
Abstract
COVID-19 is a disease caused by SARS-CoV-2 and was initially considered to cause serious damage to the respiratory system. Over time, it has been found to affect other organs due to its ability to bind to the ACE2 receptor (type 2 angiotensin-converting enzyme), which can be found in various tissues, including the central nervous system. In addition, a large formation of pro-inflammatory cytokines responsible for various lesions was observed during the evolution of this disease. Our objective was to demonstrate the molecular mechanisms involved in the infection that may demonstrate the relationship between COVID-19 and the development of depressive conditions. Based on the main medical databases (LiLacs, SciELO, Bireme, Scopus, EBSCO, and PubMed) and using the terms 'coronavirus infections' AND 'Inflammation' AND 'depression' AND 'cytokines', we conducted an integrative review of articles published in 2020. Considering this stage of Covid-19 and the inflammatory component of depression, this review showed a relationship between these two conditions based on common pathophysiological mechanisms indicating possible depressive disorders in surviving patients, especially in the most severe cases. The role of inflammatory cytokines and the presence of ACE-2 receptors on the cell surface appear to be the common pathophysiological mechanism between COVID-19 and depression.
Similar content being viewed by others


Introduction
COVID-19 was first found in Wuhan (China) in 2019, caused by SARS-CoV-2. It was considered a pandemic by the WHO, given that more than seven million cases were reported worldwide. Among the main countries affected by the disease are the USA, Brazil, and Russia [1,2,3]. The large number of infected people is worrying, considering that so far there is no specific treatment for this disease and there is a small percentage of people vaccinated. As a result, we will have a major impact on the world economy [4,5,6].
Initially considered a respiratory system disease, COVID-19 showed that it has great invasive potential to other components of the human body and can lead to serious complications. Many systemic effects have been described in the literature, among which we can cite cardiac, digestive, vascular impairment, and others not yet well elucidated, for example, its effects on the central nervous [7,8,9,10,11].
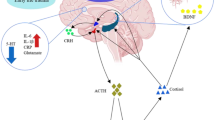
It is known that there are several types of viruses that can affect the central nervous system, including those that essentially affect its structure. Studies show that the hepatitis C virus, Zika virus, and others have this potential and may cause damage because of their invasion in this system [12, 13]. Among the resulting complications are Guillain–Barre syndrome, encephalitis, headache, meningitis, malformation, convulsions, and other manifestations. Similarly, symptoms involving memory changes, mood changes such as depression and anxiety, and schizophrenia have also been described (Fig. 1) [14]. Several studies indicate that coronaviruses have several neurotropic strains, such as MERS-CoV and SARS-CoV [15].

Neurological and psychiatric presentations associated with severe coronavirus infections
Neural manifestations may occur simultaneously with respiratory symptoms or even after a few weeks of patient recovery. This characteristic is also observed in SARS-CoV-2 because the receptor it uses to enter the respiratory cells and other tissues is also expressed in neurons and Glia cells [11, 16, 17]. Studies show this neural impairment in COVID-19 manifests itself through symptoms such as changes in smell, dizziness, and changes in consciousness, encephalitis, headache, anxiety, and depression.
Studies have shown that anxiety, memory or concentration difficulties, and depressed mood are the main neuropsychiatric manifestations in patients infected with the coronavirus, suggesting that the appearance of these symptoms may be a consequence of the viral infection. The incidence was higher in patients who required hospitalization and more evident in those who required intensive care unit admission or developed encephalopathy [7, 18]. If we consider the pathogenesis of COVID-19 and the most recent approaches on the pathophysiology of depression, it is possible that there are common mechanisms between the two conditions [16, 19,20,21].
Thus, this review aims to demonstrate the molecular mechanisms involved in the course of this infection that may demonstrate evidence of a close relationship between COVID-19 and the development of depressive conditions.
Methods
Based on the main medical databases (LiLacs, SciELO, Bireme, Scopus, EBSCO, and PubMed) and using the terms 'coronavirus infections' AND 'Inflammation' AND 'depression' AND 'cytokines', we conducted an integrative review of articles published in 2020.
Articles written in English were included. Editorials, comments, letters to the editor, review articles, articles that were not available in full or that did not contain accurate information were excluded. To guarantee the validity of these articles, the selected studies were analyzed in detail, by all authors, regarding the relationship between COVID-19 and depression. In our search, we found a total of 21 articles that made up this review.
