
Abstract
Dalbavancin is a lipoglycopeptide antibiotic recently approved by the United States Food and Drug Administration (FDA) for acute bacterial skin and skin structure infections (ABSSSIs). It is active against gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA), and minimum inhibitory concentrations (MICs) are consistently <0.125 µg/ml, much lower than most other anti-MRSA agents. Dalbavancin possesses an extended half-life of over 1 week, allowing an initial dose of 1000 mg followed by 500 mg 1 week later to complete a course of therapy for ABSSSI. It is approximately 95% protein bound and is widely distributed throughout the body, achieving concentrations similar to plasma levels in numerous tissues. Against MRSA, dalbavancin is 4–8 times more potent than vancomycin in vitro, and limited data suggest it possesses activity against MRSA with reduced susceptibility to vancomycin such as hVISA and VISA. Dalbavancin also possesses in vitro activity against streptococci and enterococci, although activity against vancomycin-resistant enterococci is lacking. In phase 3 ABSSSI studies, dalbavancin demonstrated similar activity to vancomycin and provides a more convenient dosing regimen. Limited phase 2 data suggest dalbavancin also possesses activity in catheter-related bloodstream infections. Potential further therapeutic uses include conditions that require long-term treatment such as osteomyelitis and infective endocarditis, although data are currently lacking. The extended half-life of dalbavancin, along with its in vitro activity against gram-positive organisms with reduced susceptibility to other anti-MRSA antibiotics, suggest it could have an exciting clinical role going forward.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Staphylococcus aureus is the leading cause of both community and hospital-acquired infection in the United States [1]. Among S. aureus isolates observed in the United States, between 40% and 50% are methicillin-resistant (MRSA), greatly reducing therapeutic options. MRSA are responsible for several serious infections, including endocarditis, pneumonia, catheter-associated bloodstream infections, osteomyelitis, and skin infections. Vancomycin, a glycopeptide antibiotic derived in the 1950s from Streptomyces orientalis, has been the mainstay in anti-MRSA therapy for more than 50 years [2]. Over this time, vancomycin has remained remarkably active against most gram-positive bacterial strains. A 2013 surveillance study of over 2000 MRSA bloodstream isolates found less than 0.1% possessed a vancomycin minimum inhibitory concentration (MIC) of 4 µg/ml, the MIC that establishes MRSA as a vancomycin-intermediate susceptible Staphylococcus aureus (VISA) according to the Clinical and Laboratory Standards Institute (CLSI) [3]. However, isolates with reduced susceptibility are increasing in the literature after first being reported 20 years ago [4, 5]. Even when vancomycin therapy is appropriate, the management of the antibiotic is complicated. Nephrotoxicity is often associated with vancomycin therapy, and its narrow therapeutic index makes it the only currently available antibiotic with a consensus guideline statement regarding its dosing [6]. The difficulty of vancomycin administration, increase of MRSA with reduced susceptibility to vancomycin, and toxicities associated with vancomycin use have led to the recent development of several novel anti-MRSA antibiotics.
Dalbavancin is a lipoglycopeptide antibiotic derived from teicoplanin, an analog of vancomycin [7]. Dalbavancin is approved for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible, gram-positive isolates [8]. It possesses a similar spectrum of activity to vancomycin, including activity against methicillin-susceptible S. aureus (MSSA) and MRSA [9]. Owing to its extended half-life, dalbavancin is dosed once weekly, with only two doses required for the duration of therapy [10]. This novel antibiotic possesses several qualities that make it an interesting addition to the anti-MRSA armamentarium. This review will serve to introduce dalbavancin, review pertinent in vitro and clinical data, and discuss possible future therapeutic uses for dalbavancin outside the currently FDA-approved indication.
Structure and Mechanism of Action
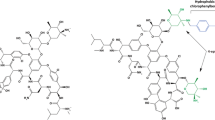
Dalbavancin is a semisynthetic lipoglycopeptide derived structurally from antibiotic A-40926, a teicoplanin-like, natural antibiotic produced by Nonomuria spp. [11]. Several structural alterations were made in an attempt to enhance activity against S. aureus as well as extend the half-life of dalbavancin [11]. Perhaps the most important addition to dalbavancin is the extended, lipophilic side chain not present in teicoplanin or A-40926. This additional side chain allows dalbavancin to anchor to the bacterial cell membrane, enhancing its potency, prolonging its half-life, and allowing for extended dosing intervals [12]. Dalbavancin also possesses an amidated carboxyl side group that enhances the agent’s anti-staphylococcal activity. The structure of dalbavancin is detailed in Fig. 1.

