
Abstract
Introduction
This study was designed to identify a group of bacteria in the human gut microbiota with specific effects on PD-1-based immunotherapy for patients with non-small cell lung cancer (NSCLC).
Methods
The study was performed in patients with advanced NSCLC, who received PD-1 monoclonal antibody (mAb) treatment for 6 months after one or several prior therapies. The combination of blood immune-related factors of the participants and their 16S rRNA gene sequencing from fecal samples at baseline was used to investigate the diversity and composition of the gut microbiota. The differences in relative abundance of gut microbiota at the genus level were compared, and the relation to blood immune-related factors was assessed using Spearman’s rank correlation coefficient analysis.
Results
The 16S rRNA gene sequencing showed a clear difference in the diversity and composition of the gut microbiota between groups with stable disease (SD) and progressive disease (PD). A comparison of differences in relative abundance at the genus level showed that the relative abundance of Escherichia-Shigella, Akkermansia and Olsenella in the SD group was significantly higher than that in the PD group. The SD group had significantly higher interleukin-12 (IL-12) and interferon γ (IFN-γ) levels than the PD group. Interestingly, the numbers of white blood cells and sorted cells in the SD group were higher than those in the PD group. Spearman’s rank correlation coefficient analysis showed that Escherichia-Shigella was positively correlated with IL-12, IFN-γ and basophils. Akkermansia was positively correlated with monocytes.
Conclusion
The response to PD-1-based immunotherapy in patients with NSCLC is affected by the diversity and composition of the gut microbiota. Escherichia-Shigella and Akkermansia may have specific effects on PD-1 inhibitory immunotherapy for NSCLC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Non-small cell lung cancer (NSCLC) patients with and without response to PD-I immunotherapy have different diversity and composition of gut microbiota. |
The relative abundances of Escherichia-Shigella, Akkermansia and Olsenella might have contributed to the different composition of gut microbiota. |
Escherichia-Shigella and Akkermansia might have specific effects on the PD-1 inhibitory immunotherapy for NSCLC, and a possible association with interleukin-12 (IL-12) and interferon gamma (IFN-γ). |
Introduction
Lung cancer is the leading cause of cancer-related deaths globally. Non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancers, and most NSCLC patients are diagnosed at an advanced stage, with a 5-year survival rate of < 5% [1,2,3]. Cancer immunotherapy has been shown to be highly effective against NSCLC [2]. Currently, the most extensively studied method for cancer immunotherapy is the blocking of the programmed cell death protein-1/programmed death-ligand 1 (PD-1/PD-L1) pathway [4,5,6,7]. Despite the clear advantages of this treatment for NSCLC patients, only 15–25% of these patients benefit from immunotherapy involving the inhibition of PD-1 [8]. Gut microbiome- and blood-based biomarkers, as well as innovative soluble genetic markers, could be helpful in predicting patient outcome and tumor response to immune-checkpoint inhibitors (ICI) [9, 10].
Recent studies have confirmed the significance of the gut microbiota in improving the effectiveness of cancer immunotherapy [11,12,13]. The administration of mouse fecal suspensions in combination with PD-L1 immunotherapy showed significantly better anti-tumor effects than immunotherapy alone, including significantly delayed tumor growth and increased response of tumor-specific T cells [1, 11, 12]. Fecal microbial transplantation (FMT) from the feces of PD-1 responder (R) and non-responder (NR) patients to sterile or antibiotic-treated mice, respectively, showed stronger fecal immune response to tumors in R patients as compared to NR patients [1, 11, 12, 14,15,16]. Increasing numbers of parallel studies have further confirmed the association of the gut microbiota with immunotherapy [16]. However, the gut microbiota is rich in species diversity, and the identification of a bacterial group with specific effects on PD-1 inhibitory immunotherapy is urgently needed.
In this study, patients with advanced NSCLC received PD-1 monoclonal antibody (mAb) treatment for 6 months after one or several prior therapies. The blood immune-related factors of the participants and 16S rRNA gene sequencing technology were combined to investigate the diversity and composition of the gut microbiota. The differences in relative abundance of gut microbiota at the genus level were studied using Spearman’s rank correlation coefficient analysis.
