
Abstract
Herpes simplex viruses are ubiquitous human pathogens represented by two distinct serotypes: herpes simplex virus (HSV) type 1 (HSV-1); and HSV type 2 (HSV-2). In the general population, adult seropositivity rates approach 90 % for HSV-1 and 20–25 % for HSV-2. These viruses cause significant morbidity, primarily as mucosal membrane lesions in the form of facial cold sores and genital ulcers, with much less common but more severe manifestations causing death from encephalitis. HSV infections in humans are difficult to study in many cases because many primary infections are asymptomatic. Moreover, the neurotropic properties of HSV make it much more difficult to study the immune mechanisms controlling reactivation of latent infection within the corresponding sensory ganglia and crossover into the central nervous system of infected humans. This is because samples from the nervous system can only be routinely obtained at the time of autopsy. Thus, animal models have been developed whose use has led to a better understanding of multiple aspects of HSV biology, molecular biology, pathogenesis, disease, and immunity. The course of HSV infection in a spectrum of animal models depends on important experimental parameters including animal species, age, and genotype; route of infection; and viral serotype, strain, and dose. This review summarizes the animal models most commonly used to study HSV pathogenesis and its establishment, maintenance, and reactivation from latency. It focuses particularly on the immune response to HSV during acute primary infection and the initial invasion of the ganglion with comparisons to the events governing maintenance of viral latency.
Similar content being viewed by others


Introduction to herpes simplex virus
Herpes simplex viruses are members of the Alphaherpesvirinae subfamily within the Herpesviridae virus family. There are two serotypes of herpes simplex virus (HSV): HSV type 1 (HSV-1), which is more frequently found in the oral mucosa and ocular areas, and HSV type 2 (HSV-2), which is most commonly encountered as the causative agent of genital tract HSV infections. HSV is a ubiquitous human pathogen, with worldwide prevalence rates approaching 90 % for HSV-1 and up to 25 % for HSV-2, depending on socioeconomic class. The virus contains a large, linear double-stranded DNA (dsDNA) genome of 150 kbp, which encodes at least 84 polypeptides and is approximately 83 % homologous between HSV-1 and HSV-2 in the coding regions (Liu 2007; Whitley and Roizman 2001). The glycoproteins gB and gC bind glycosaminoglycans within heparan sulfate moieties, which results in initial virion attachment to epithelial cells and keratinocytes at mucocutaneous sites including the mouth, eyes, and genitalia (Laquerre et al. 1998). Subsequently, gD binds one of the HSV receptors: herpes virus entry mediator (HVEM), also known as tumor necrosis factor (TNF) receptor superfamily member 14 (TNFRSF-14), nectin 1, or nectin 2 and recruits accessory glycoproteins gB, gH, and gL, which are all involved in executing membrane fusion. Upon completion of the fusion process, the nucleocapsid enters the cytoplasm and is transported to the nuclear membrane via interactions with the microtubule network. The DNA is then injected into the nucleus through nuclear pores, where it becomes a template for viral transcription and DNA synthesis (Campadelli-Fiume 2007). Following the production of HSV immediate-early regulatory proteins, a number of early enzymatic activities, and an array of early and late structural proteins, virions assemble and bud from infected cells (causing their lysis) and then spread to neighboring cells for further propagation. HSV subsequently accesses neighboring sensory nerve endings via viral envelope fusion with the neuronal plasma membrane and the nucleocapsid is transported via retrograde mechanisms to neuronal cell bodies in the corresponding ganglia (Smith 2012). From there, the virus can either initiate productive replication, which ultimately leads to destruction of the neuron, or the virus can establish a latent infection in the peripheral nervous system (PNS) neurons characterized by circularization of the viral genome and only very limited gene expression (Margolis et al. 2007). The only abundant viral RNA detected during latency is the latency-associated transcript, which is involved in maintaining viral latency (Ahmed et al. 2002). After any of a number of activating events or stimuli including stress, exposure to ultraviolet light, termination of antiviral treatment, immunosuppression, or alteration in hormonal levels, the virus reactivates from the latent state and begins transcriptional and translational processes leading to the production and release of infectious virus from the previously latently infected neuron. Reactivation events are characterized by anterograde capsid movement down the axon and productive infection at sites innervated by the same dermatome, or occasionally spread of the virus to the central nervous system (CNS). Primary HSV-1 infections are typically acquired within the first two decades of life and are usually asymptomatic. Approximately 25 % of infected individuals exhibit periodic viral reactivations from the trigeminal ganglia, which manifest as cutaneous and mucocutaneous lesions including facial herpes, cold sores in and around the mouth, and ocular involvement that can result in blindness (Piret and Boivin 2010; Simmons 2002). HSV infection of the genital tract leads to a latent infection in the sacral ganglionic neurons, from which reactivation can cause genital ulcers or asymptomatic shedding episodes. Severe cases of disease include systemic spread and infection of the CNS, which can lead to herpes simplex virus encephalitis (HSE) that can prove to be fatal. A schematic representation of the major events of HSV infection, disease, latency, and reactivation is summarized in Fig. 1.