Chemical structure of dalbavancin
Like other agents in its class, dalbavancin exerts its antimicrobial activity through interaction with terminal d-alanyl-d-alanine residues of peptidoglycan precursors [13]. The binding of dalbavancin to these terminal residues prevents both transpeptidase and transglycosylase enzymes from catalyzing peptidoglycan cross-linking and thereby destroying the integrity of the cell wall, ultimately causing cell death [12]. Recent data have demonstrated that the lipophilic side chain of dalbavancin allows dimerization of the molecule and anchors dalbavancin to lipid II in the cellular membrane, strengthening adherence to the d-alanyl-d-alanine target site and allowing for enhanced activity compared to vancomycin and teicoplanin [14].
Microbiology and Resistance
Dalbavancin possesses in vitro activity against several gram-positive pathogens, including S. aureus, Streptococcus agalactiae, Streptococcus pyogenes, Streptococcus anginosus, Enterococcus faecium, and Enterococcus faecalis, although activity against enterococci has not been observed clinically [9, 15–18]. Against staphylococci and streptococci, dalbavancin possesses 8–16-fold greater in vitro activity than vancomycin in broth microdilution MIC testing [15, 16]. Currently, the FDA has an MIC breakpoint established for only S. aureus, S. pyogenes, S. agalactiae, and S. anginosus of ≤0.125 µg/ml [8]. Population data of more than 1100 staphylococci and 300 β-hemolytic streptococci isolates from US medical centers in 2012 report dalbavancin MIC50/90 values of 0.06/0.06 and 0.06/0.12 µg/ml, respectively [15, 16]. Only three staphylococcal isolates (0.3%) and 14 streptococcal isolates (4%) possessed dalbavancin MIC values above the currently proposed FDA breakpoint. Each resistant streptococcal isolate was a member of the species S. agalactiae. Against VISA, vancomycin MIC (4–8 µg/ml) and heteroresistant VISA (hVISA), dalbavancin maintains four- to eightfold greater potency than vancomycin, although the MIC values largely fall above 0.12 µg/ml [18]. Recently, Jones and colleagues demonstrated that vancomycin susceptibility can be considered a surrogate for dalbavancin susceptibility, as greater than 99.9% of over 42,000 vancomycin-susceptible isolates were susceptible to dalbavancin [9]. Against enterococci, dalbavancin possesses higher MIC values than it does against other gram-positive species. Dalbavancin activity against enterococci is largely contingent upon vancomycin activity, as 100% of vancomycin-susceptible Enterococcus faecalis and E. faecium are inhibited at 0.125 µg/ml [15, 16]. However, vancomycin-resistant isolates of each species possess dalbavancin MIC90 values greater than 4 µg/ml.
Resistance to dalbavancin among staphylococci is rare, being reported in less than 1% of isolates [15, 16]. Mechanistically, there is precedent for reduced vancomycin susceptibility predicting reduced susceptibility to dalbavancin. Reduced susceptibility among hVISA and VISA is mediated through thickening of the cell wall and increased d-alanine-d-alanine binding sites [19–21]. These increased binding sites increase the amount of vancomycin bound to the cell wall, necessitating higher exposures for similar efficacy. Although dalbavancin is more potent at the active site than vancomycin, it is still inhibited in the same fashion. It remains to be seen whether the increased potency of dalbavancin will give it appreciable activity against these phenotypes clinically. Vancomycin-resistant S. aureus (VRSA) are similarly resistant to dalbavancin, although the mechanism for this resistance is different than for hVISA and VISA isolates. Vancomycin, and subsequently dalbavancin, resistance within VRSA is mediated through a plasmid gene VanA or VanB carried over from enterococcal species [22]. These genes are responsible for altering the d-alanyl-d-alanine target site to a d-alanyl-d-lactate, rendering glycopeptide antibiotics ineffective [23]. Interestingly, dalbavancin has in vitro activity against enterococci with VanB present, but not against those with VanA [16]. The VanB activity may be present due to dalbavancin being a derivative of teicoplanin, which also possesses activity against enterococci with VanB. Although this activity is worth noting, the vast majority of vancomycin-resistant enterococci in the United States possess VanA [24]. A complete breakdown of currently available dalbavancin MIC data against gram-positive organisms is available in Table 1.
