
Abstract
Purpose of Review
Patients suffering from Parkinson’s disease (PD) are known to experience gastrointestinal dysfunction that might precede the onset of motor symptoms by several years. Evidence suggests an important role of the gut-brain axis in PD pathogenesis. These interactions might be essentially influenced by the gut microbiota. Here, we review recent findings supporting that changes in the gut microbiota composition might be a trigger for inflammation contributing to neurodegeneration in PD.
Recent Findings
Recent research revealed that PD patients exhibit a pro-inflammatory microbiota profile in their intestinal tract that might increase gut permeability, allowing leakage of bacterial products and inflammatory mediators from the intestines. Evidence in literature indicates that alpha-synuclein deposition might start in the enteric nervous system by pro-inflammatory immune activity and then propagates to the CNS. Alternatively, the peripheral inflammatory response could impact the brain through systemic mechanisms.
Summary
A better understanding of the gut-brain interactions and the role of the intestinal microbiota in the regulation of immune responses might bring new insights in PD pathological progression and might lead to novel diagnostics and therapeutic approaches.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common age-related neurodegenerative disease, with increasing age being the greatest risk factor for its development. PD is hallmarked by the loss of dopaminergic neurons in the substantia nigra (SN), resulting in the characteristic motor impairments. These symptoms are commonly treated with dopamine-replacement medication, but these therapies do not prevent the dopaminergic neurodegeneration. Another relevant hallmark in PD is the presence of alpha-synuclein-containing inclusion bodies (Lewy pathology) in the surviving neurons in different areas of the nervous system [1,2,3].
Besides the well-known motor deficits, PD patients very often suffer from non-motor symptoms including hyposmia, anxiety, depression, impaired executive function, and most commonly gastrointestinal (GI) dysfunction [4, 5, 6•]. Some of these symptoms are very often present in pre-clinical stages, and their occurrence in healthy individuals is associated with an increased risk of developing PD [4, 7, 8].
Recent evidence suggest an important role of the GI tract and the associated enteric nervous system (ENS) in the development of PD [9,10,11,12]. The so-called gut-brain axis is a bidirectional communication system between the central nervous system (CNS) and the GI tract. The immune system might be an important interplayer by regulating immune homeostasis in both the gut and the brain [13]. It has been also lately recognized that the gut-brain interactions might be essentially influenced by the gut microbiota [14,15,16].
The cause of neuronal death in PD remains a matter of debate; however, abundant evidence suggests that inflammatory mechanism might play an important role. For instance, in the brain, microglial activation is associated with dopaminergic neuronal loss suggesting that neuroinflammation may contribute to the degenerative process [17]. Moreover, it has been reported that alpha-synuclein has an important role in the initiation and maintenance of inflammation in PD [18]. The early involvement of the GI tract in PD supports the hypothesis that the GI tract could be a source of inflammation contributing to neurodegeneration.
The Role of the GI Tract in PD
GI Dysfunction in PD
Recent evidence suggests that GI dysfunctions are consistently associated with PD. The occurrence and prevalence of different GI dysfunctions vary among patients and has been extensively reviewed [6•]. Among GI problems, constipation is the most prominent (reported in 20 to 80% of PD patients) and it is partially due to a prolonged intestinal transit time affecting both the small intestine and the colon [6•, 19]. Constipation might precede motor symptoms by over a decade [6•, 8, 19, 20] making it one of the earliest indicators of a pathological process that will ultimately lead to PD. Recently, constipation was included in the research criteria for prodromal PD diagnostics as one of the risk factors for future development of PD [21].
Alpha-Synuclein Pathological Progression
Alpha-synuclein is a protein expressed as a normal component of the CNS and the ENS and is thought to be involved in the regulation of neurotransmission and synaptic homeostasis [22, 23]. Under certain circumstances, alpha-synuclein aggregates, leading to one of the pathological hallmarks in PD, Lewy bodies in cell somata, and Lewy neurites in axons and dendrites [24, 25], were found in the brain of individuals with PD. It is not entirely clear if Lewy bodies are toxic for the neurons or if they are formed in an attempt to sequester alpha-synuclein aggregates to avoid damage of cellular components and cytotoxicity. What is clear is that the overexpression of alpha-synuclein has been proven to be sufficient to induce its aggregation and the degeneration of dopaminergic neurons [26].
