
Abstract
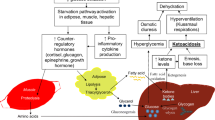
Children with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMG-CoA-LD; McKusick 24645), have inherited two areas of metabolic weakness. Firstly, they are unable to metabolize fully the carbon skeleton of leucine, and secondly, they cannot make ketone bodies in response to prolonged fasting. In the first year of life infants with HMG-CoA-LD run a high risk of developing severe hypoglycaemia which can lead to death if prompt intervention does not occur. The metabolic crisis develops when the infant is first introduced to dietary protein soon after birth, or later, when a reduced intake of glucose, often during a viral infection, results in a drain on the infant's circulating glucose levels. However, where diets are adequately adjusted to limit protein and fat intake, the metabolic handicaps of individuals with HMG-CoA-LD are not exposed and they are virtually symptomless. As children with HMG-CoA-LD grow older the incidence of hypoglycaemic attacks diminishes and they usually develop normally. This article reviews literature on cases of HMG-CoA-LD and interprets data on altered metabolism in these children.
Similar content being viewed by others


References
Atkin, B. M., Buist, N. R. M., Utter, M. F., Leiter, A. B. and Banker, B. Q. Pyruvate carboxylase deficiency and lactic acidosis in a retarded child without Leigh's disease.Pediatr. Res. 13 (1979) 109–116
Cahill, G. F., Owen, O. E. and Morgan, A. P. The consumption of fuels during prolonged starvation.Adv. Enzyme Regul. 6 (1968) 143–150
Chalmers, R. A., Stacey, T. E., Tracey, B. M., De Sousa, C., Roe, C. R., Millington, D. S. and Hoppell, C. L.l-Carnitine insufficiency in disorders of organic acid metabolism: Response tol-carnitine by patients with methylmalonic aciduria and 3-hydroxy-3-methylglutaric aciduria.J. Inher. Metab. Dis. 7, Suppl. 2 (1984) 109–110
Divry, P., Rolland, M. O., Teyssier, J., Cotte, J., Formosinho Fernandes, M. C., Tavares De Almeida, I. and Da Silveira, C. 3-Hydroxy-3-methylglutaric aciduria combined with 3-methylglutaconic aciduria: A new case.J. Inher. Metab. Dis. 4 (1981) 173–174
Duran, M., Ketting, D., Wadman, S. K., Jakobs, C., Schutgens, R. B. H. and Veder, H. A. Organic acid excretion in a patient with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency: Facts and artefacts.Clin. Chim. Acta. 90 (1978) 187–193
Duran, M., Schutgens, R. B. H., Ketel, A., Heymans, H., Berntssen, M. W. J., Ketting, D. and Wadman, S. K. 3-Hydroxy-3-methylglutaryl coenzyme A lyase deficiency: Postnatal management following prenatal diagnosis by analysis of maternal urine.J. Pediatr. 95 (1979) 1004–1007
Faull, K. F., Bolton, P. D., Halpern, B., Hammond, J. and Danks, D. M. The urinary organic acid profile associated with 3-hydroxy-3-methylglutaric aciduria.Clin. Chim. Acta 73 (1976a) 553–559
Faull, K., Bolton, P., Halpern, B., Hammond, J., Danks, D. M., Hähnel, R., Wilkinson, S. P., Wysocki, S. J. and Masters, P. L. Patient with a defect in leucine metabolism.N. Engl. J. Med. 294 (1976b) 1013
Felig, P. Amino acid metabolism in man.Ann. Rev. Biochem. 44 (1975) 933–955
Francois, B., Bachmann, C. and Schutgens, R. B. H. Glucose metabolism in a child with 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency.J. Inher. Metab. Dis. 4 (1981) 163–164
Greene, C. L., Cann, H. M., Robinson, B. H., Gibson, K. M., Sweetman, L., Holm, J. and Nyhan, W. L. 3-Hydroxy-3-methylglutaric aciduria.J. Neurogen. 1 (1984) 165–173
Hammond, J. and Wilcken, B. 3-Hydroxy-3-methylglutaric, 3-methylglutaconic and 3-methylglutaric acids can be non-specific indicators of metabolic disease.J. Inher. Metab. Dis. 7, Suppl. 2 (1984) 117–118