Dosing, Pharmacokinetics, and Pharmacodynamics
Dalbavancin is currently FDA-approved at a dose of 1000 mg on day 1 followed by 500 mg on day 8 for a complete course of therapy for ABSSSI [8]. Pharmacokinetic data from human studies demonstrate that dalbavancin possesses linear, dose-related pharmacokinetics with an extended elimination half-life of approximately 14.5 days (346 h), allowing for the extended interval between doses [28–32]. The approved dosing regimen was based on a clinical study evaluating complicated skin and skin structure infections (cSSSI) in which dalbavancin dosed at 1000 mg initially followed by 500 mg on day 8 was numerically superior to a one-time dose of 1100 mg [33]. Nearly 33% of dalbavancin is excreted in the urine unchanged, suggesting that non-renal methods of elimination play an important role in the metabolism of dalbavancin [28]. The extended half-life of dalbavancin is largely due to extensive, reversible binding to serum albumin, estimated to be roughly 95% [34]. Among both healthy subjects and those with varying degrees of renal dysfunction, the maximum concentration (C max) of dalbavancin falls between 248 and 312 µg/ml following the standard 1000 mg initial dose [28, 30]. Dalbavancin exhibits a large volume of distribution, with an initial average of 8–12 L and an ultimate volume of distribution of up to 15 L once the drug has distributed throughout the tissues [32]. Data from nine healthy male subjects given 1000 mg demonstrate that dalbavancin maintains concentrations in skin blister fluid well above current MIC90 values through 7 days, suggesting that dalbavancin will be active for the duration of ABSSSI treatment [35]. In a pharmacokinetic study of dalbavancin in patients with varying renal and hepatic function, area under the curve (AUC) values were markedly elevated over the treatment duration for patients with creatinine clearances (CrCl) of 30 ml/min relative to patients with normal renal function [30]. Because of its reduced clearance, patients with renal impairment (CrCl < 30 ml/min) should have dalbavancin doses adjusted to 750 mg initially followed by 375 mg on day 8 for a complete treatment course. Interestingly, dalbavancin is not efficiently cleared by hemodialysis and levels are similar between patients on dialysis and those with normal renal function [30]. Therefore, although it seems contradictory to data in patients with CrCl < 30 ml/min, patients on regularly scheduled hemodialysis do not require dosage adjustment. The same study found that hepatic function had no appreciable affect on dalbavancin clearance and likely does not need to be taken into account when dosing, although caution should be exercised in moderate to severe hepatic impairment, as clinical data are limited.
The pharmacokinetic parameter associated with efficacy of dalbavancin both in vitro and in vivo is the ratio of AUC to MIC, as is generally the case with glycopeptides [36]. In the initial pharmacokinetic studies, bactericidal concentrations of dalbavancin were present in serum samples up to 7 days following an initial, one-time dose of as little as 500 mg [28]. With regard to the AUC:MIC target, population pharmacokinetic modeling of dalbavancin standard dosing from three clinical trials demonstrated a likelihood of target attainment of near 100% at dalbavancin MICs up to 0.5 µg/ml [31]. Similarly, a recent study with 10,000 Monte Carlo simulations of dalbavancin 1000 mg followed by 500 mg 1 week later also showed a 100% probability of target attainment for MRSA isolates with dalbavancin MICs up to 0.12 µg/ml [37].
In Vitro and Animal Data
There are in vitro studies that have evaluated the activity of dalbavancin against gram-positive organisms, some of which examined isolates with reduced susceptibility to vancomycin. In one such study that evaluated several dalbavancin dose exposures against S. aureus, continuous concentrations of 3 µg/ml were sufficient to provide bactericidal activity against both MSSA and MRSA over 20 days [38]. Against VISA, concentrations of 15 µg/ml were bactericidal for 20 days. Dalbavancin was also studied in combination with several antibiotics against MSSA, MRSA, VISA, MRSE, vancomycin-susceptible enterococci (VSE), S. pyogenes and S. pneumoniae using broth microdilution checkerboard techniques [39]. Oxacillin was synergistic with dalbavancin against MRSA, VISA, MRSE, and VSE. Among the other agents tested, including daptomycin, clindamycin, linezolid, levofloxacin, gentamicin, quinupristin/dalfopristin, rifampicin, and vancomycin, none were antagonistic when combined with dalbavancin against these bacteria. These in vitro data are limited but suggest dalbavancin may have activity, either alone or in combination, against problem organisms such as VISA.
Dalbavancin has also been studied in several animal models of infection. When dalbavancin was evaluated against S. aureus and S. pneumoniae in neutropenic murine thigh and lung models, it was determined that AUC:MIC and C max:MIC were both correlated with dalbavancin activity [36]. These data formed the dosing regimens used in human studies.
In a study published by Lefort et al., dalbavancin possessed activity against vancomycin-susceptible S. aureus (VSSA) and VISA dependent upon dose in a rabbit model of endocarditis [40]. In the authors’ in vitro time kill studies, dalbavancin possessed superior activity to teicoplanin and vancomycin against VSSA and VISA. However, dalbavancin was not bactericidal in the endocarditis model. The authors postulated that, due to the enhanced clearance of dalbavancin in rabbits compared to people, drug exposures were not appropriate for bactericidal activity, although they may be in humans. Further studies evaluating additional dosing options in the setting of endocarditis are needed.