Several studies indicated that alpha-synuclein is also detected in higher levels in the intestines of PD patients compared to healthy controls [27,28,29,30, 31••, 32]. These alpha-synuclein accumulations are associated with damage in the enteric neurons and possibly underlie GI dysfunction [27, 32].
The spatial distribution of alpha-synuclein aggregates of PD patients is generally compatible with a staging scheme proposed in 2003 postulating that inclusions initially appear in the olfactory bulb (OB) and dorsal motor nucleus of the vagus (DMV) in the lower brainstem [33••]. The pathology then seems to spread in an ascending fashion with the subsequent involvement of monoaminergic pontine nuclei and the dopaminergic neurons of the SN. In accordance with the hypothesis, it has been proven that alpha-synuclein exhibits prion-like properties, including the ability to misfold and form aggregates, which display cell-to-cell transmission [34, 35].
Braak and colleagues postulated in their “dual-hit hypothesis” that the initial formation of alpha-synuclein aggregates occurs outside the brain, in the terminals of neurons residing in the olfactory bulb and the ENS as a consequence of exterior insults such as toxins and/or microorganisms [33••, 36,37,38]. The vagus nerve might provide a path for the spread of alpha-synuclein pathology from the ENS to the brain through the brainstem, midbrain, basal forebrain, and finally the cortical areas [36, 39], whereas the initiation of the pathological process in the OB might ultimately affect the brain via the olfactory tract [11, 37, 40]. Recent studies [31••, 41••] suggest that gut-initiated pathological processes in PD do not necessarily require an environmental pathogen and/or toxin since they can be triggered by the commensal bacteria in the gut.
Factors such as microorganisms, including nasal/gut microbiota, and toxins such as pesticides or pollutants might be the initial inflammatory trigger in the nasal and intestinal mucosa, inducing damage and the subsequent mucosal inflammation and oxidative stress, thereby initiating alpha-synuclein accumulation.
Pre-clinical and Clinical Evidence
Alpha-synuclein has the capability of spontaneous misfolding and shows prion-like properties, including cell-to-cell propagation [34, 42, 43]. Both retro- and anterograde axonal transports of alpha-synuclein fibrils have been shown to occur via the vagal nerve [44, 45], and pathological aggregates were detected in the DMV when alpha-synuclein fibrils were injected in the duodenum of rats [46]. Moreover, a single intra-peritoneal injection of alpha-synuclein fibrils in A53T transgenic mice led to marked neurological PD-like symptoms and alpha-synuclein pathology in the spinal cord and the brain [47]. In another transgenic mouse model for PD, alpha-synuclein was shown to be transmitted to engrafted neuronal precursor cells, where it created inclusions [48, 49]. Similarly, autopsies of PD patients that received fetal mesencephalic transplants displayed alpha-synuclein accumulation in the grafted neurons [50, 51].
If, indeed, the vagal nerve constitutes a major path for alpha-synuclein pathology spreading, vagotomy might then be protective against PD development. In 2015, the first epidemiological study tested whether vagotomy modifies the risk of PD. Cohorts of patients, who had undergone vagotomy between 1977 and 1995, were compared to matched controls. Full truncal vagotomy was associated with a decreased risk of developing PD compared to super-selective vagotomy (affecting only acid producing portion of gastric body) or no vagotomy [52•].
This finding has now been reproduced in a Swedish cohort where the full truncal vagotomy group exhibited a decreased risk of developing PD after more than 5 years of follow-up when compared with the background population and also when compared to the selective vagotomy group (affecting vagal branches to the stomach) and the super-selective vagotomy group [53].
