
Abstract
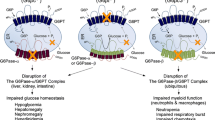
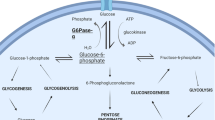
Thirty-three years after Von Gierke described the first patient with glycogen storage disease type 1 (GSD1) in 1929, the Coris detected glucose-6-phosphatase (G6Pase) deficiency. The first mutation of this enzyme was identified 41 years later and subsequently the gene was mapped to chromosome 17q21, its enzyme topology defined, a nine transmembrane helical model suggested, an enzyme deficient knockout mouse created and by infusing an adenoviral vector associated murine G6Pase gene, correction of the clinical and laboratory abnormalities was observed. A similar successful gene transfer has been performed in enzyme deficient canine puppies. To explain the function of the G6Pase complex, a multicomponent translocase catalytic model has been proposed in which different transporters shuttle glucose-6-phosphate (G6P), inorganic phosphate (Pi) and glucose across the microsomal membrane. It was suggested that GSD1b patients suffered from a G6P transporter (G6PT) defect and the first mutation in the G6PT gene subsequently recognised. The gene mapped to chromosome 11q23 and its structural organisation was defined which showed a close functional linkage between G6PT and hydrolysis. Nordlie identified the first patient with Pi transport deficiency (GSD1c). However putative GSD1c and 1d patients based on kinetic studies were found to harbour mutations in the G6PT gene so that GSD1 patients are presently divided into 1a and non-1a. G6PT deficient patients suffer from numerical and functional leucocyte defects. A mRNA leucocyte G6PT deficiency has been suggested to account for the glucose phosphorylation and subsequent calcium sequestration defects observed in theses cells. Inflammatory bowel disease which occurs frequently in GSD non-1a patients has been related to their leucocyte abnormalities. Dietary management of GSD1 patients, designed to maintain a normal blood glucose level can be achieved during the night by nocturnal gastric infusions of glucose-containing solution or by the administration of uncooked cornstarch around the clock or by a combination of both. Both therapeutic modalities, if conducted in a meticulous manner, have a major impact on the quality of life, prevention of complications and subsequent prognosis. Open questions relate to the source of endogenous glucose production in GSD1 patients which increases as a function of age from 50% to 100% of normal, concomitant with an improvement in the patients fasting tolerance. Several complications, the nature of which is incompletely understood, tend to occur after the first decade: Liver adenomata with a small risk of transforming into hepatoma, progressive renal disease, which may be related to the hyperlipidaemia observed in this disease, often leading to end stage renal failure, osteopenia apparently based on high bone turnover, growth retardation and delayed puberty.Conclusion: this review highlights the present knowledge of glycogen storage disease type 1 and subtypes, discussing unsolved questions, which reflect the limitation of our knowledge in the understanding of this intriguing group of diseases.
Similar content being viewed by others

Abbreviations
- BMD :
-
bone mineral density
- G6P :
-
glucose-6-phosphate
- G6Pase :
-
glucose-6-phosphatase
- G6PT :
-
glucose-6-phosphate transporter protein
- GSD :
-
glycogen storage disease
- IBD :
-
inflammatory bowel disease
- Pi :
-
inorganic phosphate
References
Alaupoviv P, Fernandes J (1985) The serum apoliporotein profiles of patients with glucose-6-phosphatase deficiency. Pediatr Res 19: 380–384
Alshak NS, Cocjin J, Podesta L, van de Velde R, Makowka L, Rosenthal P, Geller SA (1994) hepatocellular adenoma in glycogen storage disease type IV. Arch Pathol Lab Med 118: 88–91
Ament ME, Ochs MD (1973) Gastrointestinal manifestations of chronic granulomatous disease. N Engl J Med 288: 382–387
Annabi B, Hiraiwa H, Mansfield BC, Lei KJ, Ubagai T et al (1998) The gene for glycogen storage disease type 1b maps to chromosome 11q23. Am J Hum Genet 62: 400–405