Methods
Study Participants
This study was approved by the Ethics Committee of Hangzhou First People’s Hospital for Human Studies (protocol number 2018-16-01, approved 14 November 2018), and all the participants provided written informed consent. The study was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments. A total of 16 patients with advanced NSCLC who had undergone one or several prior therapies were recruited. The patients were treated with PD-1 mAb (3 mg/kg every 2 weeks) for 6 months. The Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST1.1) was used to evaluate the effectiveness of treatment, and patients underwent computed tomography (CT) scans of the lungs every 6–8 weeks until disease progression (PD). An increase of ≥ 20% in the sum of the maximum diameter of the target lesions or the appearance of new lesions was considered as disease progression, while a ≥ 30% reduction in the sum the of the maximum diameter of target lesions for at least 4 weeks was considered partial response (PR). When the sum of the maximum diameter of target lesions was not reduced to the PR level or increased to PD, the condition was regarded as stable disease (SD). Both fecal and blood plasma samples were collected at baseline. Inclusion criteria were as follows: (1) age between 18 and 79 years; (2) a clinical diagnosis of stage III/IV NSCLC; and (iii) treatment with anti-PD-1 ICI until PD or intolerable toxicity. The following were excluded: (1) patients with systemic metabolic diseases (hyperthyroidism/hypothyroidism, Cushing syndrome); (2) patients with infection within the past 1 month (upper sensation, pneumonia, diarrhea); (3) patients taking glucocorticoids or antibiotics within the past 3 months; (4) patients with clinically significant systemic disease (cancer, tuberculosis/chronic obstructive pulmonary disease [COPD], coronary heart disease, hypertension, hemoglobin disease, anemia); (5) patients with major cardiovascular diseases (myocardial infarction, heart failure, cardiac arrest) within the past 6 months; and (6) patients with mental illness, rendering them unable to understand the nature, scope, and possible consequences of the study.
ELISA
The concentrations of interferon γ (IFN-γ) and interleukin-12 (IL-12) in blood plasma samples were measured by enzyme-linked immunosorbent assay (ELISA) using commercially available kits (4A Biotech, China) via capture-sandwich assay, following the manufacturer’s instructions. The concentration of cytokines was calculated on the basis of standard curves provided with the kits, and the results were expressed in picograms per milliliter. For ELISA, all samples were tested in duplicate, and their average values were used in analyses.
Extraction of Fecal Genomic DNA and 16S rRNA Gene Sequencing
Fecal samples from the participants were collected at 6:30–8:30 am at the hospital, and were immediately transferred to the clinic laboratory in an ice bag using insulated polystyrene containers. The samples were divided into four aliquots of 250 mg each and immediately stored at −80 °C in 1.5-mL centrifuge tubes until further processing. Samples that stayed at room temperature for more than 2 h were discarded. DNA was extracted using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Germany), following the manufacturer’s instructions. The DNA concentration was measured using a NanoDrop spectrophotometer (Thermo Scientific), and its molecular size was assessed by 1.0% agarose gel electrophoresis on gels containing 0.5 mg/mL ethidium bromide. The extracted DNA samples were stored at −20 °C until analysis.
The extracted DNA samples were amplified with a set of primers, targeting the hypervariable V3–V4 region (338F/806R) of the 16S rRNA gene. The sequences of forward and reverse primers used in this study were 5′-ACTCCTACGGGAGGCAGCA-3′ and 5′-GGACTACHVGGGTWTCTAAT-3′, respectively. A 20-µL polymerase chain reaction (PCR) was prepared, which contained 4 µL of 5X FastPfu Buffer, 2 µL of 2.5 mM dNTPs, 5 µM of each forward and reverse primer, 0.4 µL TransStart FastPfu DNA Polymerase (TransGen Biotech, Beijing, China), and 10 ng DNA template. The following PCR conditions were used: initial denaturation at 95 °C for 2 min; 25 cycles of denaturation at 95 °C for 30 s, primer annealing at 55 °C for 30 s, and extension at 72 °C for 30 s; followed by a final extension at 72 °C for 5 min. The DNA libraries were constructed following the manufacturer’s instructions, and sequencing was performed on the Illumina MiSeq platform at Majorbio Bio-Pharm Technology Co., Ltd (Shanghai, China). The raw sequencing reads of all the samples were deposited in the GenBank Sequence Read Archive under project accession number PRJNA728213.