Dalbavancin has also been studied in a foreign-body infection model in guinea pigs both alone and in combination with rifampin [41]. Dalbavancin alone was unable to eradicate adherent MRSA, but it was able to prevent resistance to rifampin. The combination of agents was effective and superior to either agent alone, suggesting that rifampin may play an important role in therapy targeting biofilm infections.
Clinical Trials
In addition to the study that led to its approval for ABSSSI, dalbavancin has been previously evaluated clinically in the setting of complicated skin and skin structure infections (cSSSI) and bloodstream infections. In a phase II, multicenter, non-inferiority, randomized controlled trial, 2 weeks of twice daily vancomycin was compared to standard-dose dalbavancin against catheter-related bloodstream infections (CR-BSI) due to gram-positive pathogens [42]. Patients were ≥18 years old with signs of bacteremia and at least one of the following: intravascular catheter present at the start of infection, absence of any other likely source of infection, and either microbiologically documented gram-positive bacteremia or ≥2 signs of bacteremia. Patients were excluded if they had impaired renal or hepatic function, recently received immunosuppressive therapy, prolonged neutropenia, received prior antibiotic active against gram-positive bacteria within 48 h of enrollment, S. aureus bacteremia within the previous 3 months, or an alternative source of infection. The primary outcome was overall efficacy at the follow-up visit defined by clinically and microbiologically documented responses among patients with a bacterial pathogen identified at study entry. Clinical success was defined as resolution of signs and symptoms of CR-BSI such that no additional treatment was warranted. Seventy-five patients were randomized to treatment, of which 67 were included in efficacy analyses. Baseline characteristics were similar, although slightly more females were enrolled in the vancomycin group. Coagulase-negative staphylococci and S. aureus were the primary pathogens isolated, with a higher proportion of S. aureus (32.1%) reported as MRSA in the vancomycin group compared to the dalbavancin group (19.2%). The overall success rate was 87% for dalbavancin compared to 50% for vancomycin (p < 0.05), and both treatment groups were more successful with catheter removal. This effect carried across the microbiological intention to treat cohort at both end of therapy and test of cure visits, and the effect was also present among the evaluable population at both time points. Adverse effects reported by study patients were generally mild, and no patients discontinued dalbavancin due to adverse effects. In the vancomycin group, three patients withdrew therapy due to adverse effects, with one patient perhaps experiencing renal toxicity due to vancomycin. The data from this study demonstrated the efficacy of dalbavancin 1000 mg on day 1 followed by 500 mg 1 week later against uncomplicated, gram-positive bacteremia. Although not directly demonstrated by the study, the authors postulated that the increased peak levels and lipophilic tail of dalbavancin might have aided it in clearance of biofilm on infected catheters, perhaps explaining the enhanced efficacy.
Gram-positive pathogens also frequently cause skin infections, and they are a large source of healthcare expenditures with over 15 million infections annually in the United States [43]. Owing to the convenient dosing of dalbavancin and its ability to keep patients out of infusion centers, two published studies have evaluated dalbavancin for the treatment of ABSSSI against active comparators [10, 33]. In a phase II, randomized controlled study, Seltzer et al. evaluated dalbavancin given as either a single 1100 mg dose or 1000 mg followed by 500 mg 1 week later against several standard-of-care regimens for skin and soft-tissue infection (SSTI) [33]. Sixty-two patients were enrolled in the study, and patients were ≥18 years old with SSTI suspected or known to be caused by a gram-positive pathogen. Infections were required to involve deep tissue or require surgical intervention, and patients were required to have ≥2 symptoms of SSTI. Of note, patients with creatinine clearance <50 ml/min were excluded. The primary efficacy endpoint was clinical cure or improvement at follow-up visit. Failure was defined as persistence of ≥1 systemic sign or symptom of SSTI that required further therapy. The study also reported dalbavancin pharmacokinetics. At follow-up visit, a higher proportion of 2-dose dalbavancin patients achieved cure in the intention-to-treat population (19/21, 91%) compared to 1-dose dalbavancin (12/20, 60%) or comparator regimen (16/21, 76%). Dalbavancin concentrations remained >30 µg/ml for 1 week after the 1100 mg dalbavancin dose, and concentrations remained >20 µg/ml for 20 days in patients who received 1000 mg followed by 500 mg. Adverse effects were mild and considered to be unrelated to study drug. The data from this study established that dalbavancin is likely optimally dosed at 1000 mg with a supplementary 500 mg dose 1 week later for skin infections.