Observations in animal models also support that the resection of parts of the autonomic nervous system delays the progression of alpha-synuclein pathology. A pre-clinical study demonstrated that PD-like pathology could be mimicked by the oral administration of the pesticide rotenone in mice [54]. The local effect of pesticides on the ENS induced PD-like progression and reproduced the neuroanatomical and neurochemical features of PD staging from the ENS to the brain [54]. The same group showed that the progression of alpha-synuclein pathology towards the brain could be halted by the resection of sympathetic and parasympathetic nerves before the oral administration of rotenone [9]. Both the olfactory and intestinal mucosae are exposed to substances from the environment via inhalation or ingestion, and therefore, it seems probable that these environmental factors such as diets, pesticides, pollutants, and mucosal microorganisms might have an important role in triggering PD pathology, probably supported by genetic predisposition.
Gut Leakiness
Several studies have demonstrated that PD patients have increased intestinal permeability, also known as “leaky gut,” in comparison to healthy controls [31••, 55,56,57,58]. Interestingly, intestinal permeability has been shown to correlate with the levels of enteric alpha-synuclein [31••]. Alpha-synuclein aggregates might be formed as a consequence of oxidative injury [26]. Deficits in the gut barrier function and the translocation of bacteria and inflammatory bacterial products (e.g., lipopolysaccharide, LPS) might trigger an inflammatory response and the associated oxidative stress in the gut and thereby initiating alpha-synuclein accumulation in the ENS [31••, 59,60,61] that might ultimately reach the brain in a “prion-like” manner. Furthermore, gut-derived LPS can promote the disruption of the blood-brain barrier [62, 63] and thus contribute to neuroinflammation and injury in the SN.
Changes in Gut Microbiota Composition
The increased intestinal permeability has been detected in recently diagnosed PD patients [31••], but no studies have yet assess intestinal permeability in healthy individuals that will later develop PD. Therefore, we do not have evidence supporting that increased intestinal permeability is a pre-motor stage but the symptom is present at least in the early stages of the disease.
Results from different studies have shown that the gut microbiota is altered in PD patients.
Small intestinal bacterial overgrowth (SIBO) is a malabsorption syndrome associated with increased bacterial density and/or the presence of colonic-type species in the small intestine [64]. SIBO is highly prevalent in PD patients [32, 65], even when recently diagnosed [66]. The abnormalities in GI motility developed in PD patients might be responsible for the increased occurrence of SIBO. However, once SIBO is developed, it might increase intestinal permeability and contribute to bacterial translocation and therefore perpetuating the inflammatory response [67].
Recently, gut microbiota content in different cohorts of PD patients was characterized in several studies. The first study describing differences in the composition of fecal bacteria populations reported an increased abundance of Enterobacteriaceae in PD that positively correlated with the severity of postural instability and gait difficulty [68••]. They further detected lower abundance of Prevotella, prominent producers of health-promoting neuroactive short chain fatty acids (SCFA) as well as important promoters of the biosynthesis of thiamine and folate [69], which is in line with decreased levels of these vitamins in PD patients [70, 71]. The authors further indicate that a decrease in Prevotella might be related with a reduction in mucin synthesis which is associated with increased intestinal permeability [31••, 65] intensifying the translocation of bacterial antigens. In addition, a decreased abundance of Prevotella and an increased abundance of Lactobacillaceae have been associated with lower concentrations of ghrelin. Ghrelin is a gut hormone that may be involved in the maintenance and protection of normal nigrostriatal dopamine function [72], and impaired ghrelin secretion has been reported in PD patients [73].
A second study revealed differences in both mucosal and fecal microbial communities of PD patients in comparison to healthy subjects. More differences were found in fecal than in mucosal (tissue adherent) bacteria [41••]. The study showed a lower abundance of SCFA butyrate-producing bacteria associated with anti-inflammatory properties from the genera Blautia, Coprococcus, and Roseburia in PD fecal samples, thereby concluding that a reduction in SCFA might contribute to gut leakiness. In addition, metagenomics analysis indicated that genes involved in LPS biosynthesis and type III bacterial secretion systems were higher in stool samples of PD patients compared to controls. Type III secretion systems are involved in pathogenic interactions with host cells and might exacerbate bacterial product-induced inflammation [74, 75]. PD pathogenesis may be caused or exacerbated by changes in GI microbiota composition that could induce peripheral inflammatory responses and ultimately promote alpha-synuclein pathology in the intestine and the brain or by rostral to caudal cell-to-cell transfer of alpha-synuclein pathology caused by increased oxidative injury (due to an increase in pro-inflammatory bacteria).