Arion WJ, Lange AJ, Walls HE, Ballas LM (1980) Evidence for the participation of independent translocases for phosphate and glucose-6-phosphate in the microsomal glucose-6-phosphatase system. J Biol Chem 255: 10396–10406
Banhegyi G, Marcolongo P, Fulceri R, Hinds C, Burchell A, Benedetti A (1997) demonstration of metabolically active glucose-6-phosphate pool in the lumen of liver microsome vesicles. J Biol Chem 272: 13584–135490
Bashan N, Potashnik R, Peist A, Peleg N, Moran A, Moses SW (1993) Deficient glucose phosphorylation as a common denominator and its relation to abnormal leucocyte function in glycogen storage disease type 1b patients. Eur J Pediatr 152[Suppl 1]: S44-S48
Beaty RM, Jackson M, Kishnani PS, Faulkner E, Van Camp S et al (2000) Gene therapy with adenoassociated virus (AVV) vectors in canine glycogen storage disease type 1a. Am J Hum Genet 67[Suppl 2]: 432
Beaudet AL, Anderson DC, Michels VV, Arion WJ, Lange AJ (1980) Neutropenia and impaired neutrophil migration in type 1b glycoegn storage disease. J Pediatr 97: 906–910
Bianchi L (1993) Glycogen storage disease 1 and hepatocellular tumours. Eur J Pediatr 152[Suppl 1]: S63-S70
Burchell A (1998) A reevaluation of GLUT 7. Biochem J 331: 973
Burchell A, Waddell I (1991) The molecular basis of the hepatic microsomal glucose-6-phosphatase system. Biochim Biophys Acta 1092: 129–137
Burchell A, Waddell I, Zomerschoe AG, Voice MW (1992) Cloning and expression of hepatic microsomal glucose transport protein. Comparison with liver plasma glucose transport protein GLUT 2. Biochem J 286: 173–177
Chen YT (1991) Type 1 glycogen storage disease: kidney involvement, pathogenesis and treatment. Pediatr Nephrol 5: 71–76
Chen YT, Cornblath M, Sidbury JB (1984) Cornstarch therapy in type 1 glycogen storage disease. N Engl J Med 310: 171–175
Chen YT, Coleman RA, Scheinman JI, Kolbeck PC, Sidbury JB (1988) Renal disease in type 1 glycogen storage disease. N Engl J Med 318: 7–11
Chen YT, Bazarre C, Lee MM, Sidbury JB, Coleman RA (1993) Type 1 glycogen storage disease: nine years of management with cornstarch. Eur J Pediatr 152[Suppl 1]: S56-S59
Cori GT, Cori CF (1952) Glucose-6-phosphatase in the liver in glycogen storage disease. J Biol Chem 199: 661–667
Emmett M, Narias RG (1978) Renal transplantation in type 1 glycogenosis: failure to improve glucose metabolism. JAMA 239: 1642–1644
Fata F, Myers P, Addeo J, Grimberg M, Nawabi I, Cappel MS (1997) Cyclic neutropenia in Crohn's ileocolitis: efficacy of granulocyte colony-stimulating factor. J Clin Gastroenterol 24: 253–256
Fernandes J (1974) The effect of disaccharides on the hyperlacticacidaemia of glucose-6-phosphatase deficient children. Acta Paediatr Scand 63: 695–698
Gerin I, Veiga-da-Cunha M, Achoury Y, Collet JF, Van Schaftingen E (1997) Sequence of a putative glucose-6-phosphate translocase mutated in glycogen storage disease type 1b. FEBS Lett 419: 235–238
Gitzelmann R, Bosshard NU (1993) Defective neutrophil and monocyte functions in glycogen storage disease type 1b: a literature review. Eur J Pediatr 152[Suppl 1]: S33-S38
Goans RE, Weiss GH, Viera NE, Sidbury JB, Abrams SA, Yergey AL (1996) Calcium kinetics in glycogen storage disease type 1a. Calcif Tissue Int 59: 449–453.
Green H, Slonim AE, Burr IM, Moran JR (1976) Continuous nocturnal intragastric feeding for the management of type 1 glycogen storage disease. N Engl J med 294: 423–425
Guillam MT, Burcelin R, Thorens B (1998) Normal hepatic glucose production in the absence of GLUT 2 reveals an alternative pathway for glucose release from hepatocytes. proc Natl Acad Sci USA 95: 12317–12321
Havel RJ, Balasse EO, Williams HE, Kane JP, Segel N (1969) Splanchnic metabolism in Von Gierke's disease (glycogenosis type 1). Trans Assoc Am Phys 82: 305–323
Hers HG, Van Hoof F, De Barsy T (1989) Glycogen storage disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease. McGraw-Hill, New York, pp 425–452
Hiraiwa H, Pan CJ, Baochuan L, Moses SW, Chou YC (1999) Inactivation of the glucose-6-phosphate transporter causes glycogen storage disease type 1b. J Biol Chem 274: 5532–5536
Howell RR, Stevenson RE, Ben-Menahem Y, Phyliky RL, Berry H (1976) Hepatic adenomata with type 1 glycogen storage disease. JAMA 236: 1481–1484