Results and discussion
The pathophysiology of covid-19 and the inflammatory response
After entering the body, SARS-CoV-2 uses the spike glycoprotein to bind to the Angiotensin 2 Converting Enzyme (ACE2) and then enters the host cell [22]. Then, it releases viral RNA in the cell cytoplasm and the process of replication begins generating new viral particles which leads to cell death and the migration of the virus to attack other cells. The viral antigens are recognized by the antigen-presenting cells which in turn activate lymphocytes leading to the production of cytokines and chemokines. There is an increase in the production of IFN-I or IFN-α/β, inducing accumulations of mononucleated macrophages which, in turn, will produce large quantities of pro-inflammatory cytokines such as TNF-α, IL-6, and IL1-β, in addition to iNOS. In some patients, there is a large production of these mediators and this inflammatory response can lead to thrombus formation and failure of multiple organs and even death [23, 24].
There is a variation in the production of the different types of mediators. In vitro studies have shown that there is an increase in cytokine and chemokine production in respiratory epithelium cells, dendritic cells, and monocytes/macrophages in the early stages of infection. Subsequently, there is a decreased production of antiviral interferons and increased production of the pro-inflammatory mediators mentioned above, in addition to the chemokines CCL-2, CCL-3, and CCL-5 [23].
It is worth noting that this virus can attack different tissues causing several injuries to the body. This occurs due to the abundant presence of ACE-2 receptors on the cell surface. This receptor is found in cells of the pulmonary epithelium, cardiomyocyte, endothelial cells, and cells of the central nervous system, such as neurons and glia cells. The presence of this receptor in different tissues may explain the virus' ability to reach different organs and systems. However, this is not sufficient as the only explanation for this phenomenon. ACE2 is found on the surface of cells of various tissues in the human body. It is possibly an important receptor for the penetration of SARS-COV 2 into these cells. These receptors are present on the endothelium of cerebral capillaries, neurons, and glia cells. Its presence can facilitate the access of the virus to the central nervous system. We also remember that the presence of cytokines, for example, IL-6 can increase the permeability of the blood–brain barrier, favoring even more the entry of microorganisms. Once inside the cell, the virus triggers other intracellular mechanisms of activation of inflammatory response. The main function of ACE 2 is to metabolize angiotensin II into Ang1-7, which has protective effects (anti-inflammatory, antioxidant, anti-atherosclerotic, and anti-thrombotic) in different tissues, acting in specific receptors that are found in large quantities in the human brain. Once bound to the virus, ACE2 decreases these protective effects by increasing the effects of angiotensin II and this favors tissue damage, especially inflammatory, oxidative, and ischemic damages [25, 26].
The inflammatory component of depression
Depression is a multifactorial condition that for decades had its pathophysiological mechanism associated with the monoamine hypothesis. According to this theory, the decrease in neurotransmitters such as serotonin, noradrenalin, and dopamine would be the main neural alteration that would lead to the symptoms of the disease. Many findings have confirmed this hypothesis, such as the decrease in neurotransmitter metabolites in patients with depression and the decrease in neurotransmitters themselves in post-mortem studies in patients with the disease. Similarly, the emergence of pharmacological therapy of this disease aimed at increasing the concentration of neurotransmitters in the central nervous system [27, 28].
In recent decades, new hypotheses have been formulated to explain in more detail the pathophysiology of depression. Several studies have demonstrated the participation of neurotrophic substances such as the brain-derived neurotrophic factor (BDNF), the increased activity of the hypothalamus–pituitary–adrenal (HPA) axis, and that these changes are associated with increased concentration of inflammatory cytokines [29, 30].
Studies show that external conditions such as chronic stress in conjunction with genetic factors would lead to an increased production of pro-inflammatory mediators such as IL-6, Il-2, TNF-α, and IFN-y and a decrease in anti-inflammatory mediators such as IL-4 and IL-10. This increase would lead to changes in brain neurocircuits generating, for example, deregulation of HPA axis decreased the production of BDNF, and neurogenesis compromising the functioning of regions such as the frontal cortex and hippocampus, especially serotonergic transmission, thus leading to symptoms of depression [31, 32].
The increased concentration of inflammatory cytokines was verified in patients with depression, moreover, in animal experiments, the administration of these mediators led to manifestations like depressive conditions. Similarly, there is a strong relationship between the occurrence of autoimmune inflammatory diseases and the emergence of depressive symptoms, further reinforcing the link between the increase in cytokines in the body and their effects on mood. It is worth noting that this theory does not invalidate the monoamine hypothesis. In fact, it establishes a relation between the decrease in neurotransmitter levels and other mechanisms such as environmental factors and genetic predisposition, and the inflammatory process [28].
Inflammatory cytokines can be produced by cells of the immune system activated in the periphery or by neurons and glia cells, such as astrocytes, microglia, and oligodendrocytes. The effects of these cytokines may occur directly when produced in the central nervous system (CNS) itself, as well as indirectly when produced peripherally by the cells of the immune system. It is important to remember that cytokines are water-soluble substances that normally do not cross the blood–brain barrier (BBB). However, pathological conditions that increase the permeability of the barrier may allow this access. In addition, there are brain areas where permeability is facilitated by the absence of the barrier as in the choroid plexus. They can also activate receptors in afferent fibers such as the vagus nerve, which in turn promote the formation of other mediators such as prostaglandins (PGE2) and nitric oxide, promoting neuronal changes and activation of glia cells by increasing the production of cytokines in the CNS itself [33].