SCFA concentrations have also been demonstrated to be reduced in PD patients compared to age-matched controls that exceed the reductions normally associated with aging [76]. Significant reduction of acetate, propionate, and butyrate were observed in PD fecal samples. The reduction in SCFA might contribute to the reduction of peristaltic regulation by the ENS and contribute to GI dysmotility in PD.
Furthermore, SCFA butyrate has anti-inflammatory properties thought to be owing to an epigenetic mechanism or to the activation of SCFA receptors leading to anti-/microbial and anti-inflammatory effects and to a decreased intestinal barrier dysfunction [77,78,79,80].
More recently, the analysis of the gut microbiota in a German cohort revealed differences in relative abundance for four bacterial families in PD patients in comparison to controls, including an increase of Lactobacillaceae which is in accordance with the previous studies [81].
There are several variables that could affect the microbiota and cover up the disease signature. Since most PD patients are using medication, it is important to differentiate the disease effects from that of medication. In a case-only analysis, besides dysbiosis of the gut microbiota in PD patients, significant differences were found in gut microbiota composition as a function of treatment with COMT inhibitors and anticholinergics [82]. The authors indicate that the effects of COMT inhibitors and anticholinergics are independent of the PD effect because their impact on the overall microbiome was detected within patients (hence, PD was controlled for). Moreover, most of the PD-associated taxa were robustly associated with disease in patients who were not on either of these drugs. It was not possible to determine the effects of carbidopa/levodopa since 90% of the patients are taking this medication.
The gut microbiota might be involved in the metabolism of the drugs used by PD patients and might affect its absorption. The drugs might in turn affect microbiota composition.
So far, it is not possible to determine if changes in the gut microbiota are an occurrence contributing to the disease initiation or if they emerge as a consequence of PD-related pathology. However, it might still play a role in neurological dysfunction and neurodegeneration by perpetuating inflammatory cascades and oxidative injury in the brain through LPS-mediated mechanism. The microbiota, via mechanisms including metabolite production, can impact immune and inflammatory pathways leading to the peripheral and central immune activation and inflammation [83, 84]. Colonic biopsies retrieved from PD patients revealed an increased expression in the levels of pro-inflammatory cytokines, such as TNF-alpha, IFN-gamma, IL-6, and IL-1 beta as well as an increased activation of enteric glial cells [85]. Moreover, IBD and IBS, two chronic inflammatory conditions, have been reported to increase the risk of PD development [86, 87].
The importance of the intestinal microbiome in PD was recently demonstrated in a study showing that under germ-free conditions, or when bacteria are depleted with antibiotics, mice overexpressing human alpha-synuclein displayed reduced microglia activation, alpha-synuclein inclusions, and motor deficits compared to animals with a complex microbiota. These results suggest that gut microbiota is required for PD phenotype development. Moreover, reconstitution of the fecal microbiota in germ-free alpha-synuclein transgenic PD mice using stool from PD patients exacerbates motor symptoms and pathology in this model [88••].
Proposed Mechanism for PD Pathology
New evidence suggests that gut-derived inflammation may play a mechanistic role in PD pathogenesis [89,89,90,91,92,93,94,95,96,97,99]. As previously described, GI dysfunctions are very common in PD and might begin decades before the onset of motor symptoms [4, 7, 8]. The intestinal tract is a large surface area in direct contact with microorganisms [100] that have the capability to provoke inflammatory responses. Several groups reported that PD patients exhibit a pro-inflammatory microbiota profile [41••, 68••, 76] that might cause and increase in gut permeability, allowing leakage of bacterial products and inflammatory mediators from the intestines.