Time course of herpes simplex virus (HSV) infection and disease. HSV causes a productive infection of epithelial cells and keratinocytes at mucosal surfaces or within abraded skin. From there, the virus can invade the peripheral nervous system (PNS) and establish latency or crossover to the central nervous system (CNS) and cause serious disease. Periodically, in response to various stimuli, latent HSV can reactivate and cause recurrent disease at mucosal sites (genital ulcers, cold sores, keratitis, skin disease) or crossover to the CNS and cause serious disease
Parameters of HSV biology and disease studied experimentally
HSV biology and pathogenesis
Through mutagenesis approaches to disrupt gene expression, the functions of HSV-encoded mRNA species and proteins have been resolved in vitro and/or in vivo. Research in the past few decades has led to a deeper understanding of the roles of HSV-encoded products involved in entry, intracellular trafficking, nuclear import, DNA replication, envelopment, cellular egress, and immune evasion. Briefly, the HSV proteins comprising the nucleocapsid and tegument region have been elucidated, as have the multiple viral glycoproteins involved in coordinating membrane fusion and entry (Atanasiu et al. 2013; Avitabile et al. 2009). Downstream of virion entry, HSV requires the proteins encoded from the US3, US11, and US31 DNA regions, among others, for proper cellular trafficking and nuclear import and export (Greco et al. 2012; Mou et al. 2007). After cytoplasmic transport, docking of the capsid at the nuclear membrane and nuclear import of the viral genome, the immediate-early infected cell protein (ICP) gene products ICP0 and ICP4 encoded from the RL2 and RS1 gene segments, respectively, drive DNA synthesis and progeny virion assembly (Davison and McGeoch 1986; Forrester et al. 2010; Roizman 1996; Ushijima et al. 2007; Ward and Roizman 1994). Apart from structural and replicative functions, HSV encodes several proteins that are involved in evading immunologic pressures and influencing cellular viability to promote survival of the virus in the host. The viral gene products ICP34.5 and ICP47, encoded from the RL1 and US12 gene segments, respectively, are among the most well-characterized HSV proteins, and their roles in counteracting immunologic pressures and promoting neurovirulence are well appreciated (Leib et al. 2009; Tomazin et al. 1998; Ushijima et al. 2007; Ward and Roizman 1994). In addition to protein products, HSV encodes microRNA (miRNA) species that interfere with other HSV-encoded functions. For example, miRNAs have been identified that target ICP0 and ICP34.5 to regulate the replicative cycle and perhaps contribute toward the establishment of latency (Guo et al. 2010). Finally, the parameters dictating how HSV regulates its latency/reactivation cycle have remained an area of intensive research focus for more than five decades. As discussed in the subsequent sections, animal models vary in their suitability for studying these events, which makes effective mechanistic extrapolation to human disease difficult in many cases (Armien et al. 2009). Nonetheless, animal model systems have been used to study different aspects of acute HSV infection and disease including the establishment, maintenance, and reactivation of latent infection.