Thus, the increase in inflammatory cytokines correlates directly with the symptoms of depression and with the neuronal and glial alterations of the disease and is currently seen as an important component of the pathophysiology of this disease.
So, what's the possible relationship between COVID-19 and depression?
From the above and considering the pathophysiological mechanisms of COVID-19, especially the inflammatory component and the massive production of cytokines, we can suggest that there is an important relationship between the two diseases because these mediators could activate some of the mechanisms involved in depression (Fig. 2).

Inflammatory cytokines in depression: neurological mechanisms and COVID-19
Although ACE 2 does not play a direct role in the pathophysiological mechanisms of depression but considering what was described above and that the inflammatory response is currently seen as a component of the pathophysiological mechanisms of depression, we can suggest that decreased ACE 2 action could then contribute to increased depression. Since the virus SARS COV 2 probably uses this enzyme to penetrate neurons and glial cells, this could decrease its action on angiotensin II, increasing the concentrations of this mediator and favoring the development of depression due to the increased inflammatory activity. In addition, studies that show the decrease of ACE 2 expression in patients with depression, diabetes, and ischemia are already reported in the literature [25, 26, 34].
According to the literature, the changes caused by cytokines are not only of short duration and their effects may persist in different tissues including the neural tissue. It should also be noted that the production of a high level of these pro-inflammatory mediators can cause alterations in the permeability of the BBB, activating Toll-like receptors of the microglia and astrocytes, exacerbating neuro-inflammation and altering the homeostasis of the CNS, which causes cognitive and mood changes. These changes are not exclusive to SARS-CoV-2, because previous studies showed that patients with severe infections caused by SARS-CoV in the pandemic during the years 2002 and 2003 had a positive correlation between the presence of IgG antibodies against the new coronavirus and the occurrence of mental diseases, such as schizophrenic conditions and depression, among others, demonstrating the relationship between these viruses and psychiatric diseases [14, 35].
We stress that depression is a multifactorial disease and that different factors can contribute to its onset and maintenance. The isolation and hospitalization period of the patient as well as the fear of death can contribute in an important way as a stress factor. In addition, genetic predisposition can favor the onset of the disease [32].
We know that stress is a powerful stimulant of the sympathetic autonomic nervous system and a parasympathetic inhibitor. This process leads to the activation of immune cells that increase their production of mediators such as IL-6, TNF-α and IFN-γ which, as previously described, triggers the mechanisms of depression according to the inflammatory theory described above [36]. Thus, the mediators produced during the cytokine storm of COVID-19 would potentiate this process and may thus favor the emergence of depression in susceptible individuals. Unfortunately, there is still little data on the follow-up of COVID-19 survivors, especially the most serious ones, regarding the emergence of psychiatric disorders. Some studies have already shown that many patients exhibit symptoms of depression and post-traumatic stress [32].
Conclusion
The role of inflammatory cytokines and the presence of ACE-2 receptors on the cell surface appear to be the common pathophysiological mechanism between COVID-19 and depression.
References
Bulut C, Kato Y (2020) Epidemiology of COVID-19. Turk J Med Sci 50(1):563–570
Ahn DG, Shin HJ, Kim MH, Lee S, Kim HS, Myoung J et al (2020) Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). J Microbiol Biotechnol 30(3):313–324
Huang X, Wei F, Hu L, Wen L, Chen K (2020) Epidemiology and clinical characteristics of COVID-19. Arch Iran Med 23(4):268–271
Faro A, Bahiano MA, Nakano TC, Reis C, Silva BFP, Vitti LS (2020) COVID-19 e saúde mental: a emergência do cuidado. Estud Psicol (Campinas) 37:e200074
Breithaupt-Faloppa AC, Correia CJ, Prado CM, Stilhano RS, Ureshino RP, Moreira LFP (2020) 17β-Estradiol, a potential ally to alleviate SARS-CoV-2 infection. Clinics 75:e1980
Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ et al (2020) The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil Med Res 7:11