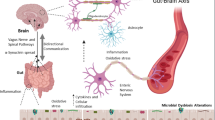
Current evidences indicate that alpha-synuclein deposition in PD might start in the ENS by pro-inflammatory immune activity [101, 102] and propagates to the CNS by transsynaptic cell-to-cell transmission. Overexpressed and aggregated alpha-synuclein would in turn stimulate pro-inflammatory responses from creating a vicious cycle [102, 103]. Alternatively, gut-derived bacterial products or the peripheral inflammatory response (e.g., cytokine production) could impact the brain through systemic mechanisms including disruption of the blood-brain barrier, which is observed in PD patients [104]. In the brain, alpha-synuclein activates microglia [102] that might already been activated due to the ongoing gastrointestinal and systemic immune responses. Within the brain, dopaminergic neurons are particularly sensitive to inflammation [102] and might begin to degenerate. When there is enough depletion of striatal dopamine as the results of the loss of these dopaminergic neurons, motor dysfunction will start manifesting (Fig. 1).

Possible pathways involved in PD pathogenesis. Changes in the gut microbiota composition might increase intestinal permeability, allowing leakage of bacterial products and inflammatory mediators from the intestines. Alpha-synuclein deposition might start in the ENS by pro-inflammatory immune activity and then propagates to the CNS. Alternatively, the peripheral inflammatory response could impact the brain through systemic mechanisms. In the brain, alpha-synuclein can activate microglia that might already been activated due to the ongoing gastrointestinal and systemic immune responses.
Conclusion
PD and possibly other neurodegenerative diseases are associated with dysbiosis in the GI tract, which can result in an unbalance of the host immune system. As a consequence, mucosal and systemic inflammation might ultimately reach the brain where they cause neurodegeneration. Lately, it has been hypothesized that PD pathology starts in the GI tract decades before progressing to the CNS. Therefore, a better understanding of the gut-brain interactions and the role of the intestinal microbiota in the regulation of immune responses might bring new insights in PD pathological progression as well as lead to novel diagnostics and therapeutic approaches. For instance, the modulation of the gut microbiota using pre-, pro-, and antibiotics or fecal transplantations might, in the future, be a viable therapeutic strategy for PD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Djaldetti R, Lev N, Melamed E. Lesions outside the CNS in Parkinson’s disease. Mov Disord Off J Mov Disord Soc. 2009;24(6):793–800.
Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol (Berl). 2010;119(6):689–702.
Del Tredici K, Braak H. Review: Sporadic Parkinson’s disease: development and distribution of α-synuclein pathology. Neuropathol Appl Neurobiol. 2016;42(1):33–50.
Chen H, Zhao EJ, Zhang W, Lu Y, Liu R, Huang X, et al. Meta-analyses on prevalence of selected Parkinson’s nonmotor symptoms before and after diagnosis. Transl Neurodegener. 2015;4(1):1.
Lee HM, Koh S-B. Many faces of Parkinson’s disease: non-motor symptoms of Parkinson’s disease. J Mov Disord. 2015;8(2):92–7.
• Fasano A, Visanji NP, Liu LWC, Lang AE, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015;14(6):625–39. Review describing the high incidence and importance of gastrointestinal dysfunction in PD
Ponsen MM, Stoffers D, Twisk JWR, Wolters EC, Berendse HW. Hyposmia and executive dysfunction as predictors of future Parkinson’s disease: a prospective study. Mov Disord Off J Mov Disord Soc. 2009;24(7):1060–5.
Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, et al. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57(3):456–62.
Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan G a, Pal A, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898.
Clairembault T, Leclair-Visonneau L, Neunlist M, Derkinderen P. Enteric glial cells: new players in Parkinson’s disease? Mov Disord Off J Mov Disord Soc. 2015;30(4):494–8.
Klingelhoefer L, Reichmann H. Pathogenesis of Parkinson disease-the gut-brain axis and environmental factors. Nat Rev Neurol. 2015;11(11):625–36.
Mulak A, Bonaz B. Brain-gut-microbiota axis in Parkinson’s disease. World J Gastroenterol WJG. 2015;21(37):10609–20.
Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701–12.
Rhee SH, Pothoulakis C, Mayer EA. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol. 2009;6(5):306–14.
Grenham S, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbe communication in health and disease. Front Physiol. 2011;2:94.