HSV immunity
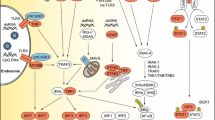
Because the virus is so prevalent in the human population, it is important to understand how each component of the mammalian innate and adaptive immune systems responds to HSV infection. A variety of pattern recognition receptors have been shown to recognize HSV structural proteins and nucleic acid moieties and trigger essential early proinflammatory responses. These include Toll-like receptors (TLRs) on cellular plasma membranes (e.g., TLR2), within endosomes (e.g., TLR3 and TLR9), and cytosolic nucleic acid receptors (e.g., DAI, IFI16, and Rig-I) (Conrady et al. 2012; Davey et al. 2010; Ishikawa et al. 2009; Kurt-Jones et al. 2004; Lund et al. 2006). A model of cutaneous HSV-1 infection is useful for studying dendritic cell migration and antigen presentation for subsequent CD8+ T cell activation. Carbone and colleagues have highlighted the impact of CD8α+ dendritic cells in this process (Allan et al. 2006). Other investigators have focused on studying the characteristics of HSV-specific T lymphocytes, including their epitope recognition repertoire and the necessary parameters to optimize their magnitude and quality. Contemporary studies are geared toward tracking the migration patterns of activated T cells to infected nervous system tissues as well as identifying their mechanisms of blocking reactivation and virus spread (Knickelbein et al. 2008; Schachtele et al. 2010). The development of HLA transgenic mice and rabbits has enabled researchers to examine the generation of T cell responses specific for HSV epitopes recognized by human lymphocytes, rather than those epitopes recognized by cells of the experimental animal host (Chentoufi et al. 2010; Hu et al. 2006). Several transgenic animals exist for the study of HSV gB- and gD-specific lymphocytes: HLA-A*0201 transgenic rabbits and HLA-A*0201 transgenic mice have been used to assess the development of antigen-specific CD8+ T cells, and HLA-DR1 and HLA-DR4 transgenic mice have been used to study HSV-specific CD4+ T cell activation (Chentoufi et al. 2010; Chentoufi et al. 2008; Dervillez et al. 2013; Zhang et al. 2008). Furthermore, studying the conformational determinants of anti-HSV antibodies and their impact on controlling virus spread is also a major goal of a number of investigators (Awasthi et al. 2014; Pan et al. 2012; Simmons and Nash 1985, 1987). Indeed, a variety of HSV epitopes have been used to induce B and T cell immune responses with the goal of either preventing new infections or reducing reactivation episodes to minimize transmission rates (Iyer et al. 2013). A detailed analysis of the current state of HSV vaccine development is outside the scope of this review; however, recent comprehensive summaries can be found elsewhere (Chung and Sen 2012; Dropulic and Cohen 2012; Lee and Ashkar 2012; Roth et al. 2013). These models have proven to be some of the most useful approaches for studying a causative agent of an infectious disease, especially from an immunological viewpoint. It should be noted that much of the knowledge accumulated over the years concerning the innate and adaptive immune systems has been derived from the study of HSV-1 infections of many different strains of mice.