Huismans D, Rake JP, Visser G, Piers DA, Smith GPA (1997) Decreased bone mineral density in glycogen storage disease type 1. J Inherit Metab Dis 20[Suppl 1]: 81
Ihara K, Nomura A, Hikino S, Takada H, Hara T (2000) Quantitative analysis of glycose-6-phosphate translocase gene expression in various human tissues and haemopoetic progenitor cells. J Inherit Metab Dis 23: 583–592
Kalhan SC, Gilfillan C, Tserng KY, Savin SM (1982) Glucose production in type 1 glycogen storage disease. J Pediatr 101: 159–160
Keane WF (1996) Lipids and progressive renal failure. Wien Klin Wochenschr 14: 420–424
Keane WF (2000) The role of lipids in renal disease: future challenges. Kidney Int 57: S27-S31
Kelsch RC, Oliver WJ (1969) Studies on dietary correction of metabolic abnormalities in hepatorenal glycogenesis. Pediatr Res 3: 160–170
Kilpatrick I, Garty B-Z, Lundquist KF, Hunter K, Stanley CA, Baker L, Douglas SD, Korchak HM (1990) Impaired metabolic function and signaling defects in phagocytic cells in glycogen storage disease type 1b. J Clin Invest 86: 196–202
Lee PJ, Patel JS, Fewtrell M, Leonard JV, Bishop NJ (1995) Bone mineralisation in type 1 glycogen storage disease. Eur J Pediatr 54: 483–487
Leese T, Farges O, Bismuth H (1988) Liver cell adenomas: a 12-year surgical experience from a specialist hepatobiliary unit. Ann Surg 205: 558–561
Leffert HL, Koch KS, Moran T, Rubalcava B (1979) Hormonal control of liver regeneration. Gastroenterology 760: 1470–1482
Lei KJ, Chen YT, Chen H, Wong LJC, Liu JL et al (1995) Genetic basis of glycogen storage disease type 1a: prevalent mutations at the glucose-6-phosphatase locus. Am J Hum Genet 57: 766–771
Lei KJ, Chen H, Pan CJ, Ward JM, Mosinger B Jr et al (1996) Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type 1a mouse. Nat Genet 13: 203–209
Levy E, Thibault LA, Roy CC, Bendayan M, Lepage G (1988) Circulating lipids and lipoproteins in glycogen storage disease type 1 with nocturnal gastric feeding. J Lipid Res 29: 215–226
Limmer J, Fleig WE, Leupold D, Bittner R, Ditschuneit H, Beger HG (1988) Hepatocellular carcinoma in type 1 glycogen storage disease. Hepatology 8: 531–537
Lin B, Hiraiwa H, Pan CJ, Nordlie RC, Chou JY (1999) Type 1c glycogen storage disease is not caused by mutations in the glucose-6-phosphate transporter gene. Hum Genet 105: 515–517
Mason DH, Anderson DH (1955) Glycogen disease of the liver (von Gierke's disease) with hepatoma: case report with metabolic studies. Pediatrics 16: 785–799
Narisawa K, Igarashi Y, Otomo H, Tada K (1978) A new variant of glycogen storage disease type 1 probably due to a defect in the glucose-6-phosphate transport system. Biochem Biophys Res Commun 83: 1360–1364
Nordlie RC, Sukalski KA, Munoz JM, Baldwin JJ (1983) Type 1c, a novel glycogenosis. Underlying mechanism. J Biol Chem 258: 9739–9744
Nordlie RC, Scott HM, Waddell I, Hume R, Burchell A (1992) Analysis of human hepatic microsomal glucose-6-phosphatase in clinical conditions where the T2 pyrophosphate/phosphate transport protein is absent. Biochem J 281: 859–863
Oda H, Keane WF (1999) Recent advances in statins and the kidney. Kidney Int 56[Suppl 7]: S2-S5
Ozen H, Ciliv G, Kocak N, Satik IN, Yuce A, Gorokan F (2000) Short term effect of captopril on microalbuminuria in children with glycogen storage disease type 1a. J Inherit Metab Dis 23: 459–463
Pan CJ, Lei KJ, Annabi B, Hemrika W, Chou JY (1998) Transmembrane topology of glucose-6-phosphatase. J Biol Chem 273: 6144–6148
Parker P, Burr I, Slonim A, Gishan FK, Green H (1981) Regression of hepatic adenoma in type 1a glycogen storage disease with dietary therapy. Gastroenterology 81: 534–536
Powell RC, Wentworth SM, Brandt JK (1981) Endogenous glucose production in type 1 glycogen storage disease. Metabolism 30: 443–450
Roe TF, Thomas DW, Gisanz V, Isaacs H Jr, Atkinson JB (1986) Inflammatory bowel disease in glycogen storage disease type 1b. J Pediatr 109: 55–59
Roe TF, Coates TD, Thomas DW, Miller JH, Gilsanz V (1992) Brief report: treatment of chronic inflammatory bowel disease in glycogen storage disease type 1b with colony stimulating factors. N Engl Med 326: 1666–1669