Analysis of 16S rRNA Gene Sequencing Data
The raw FASTQ files were de-multiplexed and mass filtered using QIIME (Quantitative Insights into Microbial Ecology) version 1.9.1 software. Briefly, the 300 bp reads were truncated in a 50 bp sliding window, with the average mass fraction at any one site < 20, and the truncated reads shorter than 50 bp were discarded. Exact barcode matches were used; where there were two-nucleotide mismatches in primers or ambiguous characters, the reads were discarded. The sequences that overlapped by more than 10 bp were assembled according to their overlapping sequences. The sequence reads that could not be assembled were discarded. UPARSE (version 7.1, http://drive5.com/uparse/) was used to cluster operational taxonomic units (OTUs), using 97% similarity as a cutoff point, and the chimeric sequences were identified and deleted using UCHIME. The RDP (Ribosomal Database Project) classifier (http://rdp.cme.msu.edu/) was used to assign the sequences in the SILVA database, with a 70% confidence threshold for each 16S rRNA gene sequence.
Statistical Analyses
The data are presented as mean ± standard deviation (SD). Categorical variables are presented as percentages. The data entry and management were performed using SPSS 22.0 software, and all P values were derived from two-tailed tests. A P value of less than 0.05 in a two-tailed test was considered statistically significant. Statistical analyses were conducted using the R platform. Continuous variables were compared between the SD and PD groups using the Wilcoxon rank-sum test. Fisher’s exact test was used to compare the categorical variables.
Results
Differences in the Diversity of the Gut Microbiota Between SD and PD Patients
Eight patients with PD and eight with SD were assessed according to their 6-month CT scan results. The diversity of gut microbiota for the two groups is shown in Fig. 1. The intergroup similarity analysis showed that the difference between the SD and PD groups was higher than that within each group, confirming the validity of the grouping. The Shannon index showed a higher diversity of gut microbiota in the SD group than the PD group (Fig. 1a). No significant within-group differences in the diversity or richness of the gut microbiota were found within in either group (Fig. 1b). Non-metric multidimensional scaling (NMDS) analysis of Bray–Curtis similarity between the gut microbiota of the two groups showed that the SD and PD groups were the most distinguished, suggesting a significant difference in the composition of the gut microbiota between the two groups (Fig. 1c). These results indicate a clear difference in the diversity of the gut microbiota between SD and PD patients, although the richness and diversity indices of their gut microbiota were largely similar.

Comparison of the diversity of gut microbiota between SD and PD patients. a Intergroup similarity analysis in the SD (blue) and PD (red) groups. b Shannon index, reflecting the alpha diversity of gut microbiota in the SD and PD groups. c Non-metric multidimensional scaling (NMDS) analysis of the beta diversity of the gut microbiota in the SD and PD groups
Differences in the Composition of the Gut Microbiota Between SD and PD Patients
The composition of the gut microbiota of the two groups is shown in Fig. 2. The Venn diagram, describing the sample similarity and overlap, shows the number of common and unique OTUs. A total of 628 and 513 OTUs were detected in samples of the SD and PD groups, respectively, among which 203 and 88 were unique OTUs. Therefore, the ratios of unique to total OTUs were 32% and 17%, respectively. A total of 425 OTUs were common to both groups (Fig. 2a). The heat-map analysis indicated differences at the genus level between the SD and PD groups. The legends above and below the heat map represent each participant. The relative abundance of each genus is indicated by a color gradient from green (low abundance) to red (high abundance). The complete linkage clustering of samples was based on the composition and abundance of each genus in the gut microbiota of SD and PD patients (Fig. 2b).

