao H (2012) The chronic protective effects of limb remote preconditioning and the underlying mechanisms involved in inflammatory factors in rat stroke. PLoS One 7:e30892. https://doi.org/10.1371/journal.pone.0030892
Weinbrenner C, Nelles M, Herzog N, Sárváry L, Strasser RH (2002) Remote preconditioning by infrarenal occlusion of the aorta protects the heart from infarction: a newly identified non-neuronal but PKC-dependent pathway. Cardiovasc Res 55:590–601. https://doi.org/10.1016/S0008-6363(02)00446-7
White SK, Frohlich GM, Sado DM, Maestrini V, Fontana M, Treibel TA, Tehrani S, Flett AS, Meier P, Ariti C, Davies JR, Moon JC, Yellon DM, Hausenloy DJ (2015) remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc Interv 8:178–188. https://doi.org/10.1016/j.jcin.2014.05.015
Wolfrum S, Nienstedt J, Heidbreder M, Schneider K, Dominiak P, Dendorfer A (2005) Calcitonin gene related peptide mediates cardioprotection by remote preconditioning. Regul Pept 127:217–224. https://doi.org/10.1016/j.regpep.2004.12.008
Wong GT, Lu Y, Mei B, Xia Z, Irwin MG (2012) Cardioprotection from remote preconditioning involves spinal opioid receptor activation. Life Sci 91:860–865. https://doi.org/10.1016/j.lfs.2012.08.037
Woo JS, Kim W, Ha SJ, Kim JB, Kim S-J, Kim WS, Seon HJ, Kim KS (2013) Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol 33:2252–2260. https://doi.org/10.1161/ATVBAHA.113.301586
Xiao Y, Hafeez A, Zhang Y, Liu S, Kong Q, Duan Y, Luo Y, Ding Y, Shi H, Ji X (2015) Neuroprotection by peripheral nerve electrical stimulation and remote postconditioning against acute experimental ischaemic stroke. Neurol Res 37:447–453. https://doi.org/10.1179/1743132815Y.0000000032
Yamamoto S, Kawana S, Miyamoto A, Ohshika H, Namiki A (1999) Propofol-induced depression of cultured rat ventricular myocytes is related to the M2-acetylcholine receptor-NO-cGMP signaling pathway. Anesthesiology 91:1712–1719. https://doi.org/10.1097/00000542-199912000-00024
Yang XM, Downey JM, Cohen MV, Housley NA, Alvarez DF, Audia JP (2017) The highly selective caspase-1 inhibitor VX-765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor. J Cardiovasc Pharmacol Ther 22:574–578. https://doi.org/10.1177/1074248417702890
Yellon DM, He Z, Khambata R, Ahluwalia A, Davidson SM (2018) The GTN patch: a simple and effective new approach to cardioprotection? Basic Res Cardiol 113:20. https://doi.org/10.1007/s00395-018-0681-2
Yu C, Liu J (2014) Protective effect of ischemia preconditioning of lower limbs on brain ischemia–reperfusion injury via neural pathways. Sichuan Da Xue Xue Bao Yi Xue Ban 45:216–220
Yu L, Huang B, Po SS, Tan T, Wang M, Zhou L, Meng G, Yuan S, Zhou X, Li X, Wang Z, Wang S, Jiang H (2017) Low-level tragus stimulation for the treatment of ischemia and reperfusion injury in patients with ST-segment elevation myocardial infarction: a proof-of-concept study. JACC Cardiovasc Interv 10:1511–1520. https://doi.org/10.1016/j.jcin.2017.04.036
Zangrillo A, Musu M, Greco T, Di Prima AL, Matteazzi A, Testa V, Nardelli P, Febres D, Monaco F, Calabrò MG, Ma J, Finco G, Landoni G (2015) Additive effect on survival of anaesthetic cardiac protection and remote ischemic preconditioning in cardiac surgery: a bayesian network meta-analysis of randomized trials. PLoS One 10:e0134264. https://doi.org/10.1371/journal.pone.0134264
Zhao JJ, Xiao H, Zhao WB, Zhang XP, Xiang Y, Ye ZJ, Mo M-M, Peng XT, Wei L (2018) Remote ischemic postconditioning for ischemic stroke: a systematic review and meta-analysis of randomized controlled trials. Chin Med J (Engl) 131:956–965. https://doi.org/10.4103/0366-6999.229892
Zhou C, Liu Y, Yao Y, Zhou S, Fang N, Wang W, Li L (2013) β-blockers and volatile anesthetics may attenuate cardioprotection by remote preconditioning in adult cardiac surgery: a meta-analysis of 15 randomized trials. J Cardiothorac Vasc Anesth 27:305–311. https://doi.org/10.1053/j.jvca.2012.09.028
Zhou Y, Fathali N, Lekic T, Ostrowski RP, Chen C, Martin RD, Tang J, Zhang JH (2011) Remote limb ischemic postconditioning protects against neonatal hypoxic–ischemic brain injury in rat pups by the opioid receptor/akt pathway. Stroke 42:439–444. https://doi.org/10.1161/STROKEAHA.110.592162
Zhou ZH, Peng J, Ye F, Li NS, Deng HW, Li YJ (2002) Delayed cardioprotection induced by nitroglycerin is mediated by alpha-calcitonin gene-related peptide. Naunyn Schmiedebergs Arch Pharmacol 365:253–259. https://doi.org/10.1007/s00210-002-0537-y
Acknowledgements
We acknowledge funding support from the NIHR Biomedical Research Council (SD), the British Heart Foundation PG/15/52/31598 (SD), and the Hatter Foundation (MB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Basalay, M.V., Davidson, S.M., Gourine, A.V. et al. Neural mechanisms in remote ischaemic conditioning in the heart and brain: mechanistic and translational aspects. Basic Res Cardiol 113, 25 (2018). https://doi.org/10.1007/s00395-018-0684-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-018-0684-z