Rother KI, Schwenk WF (1995) Glucose production in glycogen storage disease 1 is not associated with increased cycling through hepatic glycogen. Am J Physiol 269: E774-E778
Sadeghi-Nejad A, Hochmann H, Senior B (1974) Studies in type 1 glycogenosis of the liver. J Pediatr 85: 49–54
Schoenheimer R (1929) Über eine eigenartige Störung des Kohlenhydratstoffwechsels. Z. Physiol Chem 182: 149–151
Schulze H, Nolte B, Kannler R (1986) Evidence for changes in the conformational status of rat liver microsomal glucose-6-phosphate: phosphohydrolase during detergent dependent modification. J Biol Chem 261: 16571–16578
Selby R, Starzl TE, Yunis E, Todo S, Tzakis AG, Brown BI, Kendall RS (1993) Liver transplantation for type 1 and type IV glycogen storage disease. Eur J Pediatr 152[Suppl 1]: S71-S76
Senior B, Loridan L (1968) Studies of liver glycogenosis, with particular reference to the metabolism of intravenously administered glycerol. N Engl J med 279: 958–970
Shanahan F, Bernstein CN (1993) Odd forms of inflammatory bowel disease: what can they tell us?, Gastroenterology 104: 327–329
Smith GPA (1993) results of the European Study on Glycogen Storage Type 1. Eur J Pediatr 152 [Suppl 1]: S52-S55
Srivastava T, Alon US (1999) Biphosphonates: from grand-parents to gradchildren. Clin Pediatr 38: 687–702
Starzl TF, Putman CV, Porter KA, Halgrimson CG, Corman J et al (1973) Portal diversion for the treatment of glycogen storage disease in humans. Ann Surg 178: 525–539
Steim H, Zollinger HU (1967) Tödliche Schrumpfniere bei Glykogenspeicherkrankheit typ Von Gierke. Klin Wochenschr 45: 295–299
Talente GM, Coleman GA, Alter C, Baker L, Brown BI, Cannon RA et al (1994) Glycogen storage disease in adults. Ann Intern Med 120: 218–226
Tsalikian E, Simmons P, Gerich JE, Howard C, Haymond MW (1984) Glucose production and utilisation in children with glycogen storage disease type 1. Am J Physiol 247: E513-E519
Van Hoof F, Hue L, De Barsy T, Jacquemin P, Devos P, Hers HG (1972) Glycogen storage diseases. Biochimie 54: 745–752
Veiga-da-Cunha M, Gerin M, Chen YT, Lee PJ, Leonard JV, Maire I, Wendel U, Vikkula M, Van Schaftingen E (1999) The putative glucose-6-phosphatase gene is mutated essentially in all cases of glycogen storage disease type 1 non-a. Eur J Hum Genet 7: 717–723
Verhoeven AJ, Visser G, Zwieten RV, Gruszczynska B, Pol-The BT, Smith GPA (1999) A convenient diagnostic function test of peripheral blood neutrophils in glycogen storage disease 1b. Pediatr Res 45: 881–885
Visser G, Rake JP, Fernandes J, Labrune P, Leonard JV, Moses S, Ullrich K, Smit GPA (2000) Neutropenia, neutrophil dysfunction and inflammatory bowel disease in glycogen storage disease type 1b. J Pediatr 137: 187–191
Von Gierke E (1929) Hepato-nephro-megalia-glycogenica (Glykogenspeicherkrankheit der Leber und Nieren). Beitr Pathol Anat 82: 497–513
Waddell I, Scott H, Grant A, Burchell A (1991) Identification and characterisation of a rat microsomal glucose transport protein. Glucose-6-phosphatase T3? Biochem J 285: 173–177
Wallace JL, McKnight W, Asfaha S, Lin DY (1998) Reduction od acute and reactive colitis in rats by an inhibitor of neutrophil activation. Am J Physiol 274: G802-G808
Weinstein DA, Somer MJG, Wolsdorf JI (2000) Inverse correlation between urinary citrate excretion and age in type 1a glycogen storage disease: pathophysiology and implications for renal complications. Pediatr Res 47[Suppl 454]: A2686
Wolsforf JI, Crigler JF Jr (1999) Effect of continuous glucose therapy begun in infancy on the long term clinical course of patients with type 1 glycogen storage disease. J Pediatr Gastroenterol Nutr 29: 136–143.
Zingone A, Hiraiwa H, Pan CJ, Lin B, Chen H, Ward JM, Chou YC (2000) Correction of glycogen storage disease type 1a in a mouse model by gene therapy. J Biol Chem 275: 828–832
Author information
Authors and Affiliations
Additional information
Published online: 23 August 2002
Rights and permissions
About this article

Cite this article
Moses, S.W. Historical highlights and unsolved problems in glycogen storage disease type 1. Eur J Pediatr 161, S2–S9 (2002). https://doi.org/10.1007/BF02679988
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF02679988