Comparison of the composition of gut microbiota between SD and PD patients. a Number of OTUs, presented in a Venn diagram, show the composition of gut microbiota in the SD (blue) and PD groups (red). b The heat map shows similarities and differences in the composition of the gut microbiota at the genus level in the SD (blue) and PD groups (red)
Changes in the Species Abundance of the Gut Microbiota Between SD and PD Patients
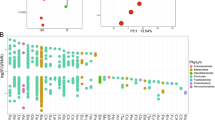
Differences in the relative abundance of gut microbiota at the genus level between the SD and PD groups are shown in Fig. 3. The relative abundance of Escherichia-Shigella, Akkermansia and Olsenella in the SD group was significantly higher than that in the PD group (P < 0.05), while the relative abundance of Anaeroglobus in the SD group was significantly lower than that in the PD group (P < 0.05) (Fig. 3a). Multi-level species difference discriminant analysis (LEfSe) identified the most differentially abundant species between SD and PD patients. The taxonomic cladogram obtained from LEfSe of 16S rRNA gene sequences (relative abundance > 0.5%) shows the enriched taxa in the SD and PD groups in blue and red, respectively. The brightness of each dot is proportional to the size of its effect (Fig. 3b). The enriched taxa of the SD and PD groups are indicated with positive (blue) and negative (red) linear discriminant analysis (LDA) scores. Only the taxa meeting an LDA significance threshold of 2 are shown (Fig. 3c).

Differences in the relative abundance of genera in SD and PD patients. a Kruskal–Wallis H test analysis for the assessment of significance in differences in relative abundance at the genus level in the SD (blue) and PD groups (red). b Multi-level species difference discriminant analysis (LEfSe) identifies the features of different abundance and associated categories in the SD (blue) and PD groups (red). c Linear discriminant analysis (LDA) identifies communities or species with a significant differential impact on sample delineation in SD (blue) and PD groups (red). *P < 0.05; **P < 0.01; ***P < 0.001
Heat-Map Correlation Analysis Between Immune-Related Factors and Gut Microbiota
The levels of immune-related factors in the two groups are shown in Fig. 4. The SD group showed significantly higher levels of IL-12 and IFN-γ than the PD group (P < 0.05) (Fig. 4a and b). Interestingly, the number of white blood cells and sorted cells in the SD group was higher than that in the PD group (Fig. 4c). The correlation between immune-related factors (IL-12, IFN-γ, white blood cells [WBC], neutrophils [N], lymphocytes [L], monocytes [M], eosinophils [EO], basophils [BASO]) and the 50 most abundant genera in each group was evaluated using a Spearman’s rank correlation heat map (Fig. 4d, e). Escherichia-Shigella were positively correlated with IL-12 (0.56 < R < 0.75), IFN-γ (0.57 < R < 0.77) and BASO (0.57 < R < 0.77). Akkermansia was positively correlated with monocytes (0.56 < R < 0.75).

Spearman’s rank correlation coefficient analysis of the species abundance at the genus level in relation to immune-related factors. Red and green indicate positive and negative correlations, respectively. *P < 0.05; **P < 0.01; ***P < 0.001
Discussion
This study examined patients with advanced NSCLC who received PD-1/PD-L1 mAb treatment for 6 months after one or several prior therapies (Fig. 1). The 16S rRNA gene sequencing technology was used to identify the composition and diversity of the gut microbiota in the SD and PD groups. Intergroup similarity analysis showed that the grouping was valid. Although the Shannon index showed that the differences in diversity of gut microbiota was not significant between the SD and PD groups (Fig. 1b), the NMDS analysis of the Bray–Curtis similarity suggested significant differences between the two groups. This indicates that the PD-1/PD-L1 treatment altered the composition and diversity of the gut microbiota.
In order to observe the contribution of specific genera to the effects of PD-1/PD-L1 mAb, the differences in relative abundance of gut microbiota at the genus level after treatment with PD-1/PD-L1 mAb for 6 months were compared (Fig. 3). Interestingly, the results showed that the relative abundance of Escherichia-Shigella, Akkermansia and Olsenella in the SD group was significantly higher than that in the PD group (Fig. 3b and c). A significant positive correlation was also found between Escherichia-Shigella and immune-related factors (IL-12, IFN-γ) (P < 0.05) and between Akkermansia and mononuclear cells (M) (Fig. 4). Escherichia-Shigella might trigger dendritic cells (DCs) to produce interleukin (IL)-10, IL-12 and tumor necrosis factor (TNF)-α [17]. Akkermansia was abundantly present in the gut microbiota of NSCLC patients, who benefited from PD-1 inhibition [15]. Oral supplementation with Akkermansia after FMT in NR was found to restore the efficacy of PD-1 blockade in an IL-12-dependent manner in mouse models [15]. This study suggests that the diversity of gut microbiota might be an important factor in the treatment of NSCLC using PD-1/PD-L1 mAb. In addition to diversity, the composition of the gut microbiota is related to favorable responses to PD-1-based immunotherapy. It is widely accepted that the immune responses can be reshaped by the gut microbiota. The limitation of this study is the small number of patients, which may have led to the low Shannon diversity index.