Jauregui et al. were the first to evaluate dalbavancin at 1000 mg followed by 500 mg 1 week later in a phase III trial for the treatment of cSSSI [44]. The study was a multicenter, non-inferiority trial comparing dalbavancin to linezolid for 14 days. Enrollment criteria were similar to the study by Seltzer and colleagues. The primary end point was clinical success, defined as the lack of necessary further antibacterial therapy. The treatment groups were similar at baseline except for a higher proportion of vascular disease in the dalbavancin group. The average age was 47 years, and 62% of patients were male. Abscess (32%) and cellulitis (28%) were the primary types of infection. Efficacy at test-of-cure for dalbavancin was 88.9% and was 91.2% for linezolid (lower limit CI −7.28%), which achieved non-inferiority. The vast majority of cultured isolates were from abscesses, with 90% of those isolates being S. aureus, and dalbavancin and linezolid cured 91% and 89% of these isolates, respectively. Adverse events were generally mild, and adverse events related to treatment were higher in the linezolid arm compared to dalbavancin (32.2–25.4%, respectively).
Boucher and colleagues evaluated dalbavancin dosed at 1000 mg followed by 500 mg in two phase III, randomized controlled trials comparing dalbavancin to standard therapy against ABSSSI, DISCOVER 1 and DISCOVER 2 [10]. Standard therapy consisted of at least 3 days of intravenous vancomycin followed by an optional switch to oral linezolid for the completion of 10–14 days of therapy. The two trials were identically designed, double-dummy, international, non-inferiority studies, allowing for pooling of data. Patients were included who were ≥18 years old and were diagnosed with ABSSSI. The diagnosis of ABSSSI was based on cellulitis, a major abscess, or wound infection with an area of at least 75 cm2, which is about the area of a modern smart phone. Patients were eligible if they were likely to require at least 3 days of intravenous antibiotics and possessed at least one systemic sign of infection. In the DISCOVER trials, enrollment was limited to a maximum of 30% of patients with abscess, owing to high cure rates in these patients with incision and drainage alone, and at least 25% of patients had fever, establishing a less healthy population than previous studies. Per recent FDA guidance, the primary endpoint was cessation of spread of erythema and a temperature of ≤37.6 °C at 48–72 h. More than 85% of patients enrolled possessed temperatures ≥38 °C, and the median size of infection was 343 cm2. The majority of patients were white (89.3%) with a mean age of 49.5 years. Greater than 50% of patients met SIRS criteria, and <26% of patients had abscesses.
In the pooled dalbavancin group, 525 of 659 patients (79.7%) had a successful outcome compared to 521 of 653 patients (79.8%) in the standard therapy group (95% CI −4.5 to 4.2), meeting the criteria for non-inferiority. Missing temperature data was the primary reason for failure in both treatment arms and was the primary treatment-related reason for failure. The two arms were similarly successful in patients with bacteremia, as 23 of 23 patients (100%) in the dalbavancin arm had negative cultures at follow-up and 12 of 14 patients (85.7%) in the standard therapy arm had negative cultures. Clinical success was similar between the arms according to infection type, severity of illness, and severity of infection. Each treatment regimen was >97% successful against documented MRSA skin infection.
More patients in the standard therapy arm experienced an adverse event (645 vs. 540, p = 0.05) compared to patients in the dalbavancin arm, and the total number of treatment-related adverse events was higher in the standard therapy arm as well (183 vs. 139, p = 0.02). However, serious adverse events were similar between the groups. The most common adverse events were nausea, diarrhea, and pruritus, and each occurred in <3% of patients. Only two patients in the study, both in the vancomycin-linezolid arm, experienced nephrotoxic adverse effects.
The data from this study suggest that dalbavancin is similarly efficacious to standard therapy against ABSSSI with regard to the newly established FDA criteria. The patients in this study were quite sick, with >85% possessing fever and more than half meeting SIRS criteria, demonstrating that dalbavancin can be an effective alternative to therapy with vancomycin and linezolid, especially in the case of suspected MRSA infection. The incidence of serious adverse effects is low and has been similar to all comparator arms.
Currently, dalbavancin is being evaluated in a phase 3b study to compare the FDA-approved dose against a one-time dose of 1500 mg for ABSSSI (ClinicalTrials.gov NCT02127970). The results have yet to be published, but preliminary data presented in a press release suggest that the one-time dose of 1500 mg is similar to the two-dose regimen with regard to ≥20% reduction in ABSSSI lesion size at 48–72 h. Although conclusions cannot be made without the full breadth of data available, it does appear that dalbavancin may be efficacious and safe with a one-time dose, further enhancing its therapeutic ease of use.