Borre YE, Moloney RD, Clarke G, Dinan TG, Cryan JF. The impact of microbiota on brain and behavior: mechanisms & therapeutic potential. Adv Exp Med Biol. 2014;817:373–403.
Schapansky J, Nardozzi JD, LaVoie MJ. The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson’s disease. Neuroscience. 2015;302:74–88.
Jellinger KA. Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord. 2012;27(1):8–30.
Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Parkinsonism Relat Disord. 2011 Jan;17(1):10–5.
Savica R, Carlin JM, Grossardt BR, Bower JH, Ahlskog JE, Maraganore DM, et al. Medical records documentation of constipation preceding Parkinson disease: a case-control study. Neurology. 2009;73(21):1752–8.
Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord Off J Mov Disord Soc. 2015;30(12):1600–11.
Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663–7.
Dikiy I, Eliezer D. Folding and misfolding of alpha-synuclein on membranes. Biochim Biophys Acta. 2012;1818(4):1013–8.
Gibb WR, Lees AJ. The significance of the Lewy body in the diagnosis of idiopathic Parkinson’s disease. Neuropathol Appl Neurobiol. 1989;15(1):27–44.
Braak H, Sandmann-Keil D, Gai W, Braak E. Extensive axonal Lewy neurites in Parkinson’s disease: a novel pathological feature revealed by alpha-synuclein immunocytochemistry. Neurosci Lett. 1999;265(1):67–9.
Shults CW. Lewy bodies. Proc Natl Acad Sci U S A. 2006;103(6):1661–8.
Gold A, Turkalp ZT, Munoz DG. Enteric alpha-synuclein expression is increased in Parkinson’s disease but not Alzheimer’s disease. Mov Disord Off J Mov Disord Soc. 2013;28(2):237–40.
Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord Off J Mov Disord Soc. 2012;27(6):716–9.
Braak H, de Vos RAI, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396(1):67–72.
Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord Off J Mov Disord Soc. 2012;27(6):709–15.
•• Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One. 2011;6(12):e28032. Study demonstrating the possible link between intestinal permeability and alpha-synuclein in the enteric nervous system
Sánchez-Ferro Á, Rábano A, Catalán MJ, Rodríguez-Valcárcel FC, Fernández Díez S, Herreros-Rodríguez J, et al. In vivo gastric detection of α-synuclein inclusions in Parkinson’s disease. Mov Disord Off J Mov Disord Soc. 2015;30(4):517–24.
•• Braak H, Rüb U, Gai WP, De1 Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm Vienna Austria 1996. 2003;110(5):517–36. First publication suggesting that PD might start outside the central nervous system
Goedert M, Masuda-Suzukake M, Falcon B. Like prions: the propagation of aggregated tau and α-synuclein in neurodegeneration. Brain J Neurol. 2017;140(Pt 2):266–78.
Uchihara T, Giasson BI. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol (Berl). 2016;131(1):49–73.
Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33(6):599–614.
Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci. 2009;1170:615–22.
Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJA, Kraneveld AD. Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol. 2017;8:37.
Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110(5):517–36.
Hawkes CH, Del Tredici K, Braak H. A timeline for Parkinson’s disease. Parkinsonism Relat Disord. 2010;16(2):79–84.
•• Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in Parkinson’s disease. Mov Disord Off J Mov Disord Soc. 2015;30(10):1351–60. Important publication describing changes in microbiota composition in PD patients
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53.
Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain J Neurol. 2013;136(Pt 4):1128–38.
Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, et al. Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med. 2013;5(7):1119–27.
Ulusoy A, Phillips RJ, Helwig M, Klinkenberg M, Powley TL, Di Monte DA. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol (Berl). 2017;133(3):381–93.
Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol (Berl). 2014;128(6):805–20.
Breid S, Bernis ME, Babila JT, Garza MC, Wille H, Tamgüney G. Neuroinvasion of α-synuclein prionoids after intraperitoneal and intraglossal inoculation. J Virol. 2016;90(20):9182–93.
Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106(31):13010–5.
Brundin P, Li J-Y, Holton JL, Lindvall O, Revesz T. Research in motion: the enigma of Parkinson’s disease pathology spread. Nat Rev Neurosci. 2008;9(10):741–5.