Jakobs, C., Bojasch, M., Duran, M., Ketting, D., Wadman, S. K. and Leupold, D. 3-Methyl-3-butenoic acid: An artefact in the urinary metabolic pattern of patients with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency.Clin. Chim. Acta 106 (1980) 85–89
Ketel, J. A., Ket, J. L., Schutgens, R. B. H., Duran, M. and Wadman, S. K. Clinical and biochemical observations on a child with a deficiency of 3-hydroxy-3-methylglutaryl coenzyme A lyase.J. Inher. Metab. Dis. 3 (1980) 89–90
Kofranyi, E., Jekat, F. and Müller-Wecker, H. The minimum protein requirement of humans, tested with mixtures of whole egg plus potato and maize plus beans.Hoppe-Seyler's Z. Physiol. Chem. 351 (1970) 1485–1493
Leonard, J. V., Seakins, J. W. T. and Griffin, N. K. β-Hydroxy-β-methylglutaric aciduria presenting as Reye's syndrome.Lancet 1 (1979) 680
Leupold, D., Bojasch, M. and Jacobs, C. 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency in an infant with macrocephaly and mild metabolic acidosis.Eur. J. Pediatr. 138 (1982) 73–76
McGarry, J. D. and Foster, D. W. Regulation of hepatic fatty acid oxidation and ketone body production.Ann. Rev. Biochem. 49 (1980) 395–420
Norman, E. J., Drue Denton, M. and Berry, H. K. Gas chromatographic/mass spectrometric detection of 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in double first cousins.Clin. Chem. 28 (1982) 137–140
Owen, O. E., Morgan, A. P., Kemp, H. G., Sullivan, J. M., Herrera, M. G. and Cahill, G. F. Brain metabolism during fasting.J. Clin. Invest. 46 (1967) 1589–1595
Robinson, B. H., Oei, J., Sherwood, W. G., Slyper, A. H., Heininger, J. and Mamer, O. A. Hydroxymethylglutaryl CoA lyase deficiency: Features resembling Reye syndrome.Neurology 30 (1980) 714–718
Schutgens, R. B. H., Heymans, H., Ketel, A., Veder, H. A., Duran, M., Ketting, D. and Wadman, S. K. Lethal hypoglycemia in a child with a deficiency of 3-hydroxy-3-methylglutaryl coenzyme A lyase.J. Pediatr. 94 (1979) 89–91
Shilkin, R., Wilson, G. and Owles, E. 3-Hydroxy-3-methylglutaryl coenzyme A lyase deficiency.Acta Paediatr. Scand. 70 (1981) 265–268
Sovik, O., Sweetman, L., Gibson, K. M. and Nyhan, W. L. Genetic complementation analysis of 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency in cultured fibroblasts.Am. J. Hum. Genet. 36 (1984) 791–801
Tracey, B. M., Stacey, T. E. and Chalmers, R. A. Urinary and plasma organic acids in dizygotic twin siblings with 3-hydroxy-3-methylglutaric aciduria, studied by gas chromatography and mass spectrometry using fused silica capillary columns.J. Inher. Metab. Dis. 6, Suppl. 2 (1983) 125–126
Wilson, W. G., Cass, M. B., Sovik, O., Gibson, K. M. and Sweetman, L. A child with acute pancreatitis and recurrent hypoglycemia due to 3-hydroxy-3-methylglutaryl-CoA lyase deficiency.Eur. J. Pediatr. 142 (1984) 289–291
Wysocki, S. J. and Hähnel, R. 3-hydroxy-3-methylglutaric aciduria: Deficiency of 3-hydroxy-3-methylglutaryl coenzyme A lyase.Clin. Chim. Acta 71 (1976a) 349–351
Wysocki, S. J. and Hähnel, R. 3-Hydroxy-3-methylglutaric aciduria: 3-hydroxy-3-methylglutaryl-coenzyme A lyase levels in leucocytes.Clin. Chim. Acta 73 (1976b) 373–375
Wysocki, S. J. and Hähnel, R. 3-Methylcrotonylglycine excretion in 3-hydroxy-3-methylglutaric aciduria.Clin. Chim. Acta 86 (1978) 101–108
Wysocki, S. J., Hähnel, R., Truscott, R. J. W., Halpern, B. and Wilcken. B. Hyperammonaemia and urinary organic acids.Lancet 2 (1979) 371–372
Wysocki, S. J., Wilkinson, S. P., Hähnel, R., Wong, C. Y. B. and Panegyres, P. K. 3-Hydroxy-3-methylglutaric aciduria, combined with 3-methylglutaric aciduria.Clin. Chim. Acta 70 (1976) 399–406
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Wysocki, S.J., Hähnel, R. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: A review. J Inherit Metab Dis 9, 225–233 (1986). https://doi.org/10.1007/BF01799652
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF01799652