Safety and Drug Interactions
The safety of dalbavancin has been evaluated in several clinical and pre-clinical studies. Combined phase II and phase III data thus far have revealed the most common adverse effects to be nausea (5.5%), headache (4.7%), diarrhea (4.4%), vomiting (2.8%), rash (2.7%), and pruritus (2.1%) [10, 28–30, 33, 42]. In the phase II study of CR-BSI, dalbavancin patients experienced more hypokalemia (6/33, 18%) and hypotension (7/33, 21%) than those treated with vancomycin, although these trends did not continue across other studies [42]. Additionally, dalbavancin has demonstrated a 0.8% rate of ALT levels greater than three times the upper limit of normal, although these have been reversible with treatment cessation [8]. In the phase III clinical trials, treatment-related adverse events were fewer in the dalbavancin arms compared to treatment with vancomycin or linezolid [10, 44]. In contrast to the nephrotoxic effects often associated with glycopeptides, dalbavancin has not been associated with nephrotoxicity to date. Although increased nephrotoxicity was not demonstrated in the vancomycin arms of comparator trials, nephrotoxicity is a historically demonstrated adverse effect of vancomycin, and the lack of reduced renal function with dalbavancin therapy is encouraging. Although further safety data are needed, it appears that dalbavancin has a favorable renal safety profile. Due to potential concern for QTc prolongation with telavancin, another lipoglycopeptide antibiotic, dalbavancin has recently been studied at therapeutic and supratherapeutic doses, and dalbavancin caused no increase in QTc interval [45, 46]. Because dalbavancin has such an extended half-life, there has been some concern regarding the potential for long-term adverse effects. To date, no adverse effects have been demonstrated up to 28 days after treatment cessation [10]. However, long-term data are still limited, and post-approval adverse effect reporting will shed more light on any long-term effects.
Several studies have evaluated dalbavancin pharmacokinetics in the presence of known cytochrome P450 inducers, substrates, and inhibitors to determine the presence of significant drug interactions [28, 32]. Unsurprisingly, dalbavancin was not affected by co-administration of other agents, as dalbavancin is not cleared by the P450 metabolic pathway. The available evidence suggests that dalbavancin can be administered without regard to agents known to affect the P450 pathway.
Conclusion
With options for the treatment of resistant staphylococcal infections limited and vancomycin susceptibility demonstrating signs of decline, dalbavancin presents an exciting therapeutic alternative. It has evidenced an excellent safety and efficacy profile in the setting of ABSSSI and has gained FDA approval for this indication. ABSSSI infections continue to be a large healthcare burden, and the availability of a two-dose therapeutic regimen has important implications for patient adherence, potential hospital avoidance, and cost savings versus inpatient therapy. Emergency departments and observational care units stand to benefit especially, as patients will be able to be easily transitioned back into the community after being seen for only a limited amount of time. Perhaps most exciting are the clinical opportunities for dalbavancin use outside of its current approved indication. Dalbavancin has already demonstrated efficacy in CR-BSI, and further studies will be important to determine its place in deep-seated infections such as pneumonia, osteomyelitis, and endocarditis. Preliminary data are promising, as dalbavancin achieves concentrations in bone tissue well above staphylococcal MIC90 values 14 days after a 1000 mg dose [47]. Going forward, it will be imperative to determine optimal dosing strategies for infections that require weeks-long durations of therapy. Owing to the activity dalbavancin possesses in vitro and in vivo against S. aureus with reduced vancomycin susceptibility, further study involving dalbavancin against these isolates is also intriguing and warranted. It remains to be seen whether antibiotic combinations with dalbavancin will display similar additive effects as they do with vancomycin and daptomycin against gram-positive pathogens. Dalbavancin has the potential to be an effective agent to combat resistant gram-positive organisms, and expanded use of this agent post-approval will help determine its ultimate place in therapy.
References
Sievert DM, Ricks P, Edwards JR, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol. 2013;34:1–14.
Moellering RC Jr. Vancomycin: a 50-year reassessment. Clin Infect Dis. 2006;42(Suppl 1):S3–4.
Sader HS, Farrell DJ, Flamm RK, Jones RN. Activity of ceftaroline and comparator agents tested against Staphylococcus aureus from patients with bloodstream infections in US medical centres (2009–13). J Antimicrob Chemother. 2015;70(7):2053–56.
Hiramatsu K. The emergence of Staphylococcus aureus with reduced susceptibility to vancomycin in Japan. Am J Med. 1998;104:7S–10S.
Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother. 1997;40:135–6.
Rybak M, Lomaestro B, Rotschafer JC, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health-Syst Pharm. 2009;66:82–98.
Kim A, Kuti JL, Nicolau DP. Review of dalbavancin, a novel semisynthetic lipoglycopeptide. Expert Opin Investig Drugs. 2007;16:717–33.
Dalvance (dalbavancin) for injection [package insert]. Chicago, IL: Durata; Rev 05/14.
Jones RN, Farrell DJ, Flamm RK, Sader HS, Dunne MW, Mendes RE. Surrogate analysis of vancomycin to predict susceptible categorization of dalbavancin. Diagn Microbiol Infect Dis. 2015;82(1):73–7.