Conclusion
In summary, this study showed that the response to PD-1-based immunotherapy for NSCLC is affected by the diversity and composition of the gut microbiota. Escherichia-Shigella and Akkermansia may have specific effects on PD-1 inhibitory immunotherapy in NSCLC.
References
Derosa L, et al. The intestinal microbiota determines the clinical efficacy of immune checkpoint blockers targeting PD-1/PD-L1. Oncoimmunology. 2018;7(6):e1434468.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–54.
Hsu PP, Shaw AT. Lung cancer: a wily genetic opponent. Cell. 2017;169(5):777–9.
Doroshow DB, et al. Immunotherapy in non-small cell lung cancer: facts and hopes. Clin Cancer Res. 2019;25(15):4592–602.
Forde PM, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378(21):1976–86.
Sgambato A, et al. Anti PD-1 and PDL-1 immunotherapy in the treatment of advanced non- small cell lung cancer (NSCLC): a review on toxicity profile and its management. Curr Drug Saf. 2016;11(1):62–8.
Xia L, Liu Y, Wang Y. PD-1/PD-L1 blockade therapy in advanced non-small-cell lung cancer: current status and future directions. Oncologist. 2019;24(Suppl 1):S31-s41.
Maier B, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. 2020;580(7802):257–62.
Brueckl WM, Ficker JH, Zeitler G. Clinically relevant prognostic and predictive markers for immune-checkpoint-inhibitor (ICI) therapy in non-small cell lung cancer (NSCLC). BMC Cancer. 2020;20(1):1185.
Hakozaki T, et al. The gut microbiome associates with immune checkpoint inhibition outcomes in patients with advanced non-small cell lung cancer. Cancer Immunol Res. 2020;8(10):1243–50.
Gopalakrishnan V, et al. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33(4):570–80.
Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103.
Li W, et al. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019;447:41–7.
Adachi K, Tamada K. Microbial biomarkers for immune checkpoint blockade therapy against cancer. J Gastroenterol. 2018;53(9):999–1005.
Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7.
Sears CL, Pardoll DM. The intestinal microbiome influences checkpoint blockade. Nat Med. 2018;24(3):254–5.
Moreno AC, et al. Recognition of enteroinvasive Escherichia coli and Shigella flexneri by dendritic cells: distinct dendritic cell activation states. Mem Inst Oswaldo Cruz. 2012;107(1):138–41.
Acknowledgements
Funding
This work is financially supported by Zhejiang Province Health Technology Grants No. 2019RC235, 2019ZD014 and 2020KY209 and Hangzhou Health Science and Technology Grant No. A2020125. The Rapid Service Fee was funded by the authors.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
Experiment conception and design: Daqiang He and Xianjun Wang. Xing Li, Rui An, Lihong Wang, Yun Wang, Song Zheng and Xueqing Chen performed the experiments. Data Analysis: Daqiang He. Manuscript drafting: Daqiang He and Xianjun Wang.
Disclosures
Daqiang He, Xing Li, Rui An, Lihong Wang, Yun Wang, Song Zheng, Xueqing Chen and Xianjun Wang declare that no competing interests exist.
Compliance with Ethics Guidelines
This study was approved by the Ethics Committee of Hangzhou First People’s Hospital for Human Studies (protocol number 2018-16-01, approved 14 November 2018), and all the participants provided written informed consent. The study was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments.
Data Availability
The raw sequencing reads of all the samples during the current study are available in the GenBank Short Read Archive under the project accession number PRJNA728213.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
He, D., Li, X., An, R. et al. Response to PD-1-Based Immunotherapy for Non-Small Cell Lung Cancer Altered by Gut Microbiota. Oncol Ther 9, 647–657 (2021). https://doi.org/10.1007/s40487-021-00171-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-021-00171-3