Rogers JP, Chesney E, Oliver D, Pollak TA, McGuire P, Fusar-Poli P et al (2020) Psychiatric and neuropsychiatric presentations associated with severe coronavirus infections: a systematic review and meta-analysis with comparison to the COVID-19 pandemic. Lancet Psychiatry 7(7):611–627
Harenberg J, Favaloro E (2020) COVID-19: progression of disease and intravascular coagulation - present status and future perspectives. Clin Chem Lab Med 58(7):1029–1036
Wong SH, Lui RN, Sung JJ (2020) Covid-19 and the digestive system. J Gastroenterol Hepatol 35(5):744–748
Troyer EA, Kohn JN, Hong S (2020) Are we facing a crashing wave of neuropsychiatric sequelae of COVID-1? Neuropsychiatric symptoms and potential immunologic mechanism. Brain Behav Immun 87:34–39
Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L et al (2020) Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun 87:18–22
Shao O, Herrlinger S, Zhu YN, Yang M, Goodfellow F, Stice S et al (2017) The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 144(22):4114–4124
Fletcher NF, McKeating JA (2012) Hepatitis C virus and the brain. J Viral Hepat 19(5):301–306
Severance EG, Dickerson FB, Yolken RH (2018) Autoimmune phenotypes in schizophrenia reveal novel treatment targets. Pharmacol Ther 189:184–198
Hascup ER, Hascup KN (2020) Does SARS-CoV-2 infection cause chronic neurological complications? Geroscience 42(4):1083–1087
Li H, Xue Q, Xu X (2020) Involvement of the Nervous System in SARS-CoV-2 Infection. Neurotox Res. https://doi.org/10.1007/s12640-020-00219-8
Serrano-Castro PJ, Estivill-Torrús G, Cabezudo-García P, Reyes-Bueno JÁ, Petersen NC, Aguilar-Castillo MJ et al (2020) Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: a delayed pandemic? Neurologia 35(4):245–251
Taquet M, Geddes JR, Husain M, Luciano S (2021) 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry 8(5):416–427
Asadi-Pooya AA, Simani L (2020) Central nervous system manifestations of COVID-19: A systematic review. J Neurol Sci 413:116832
Singha T (2020) A Review of Coronavirus Disease-2019 (COVID-19). Indian J Pediatr 87(4):281–286
Tian S, Hu N, Lou J, Chen K, Kang X, Xiang Z et al (2020) Characteristics of COVID-19 infection in Beijing. J Infect 80(4):401–406
Jin Y, Yang H, Ji W, Wu W, Chen S, Zhang W et al (2020) Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 12(4):372
Ye Q, Wang B, Mao J (2020) The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect 80(6):607–613
Soy M, Keser G, Atagündüz P, Tabak F, Atagündüz I, Kayhan S (2020) Cytokine storm in COVID-19: pathogenesis and overview of anti-inflammatory agents used in treatment. Clin Rheumatol 39(7):2085–2094
Haidar MA, Jourdi H, Hassan ZH, Ashekyan O, Fardoun M, Wehbe Z et al (2020) Neurological and neuropsychological changes associated with SARS-CoV-2 infection: New observations, new mechanisms. Neuroscientist. https://doi.org/10.1177/1073858420984106
Mahalakshmi AM, Ray B, Tuladhar S, Bhat A, Paneyala S, Patteswari D et al (2021) Does COVID-19 contribute to development of neurological disease? Immun Inflamm Dis 9(1):48–58
Malhi GS, Mann JJ (2018) Depression. Lancet 392:2299–2312
Dean J, Keshavan M (2017) The neurobiology of depression: An integrated view. Asian J Psychiatr 27:101–111
Kim YK, Suh IB, Kim H, Han CS, Lim CS, Choi SH et al (2002) The plasma levels of interleukin-12 in schizophrenia, major depression, and bipolar mania: effects of psychotropic drugs. Mol Psychiatry 7(10):1107–1114
Nestler E, Barrot M, Di Leone R, Eisch A, Gold S, Monteggia L (2002) Neurobiology of depression. Neuron 34(1):13–25
Miller AH, Maletic V, Raison CL (2009) Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol Psychiatry 65(9):732–741
Halaris A (2019) Inflammation and depression but where does the inflammation come from? Curr Opin Psychiatry 32(5):422–428
Rhie SJ, Jung EY, Shim I (2020) The role of neuroinflammation on pathogenesis of affective disorders. J Exerc Rehabil 16(1):2–9
Vian J, Pereira C, Chavarria V, Köhler C, Stubbs B, Quevedo J et al (2017) The renin–angiotensin system: A possible new target for depression. BMC Med 15(1):144
Velasco-Fargas R, Martínez-Hernández E (2020) Mecanismos fisiopatogénicos de afectación neurológica. In: Porta-Etessam J et al. Manual COVID-19 para el neurólogo general. Madrid: Ediciones SEN.
Felger JC, Lotrich FE (2013) Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience 246:199–229
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals perfomed by any of the authors.
Informed consent
For this type of study formal consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
da Silva Lopes, L., Silva, R.O., de Sousa Lima, G. et al. Is there a common pathophysiological mechanism between COVID-19 and depression?. Acta Neurol Belg 121, 1117–1122 (2021). https://doi.org/10.1007/s13760-021-01748-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-021-01748-5