Boucher HW, Wilcox M, Talbot GH, Puttagunta S, Das AF, Dunne MW. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370:2169–79.
Malabarba A, Goldstein BP. Origin, structure, and activity in vitro and in vivo of dalbavancin. J Antimicrob Chemother. 2005;55(Suppl 2):ii15–20.
Zhanel GG, Calic D, Schweizer F, et al. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs. 2010;70:859–86.
Nagarajan R. Structure–activity relationships of vancomycin-type glycopeptide antibiotics. J Antibiot. 1993;46:1181–95.
Trevino J, Bayon C, Arda A, et al. New insights into glycopeptide antibiotic binding to cell wall precursors using SPR and NMR spectroscopy. Chemistry. 2014;20:7363–72.
Jones RN, Flamm RK, Sader HS. Surveillance of dalbavancin potency and spectrum in the United States (2012). Diagn Microbiol Infect Dis. 2013;76:122–3.
Jones RN, Sader HS, Flamm RK. Update of dalbavancin spectrum and potency in the USA: report from the SENTRY Antimicrobial Surveillance Program (2011). Diagn Microbiol Infect Dis. 2013;75:304–7.
Chong YP, Park SJ, Kim HS, et al. In vitro activities of ceftobiprole, dalbavancin, daptomycin, linezolid, and tigecycline against methicillin-resistant Staphylococcus aureus blood isolates: stratified analysis by vancomycin MIC. Diagn Microbiol Infect Dis. 2012;73:264–6.
Citron DM, Tyrrell KL, Goldstein EJ. Comparative in vitro activities of dalbavancin and seven comparator agents against 41 Staphylococcus species cultured from osteomyelitis infections and 18 VISA and hVISA strains. Diagn Microbiol Infect Dis. 2014;79:438–40.
Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139.
Howden BP, Peleg AY, Stinear TP. The evolution of vancomycin intermediate Staphylococcus aureus (VISA) and heterogenous-VISA. Infect Genet Evol. 2013;21:575–82.
Ortwine JK, Werth BJ, Sakoulas G, Rybak MJ. Reduced glycopeptide and lipopeptide susceptibility in Staphylococcus aureus and the “seesaw effect”: taking advantage of the back door left open? Drug Resist Updates: Rev Comment Antimicrob Anticancer Chemother. 2013;16:73–9.
Sievert DM, Rudrik JT, Patel JB, McDonald LC, Wilkins MJ, Hageman JC. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin Infect Dis. 2008;46:668–74.
Rossi F, Diaz L, Wollam A, et al. Transferable vancomycin resistance in a community-associated MRSA lineage. N Engl J Med. 2014;370:1524–31.
Deshpande LM, Fritsche TR, Moet GJ, Biedenbach DJ, Jones RN. Antimicrobial resistance and molecular epidemiology of vancomycin-resistant enterococci from North America and Europe: a report from the SENTRY antimicrobial surveillance program. Diagn Microbiol Infect Dis. 2007;58:163–70.
McCurdy SP, Jones RN, Mendes RE, Puttagunta S, Dunne MW. In vitro activity of dalbavancin against drug resistant Staphylococcus aureus from a Global Surveillance Program. Antimicrob Agents Chemother. 2015;59(8):5007–9.
Jones RN, Stilwell MG. Comprehensive update of dalbavancin activity when tested against uncommonly isolated streptococci, Corynebacterium spp., Listeria monocytogenes, and Micrococcus spp. (1357 strains). Diagn Microbiol Infect Dis. 2013;76:239–40.
Karlowsky JA, Adam HJ, Poutanen SM, Hoban DJ, Zhanel GG. In vitro activity of dalbavancin and telavancin against staphylococci and streptococci isolated from patients in Canadian hospitals: results of the CANWARD 2007–2009 study. Diagn Microbiol Infect Dis. 2011;69:342–7.
Leighton A, Gottlieb AB, Dorr MB, et al. Tolerability, pharmacokinetics, and serum bactericidal activity of intravenous dalbavancin in healthy volunteers. Antimicrob Agents Chemother. 2004;48:940–5.
Dorr MB, Jabes D, Cavaleri M, et al. Human pharmacokinetics and rationale for once-weekly dosing of dalbavancin, a semi-synthetic glycopeptide. J Antimicrob Chemother. 2005;55(Suppl 2):ii25–30.
Marbury T, Dowell JA, Seltzer E, Buckwalter M. Pharmacokinetics of dalbavancin in patients with renal or hepatic impairment. J Clin Pharmacol. 2009;49:465–76.
Dowell JA, Goldstein BP, Buckwalter M, Stogniew M, Damle B. Pharmacokinetic-pharmacodynamic modeling of dalbavancin, a novel glycopeptide antibiotic. J Clin Pharmacol. 2008;48:1063–8.
