
Summary
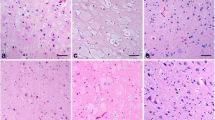
An adult-onset lysosomal storage disorder was diagnosed in a 5-year-old Schipperke dog with progressive cerebellar and central vestibular signs. It was characterized by cerebellar atrophy with extensive loss of Purkinje and granular cells, and hydrocephalus. Enlarged and vacuolated neurons were observed in spinal cord and brain; pancreatic centrolobular and islet cells were also vacuolated. Ultrastructurally, enlarged secondary lysosomes laden with lamellated membrane structures were present in neurons and empty enlarged vacuoles were found in pancreatic centroacinar, ductal, and islet cells. On frozen sections neurons stained with Ricinus communis agglutinin-I and wheat germ agglutinin. On paraffin sections neurons stained with luxol fast blue, periodic acid-Schiff, Concanavalia ensiformis agglutinin, and were autofluorescent. These findings indicate an accumulation of glycolipids containing terminal β-galactosyl and α-sialyl residues, and N-linked oligosaccharides. Tissue activity of lysosomal β-galactosidase was 50% of normal and the activity of β-hexosaminidase was elevated. Brain lipid-bound sialic acid was twice normal, with a small increase of GM1-ganglioside, but there was a significant elevation of GM2 (GD2) and GM3 (GD3). In addition, significant elevations of sialylated and non-sialylated oligosaccharides were noted. These clinical, biochemical and pathological findings are similar to those observed in human patients with adult-onset galactosialidosis.
Similar content being viewed by others


References
Alroy J, Ucci AA, Periera MEA (1984) Lectin: histochemical probes for specific carbohydrate residues. In: DeLellis RA (ed) Diagnostic Immunohistochemistry, vol 2. Masson Inc, New York, pp 67–88
Alroy J, Ucci AA, Goyal V, Woods W (1986) Lectin histochemistry of glycolipid storage diseases on frozen and paraffinembedded tissue sections. J Histochem Cytochem 34:501–505
Alroy J, Warren CD, Raghavan SS, Kolodny EH (1989) Animal models for lysosomal storage diseases: their past and future contribution. Hum Pathol 20:823–826
Amano N, Yokoi S, Akagi M, Sakai M, Yagishita S, Nakata K (1983) Neuropathological findings of an autopsy case of adult β-galactosidase and neuraminidase deficiency. Acta Neuropathol (Berl) 61:283–290
Ando S (1983) Gangliosides in the nervous system. Neurochem Int 5:507–537
Cummings JF, Wood PA, Walkley SU, deLahunta A, DeForest ME (1985) GM2 gangliosidosis in a Japanese spaniel. Acta Neuropathol (Berl) 67:247–253
Dorling PR (1984) Lysosomal storage diseases in animals. In: Dingel JT, Dean RT, Sly W (eds) Lysosomes in biology and pathology. Elsevier, Amsterdam, pp 347–379
Dyke PR (1988) Reconsideration in the classification of the neuronal ceroidlipofuscinosis. Am J Med Genet [Suppl] 5:69–84
Folch J, Lees M, Sloane-Stanley GH (1957) A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 226:497–509
Gahl WA, Renlund M, Thoene JG (1989) Lysosomal transport disorders: cystinosis and sialic acid storage disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 2619–2647
Galjaard H, Willemsen R, Hoogeveen AT, Mancini GM, Palmeri S, Verheijen FW, D'Azzo A (1987) Molecular heterogeneity in human beta-galactosidase and neuraminidase deficiency. Enzyme 38:132–143
Hannun YA, Bell RM (1987) Lysosphingolipids inhibit protein kinase C: implications for sphingolipidoses. Science 235:670–674
Kase R, Itoh K, Takiyama N, Oshima A, Sakuraba H, Suzuki Y (1990) Galactosialidosis: simultaneous deficiency of esterase, carboxy-terminal deamidase and acid carboxypeptidase activities. Biochem Biophys Res Commun 172:1175–1179
Kornfeld S (1986) Trafficking of lysosomal enzymes in normal and disease states. J Clin Invest 77:1–6
Ishikawa Y, Li S-C, Wood PA, Li Y-T (1987) Biochemical basis of type AB GM2 gangliosidosis in a Japanese spaniel. J Neurochem 48:860–864
Leeden RW, Yu RK (1982) Gangliosides: Structures, isolation and analysis. Methods Enzymol 83:139–192
Morreau H, Galiart NJ, Willemsen R, Gillemans N, Zhou XY, d'Azzo A (1992) Human lysosomal protective protein: glycosylation, intracellular transport, and association with β-galactosidase in the endoplasmic reticulum. J Biol Chem 267:17949–17956
Neufeld EF (1991) Lysosomal storage diseases. Annu Rev Biochem 60:257–280
Nolan CM, Sly WS (1989) I-cell diseases and pseudo-Hurler polydystrophy: disorders of lysosomal enzyme phosphorylation and location. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 1589–1601
O'Brien JS (1989) β-galactosidase deficiency (GM1 gangliosidosis, galactosialidosis, and Morquio syndrome type B); ganglioside sialidase deficiency (mucolipidosis IV). In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 1797–1806
O'Brien JS, Kishimoto Y (1991) Saposin proteins: structure, function, and role in human lysosomal storage disorders. FASEB J 5:301–308
Ogawa-Goto K, Kunamoto N, Abe T, Nagashima K (1990) Different ceramide compositions of gangliosides between human motor and sensory nerves. J Neurochem 55:1486–1493
Pearse AGE (1972) Fluorescence microscopy. In: Histochemistry: theoritical and applied, 3rd edn, vol 2. Churchill Livingstone, Edinburgh, pp 1172–1206
Sakuraba H, Suzuki Y, Akagi M, Sakai M, Amano N (1983) β-Galactosidase-neuraminidase deficiency (galactosialidosis): clinical, pathological, and enzymatic studies in a postmortem case. Ann Neurol 13:497–503
Scriver CR, Beaudet AL, Sly WS, Valle D (eds) (1989) In: The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 1565–1839
Stutelsky E, Alroy J, Ucci AA, Carpenter JL, Moore FM (1987) Modulation of carbohydrate residues in regenerative nodules and neoplasms of canine and feline pancreas. Am J Pathol 126:25–32
Slatter D (1990) Fundamantals of veterinary ophthalmology, 2nd edn. W.B. Saunders, Philadelphia, p 403
Trachemontagne J, Michud L, Potier M (1990) Deficient lysosomal carboxypeptidase activity in galactosialidosis. Biochem Biophys Res Commun 168:22–29
Wenger DA, Williams C (1991) Screening for lysosomal disorderts. In: Homes FA (ed) Techniques in diagnostic human biochemical genetics. Wiley-Liss, New York, pp 587–617
Yoshino H, Miyashita K, Miyatani N, Ariga T, Hashimoto Y, Tsuji S, Oyanagi K, Ohama E, Ikuta F, Suzuki A, Miyatake T (1990) Abnormal glycosphinogolipid metabolism in nervous system galactosialidosis. J Neurol Sci 97:53–65
Yu RK, Saito M (1989) Structure and localization of gangliosides. In: Margolis RU, Margolis RK (eds) Neurobiology of glycoconjugates. Plenum Press, New York, pp 1–42
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Knowles, K., Alroy, J., Castagnaro, M. et al. Adult-onset lysosomal storage disease in a Schipperke dog: clinical, morphological and biochemical studies. Acta Neuropathol 86, 306–312 (1993). https://doi.org/10.1007/BF00304147
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00304147