Li J-Y, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14(5):501–3.
Kordower JH, Brundin P. Lewy body pathology in long-term fetal nigral transplants: is Parkinson’s disease transmitted from one neural system to another? Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2009;34(1):254.
• Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78:522–9. Study demonstrating a decrease risk of developing PD in truncal vagotomized subjects
Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, et al. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology. 2017;88(21):1996–2002.
Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One. 2010;5(1) https://doi.org/10.1371/journal.pone.0008762.
Shaikh M, Rajan K, Forsyth CB, Voigt RM, Keshavarzian A. Simultaneous gas-chromatographic urinary measurement of sugar probes to assess intestinal permeability: use of time course analysis to optimize its use to assess regional gut permeability. Clin Chim Acta Int J Clin Chem. 2015;442:24–32.
Clairembault T, Leclair-Visonneau L, Coron E, Bourreille A, Le Dily S, Vavasseur F, et al. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol Commun. 2015;3:12.
Hasegawa S, Goto S, Tsuji H, Okuno T, Asahara T, Nomoto K, et al. Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One. 2015;10(11):e0142164.
Salat-Foix D, Tran K, Ranawaya R, Meddings J, Suchowersky O. Increased intestinal permeability and Parkinson disease patients: chicken or egg? Can J Neurol Sci J Can Sci Neurol. 2012;39(2):185–8.
Quigley EMM, Quera R. Small intestinal bacterial overgrowth: roles of antibiotics, prebiotics, and probiotics. Gastroenterology. 2006;130(2 Suppl 1):S78–90.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–34.
Perez-Pardo P, Kliest T, Dodiya HB, Broersen LM, Garssen J, Keshavarzian A, et al. The gut-brain axis in Parkinson’s disease: possibilities for food-based therapies. Eur J Pharmacol. 2017; https://doi.org/10.1016/j.ejphar.2017.05.042.
Banks WA, Dohgu S, Lynch JL, Fleegal-DeMotta MA, Erickson MA, Nakaoke R, et al. Nitric oxide isoenzymes regulate lipopolysaccharide-enhanced insulin transport across the blood-brain barrier. Endocrinology. 2008;149(4):1514–23.
Banks WA, Erickson MA. The blood–brain barrier and immune function and dysfunction. Spec Issue Blood Brain Barrier. 2010;37(1):26–32.
Gasbarrini A, Lauritano EC, Gabrielli M, Scarpellini E, Lupascu A, Ojetti V, et al. Small intestinal bacterial overgrowth: diagnosis and treatment. Dig Dis Basel Switz. 2007;25(3):237–40.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6(10):e25792.
Tan AH, Mahadeva S, Thalha AM, Gibson PR, Kiew CK, Yeat CM, et al. Small intestinal bacterial overgrowth in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(5):535–40.
Chen WC, Quigley EMM. Probiotics, prebiotics & synbiotics in small intestinal bacterial overgrowth: opening up a new therapeutic horizon! Indian J Med Res. 2014;140(5):582–4.
•• Scheperjans F, Aho V, Pereira PAB, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord Off J Mov Disord Soc. 2015;30(3):350–8. First study demonstrating changes in gut microbiota composition in PD patients
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–80.
dos Santos EF, Busanello ENB, Miglioranza A, Zanatta A, Barchak AG, Vargas CR, et al. Evidence that folic acid deficiency is a major determinant of hyperhomocysteinemia in Parkinson’s disease. Metab Brain Dis. 2009;24(2):257–69.
Luong KVQ, Nguyễn LTH. The beneficial role of thiamine in Parkinson disease. CNS Neurosci Ther. 2013;19(7):461–8.
Andrews ZB, Erion D, Beiler R, Liu Z-W, Abizaid A, Zigman J, et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci. 2009;29(45):14057–65.
Unger MM, Möller JC, Mankel K, Eggert KM, Bohne K, Bodden M, et al. Postprandial ghrelin response is reduced in patients with Parkinson’s disease and idiopathic REM sleep behaviour disorder: a peripheral biomarker for early Parkinson’s disease? J Neurol. 2011;258(6):982–90.
Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev MMBR. 1998;62(2):379–433.
Galán JE, Collmer A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 1999;284(5418):1322–8.
Unger MM, Spiegel J, Dillmann K-U, Grundmann D, Philippeit H, Bürmann J, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord. 2016;32:66–72.
Forsythe P, Kunze WA. Voices from within: gut microbes and the CNS. Cell Mol Life Sci CMLS. 2013;70(1):55–69.
Ganapathy V, Thangaraju M, Prasad PD, Martin PM, Singh N. Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr Opin Pharmacol. 2013;13(6):869–74.
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014 16;40(1):128–39.
Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nat Immunol. 2011;12(1):5–9.
Hopfner F, Künstner A, Müller SH, Künzel S, Zeuner KE, Margraf NG, et al. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 2017;1667:41–5.
Hill-Burns EM, Debelius JW, Morton JT, Wissemann WT, Lewis MR, Wallen ZD, et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord Off J Mov Disord Soc. 2017;32(5):739–49.
Lyte M. Microbial endocrinology in the microbiome-gut-brain axis: how bacterial production and utilization of neurochemicals influence behavior. PLoS Pathog. 2013;9(11):e1003726.
Cryan JF, O’Mahony SM. The microbiome-gut-brain axis: from bowel to behavior. Neurogastroenterol Motil Off J Eur Gastrointest Motil Soc. 2011;23(3):187–92.
Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50(1):42–8.
Lin J-C, Lin C-S, Hsu C-W, Lin C-L, Kao C-H. Association between Parkinson’s disease and inflammatory bowel disease: a nationwide Taiwanese retrospective cohort study. Inflamm Bowel Dis. 2016;22(5):1049–55.
Lai S-W, Liao K-F, Lin C-L, Sung F-C. Irritable bowel syndrome correlates with increased risk of Parkinson’s disease in Taiwan. Eur J Epidemiol. 2014;29(1):57–62.
•• Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167(6):1469–1480.e12. Study demonstrating the importance of the gut microbiota for the genesis of PD phenotype in mice
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–97.
Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S210–2.
Xanthos DN, Sandkühler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15(1):43–53.
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180(2):147–50.
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165(1–2):208–10.
Garcia-Esparcia P, Llorens F, Carmona M, Ferrer I. Complex deregulation and expression of cytokines and mediators of the immune response in Parkinson’s disease brain is region dependent. Brain Pathol Zurich Switz. 2014;24(6):584–98.
Blum-Degen D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202(1–2):17–20.
Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett. 1996;211(1):13–6.
Brodacki B, Staszewski J, Toczyłowska B, Kozłowska E, Drela N, Chalimoniuk M, et al. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci Lett. 2008;441(2):158–62.
Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, et al. Peripheral cytokines profile in Parkinson’s disease. Brain Behav Immun. 2009;23(1):55–63.
Houser MC, Tansey MG. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Park Dis. 2017;3:3.
Helander HF, Fändriks L. Surface area of the digestive tract—revisited. Scand J Gastroenterol. 2014;49(6):681–9.
Kelly LP, Carvey PM, Keshavarzian A, Shannon KM, Shaikh M, Bakay RAE, et al. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord Off J Mov Disord Soc. 2014;29(8):999–1009.
Lema Tomé CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P. Inflammation and α-synuclein’s prion-like behavior in Parkinson’s disease—is there a link? Mol Neurobiol. 2013;47(2):561–74.
Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJ, Anthony DC. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation. 2011;8(1):166.
Guan J, Pavlovic D, Dalkie N, Waldvogel HJ, O’Carroll SJ, Green CR, et al. Vascular degeneration in Parkinson’s disease. Brain Pathol Zurich Switz. 2013;23(2):154–64.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Brain and Microbiome
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Perez-Pardo, P., Hartog, M., Garssen, J. et al. Microbes Tickling Your Tummy: the Importance of the Gut-Brain Axis in Parkinson’s Disease. Curr Behav Neurosci Rep 4, 361–368 (2017). https://doi.org/10.1007/s40473-017-0129-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40473-017-0129-2