Buckwalter M, Dowell JA. Population pharmacokinetic analysis of dalbavancin, a novel lipoglycopeptide. J Clin Pharmacol. 2005;45:1279–87.
Seltzer E, Dorr MB, Goldstein BP, Perry M, Dowell JA, Henkel T. Once-weekly dalbavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infections. Clin Infect Dis. 2003;37:1298–303.
Cavaleri M, Riva S, Valagussa A, et al. Pharmacokinetics and excretion of dalbavancin in the rat. J Antimicrob Chemother. 2005;55(Suppl 2):ii31–5.
Nicolau DP, Sun HK, Seltzer E, Buckwalter M, Dowell JA. Pharmacokinetics of dalbavancin in plasma and skin blister fluid. J Antimicrob Chemother. 2007;60:681–4.
Andes D, Craig WA. In vivo pharmacodynamic activity of the glycopeptide dalbavancin. Antimicrob Agents Chemother. 2007;51:1633–42.
Salem AH, Zhanel GG, Ibrahim SA, Noreddin AM. Monte Carlo simulation analysis of ceftobiprole, dalbavancin, daptomycin, tigecycline, linezolid and vancomycin pharmacodynamics against intensive care unit-isolated methicillin-resistant Staphylococcus aureus. Clin Exp Pharmacol Physiol. 2014;41:437–43.
Bowker KE, Noel AR, MacGowan AP. Pharmacodynamics of dalbavancin studied in an in vitro pharmacokinetic system. J Antimicrob Chemother. 2006;58:802–5.
Johnson DM, Fritsche TR, Sader HS, Jones RN. Evaluation of dalbavancin in combination with nine antimicrobial agents to detect enhanced or antagonistic interactions. Int J Antimicrob Agents. 2006;27:557–60.
Lefort A, Pavie J, Garry L, Chau F, Fantin B. Activities of dalbavancin in vitro and in a rabbit model of experimental endocarditis due to Staphylococcus aureus with or without reduced susceptibility to vancomycin and teicoplanin. Antimicrob Agents Chemother. 2004;48:1061–4.
Baldoni D, Furustrand Tafin U, Aeppli S, et al. Activity of dalbavancin, alone and in combination with rifampicin, against meticillin-resistant Staphylococcus aureus in a foreign-body infection model. Int J Antimicrob Agents. 2013;42:220–5.
Raad I, Darouiche R, Vazquez J, et al. Efficacy and safety of weekly dalbavancin therapy for catheter-related bloodstream infection caused by Gram-positive pathogens. Clin Infect Dis. 2005;40:374–80.
Ray GT, Suaya JA, Baxter R. Incidence, microbiology, and patient characteristics of skin and soft-tissue infections in a U.S. population: a retrospective population-based study. BMC Infect Dis. 2013;13:252.
Jauregui LE, Babazadeh S, Seltzer E, et al. Randomized, double-blind comparison of once-weekly dalbavancin versus twice-daily linezolid therapy for the treatment of complicated skin and skin structure infections. Clin Infect Dis. 2005;41:1407–15.
Barriere S, Genter F, Spencer E, Kitt M, Hoelscher D, Morganroth J. Effects of a new antibacterial, telavancin, on cardiac repolarization (QTc interval duration) in healthy subjects. J Clin Pharmacol. 2004;44:689–95.
Dunne MW, Zhou M, Darpo B. A thorough QT study with dalbavancin: a novel lipoglycopeptide antibiotic for the treatment of acute bacterial skin and skin-structure infections. Int J Antimicrob Agents. 2015;45:393–8.
Dunne MW, Puttagunta S, Sprenger CR, Rubino C, Van Wart S, Baldassarre J. Extended-duration dosing and distribution of dalbavancin into bone and articular tissue. Antimicrob Agents Chemother. 2015;59:1849–55.
Acknowledgments
No sponsorship or funding was received for this publication. The authors would like to acknowledge Jeffrey M. Rybak for his assistance with the drawing of the chemical structure of dalbavancin. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Conflict of interest
Jordan R. Smith and Karrine D. Roberts have nothing to disclose. Michael J. Rybak has been a speaker and/or consultant and obtained research support from Actavis, Cubist, and Theravance; speaker and/or consultant for Bayer, Durata, Novartis, The Medicines Company, and Sunovian; and obtained research and partial salary support from NIH R21 AI109266-01.
Compliance with ethics guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Smith, J.R., Roberts, K.D. & Rybak, M.J. Dalbavancin: A Novel Lipoglycopeptide Antibiotic with Extended Activity Against Gram-Positive Infections. Infect Dis Ther 4, 245–258 (2015). https://doi.org/10.1007/s40121-015-0077-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-015-0077-7