Animal species used in experimental models
Multiple animal models have been used to study HSV-induced immune responses and pathogenesis. The species most commonly studied are mice, guinea pigs, rats, and rabbits. Mice are widely used to study HSV-1-induced immune responses because their genetic systems have been well characterized; immunologic and molecular biologic reagents are widely obtainable; and the number of genetic knockout strains available for analysis is greater than for other species. HSV-1 reproducibly establishes latency in the mouse model within the earliest stages of acute infection, with the viral genome reaching the neuronal ganglia within the first 24 h of infection (Steiner et al. 1990). This provides a useful model system for studying the interconnectivity of primary infection, the immune response, neuroinvasion, establishment of latent infection, and the early phase revolving around the maintenance of viral latency. HSV infection of the mouse brain exhibits a diffuse pattern of viral spread and neuroinflammatory lesions compared to the more focal patterns of viral and inflammatory lesions in the human brain (Fig. 2) (Barker et al. 2014; Meyding-Lamadé et al. 2003). The strength of the rabbit and guinea pig models rests in their utility to study the processes of HSV reactivation, which do not appear to occur spontaneously in mice (Valencia et al. 2013; Wagner and Bloom 1997). The guinea pig model closely simulates acute genital tract infection as well as recurrent ulcerative disease (Hsiung et al. 1984). This model has also been widely used to assess the efficacy of anti-HSV vaccines with respect to controlling the recurrent phase of disease (Iyer et al. 2013). Other species less commonly used to evaluate parameters of HSV disease include cotton rats, owl monkeys, and rhesus macaques. This review focuses on the models that are most commonly used to study the pathogenesis and immune response of HSV infection and disease, with primary emphasis on HSV-1 mouse studies and periodic discussion of the experimental results realized from guinea pig and rabbit infection models. Table 1 summarizes the specific details relating to the array of experimental HSV infection models including routes of infection, animals species used, and commonly assessed parameters of HSV pathogenesis and immunity. We have not attempted to review the literature with respect to the many interesting observations brought to light concerning the disease pathogenesis and the processes of establishment, maintenance, and reactivation of latent infection of endogeneous herpesviruses of non-human organisms.

Magenetic resonance image (MRI) of an HSV-infected human brain. Axial brain MRI from a 64-year-old male with confirmed HSV-2 encephalitis. The image shows signal change and swelling of the left anterior temporal lobe and left gyrus rectus. Reprinted from the Journal of Clinical Virology, volume 59, Barker et al., Encephalitis in an immunocompetent man, 1–3, 2014, with permission from Elsevier
Monitoring pain and distress in infected animals
As previously discussed, experimental HSV infections can be used to study mechanisms governing the establishment and maintenance of viral latency. These experiments require the animals to be maintained for at least 3 to 4 weeks in most cases and longer in certain circumstances. In other types of research, immunodeficient or young animals with reduced HSV resistance are used. Inevitably, for these reasons and others discussed in the subsequent sections, a portion of infected animals will experience significant morbidity and may progress to a fatal outcome, either from the acute spread of the virus prior to the establishment of latency or from reactivation of latent virus. For humane and frequently legal reasons, establishing clear endpoints of the study is imperative in order to reduce animal pain and suffering (Hawkins et al. 2011; Toth 2000). Importantly, the state of the animal’s welfare may rely on the investigator’s subjective assessment and opinion; thus, it has been proposed that concrete interventional endpoint criteria should be formulated and agreed upon by the investigators, veterinarians, and personnel from the institutional animal care and use committee and applied in each unique experimental design (Hankenson et al. 2013; Hawkins et al. 2011; Toth 2000). The selection of endpoints may also reduce the number of animals that experience spontaneous mortality (prior to the investigator’s knowledge), which would preclude the reliable use of their body tissues for analysis or unknowingly impact the quality of the dataset (Hankenson et al. 2013).
The most widely used approach to assess the health of experimental animals is to measure their core body temperature (Toth 2000). Pilot experiments are suggested to establish the species- and strain-specific baseline temperatures for each investigation so that deviations may be observed and used to inform the decision to euthanize specific animals (Hankenson et al. 2013). Importantly, hyperthermic stress may be employed to induce HSV-1 reactivation in mice. In this case, measuring core body temperature would not be informative of the animal’s well-being. Other components of the animal’s physical welfare state include its coat appearance, posture, ability to ambulate, and excessive attention to manipulated body sites (Hawkins et al. 2011; Toth 2000). Together, these parameters can be used to visually distinguish moribund animals from those appearing normal. In our HSV-1 infection models involving peripheral viral inoculation (namely, the hind footpad injection and lip scarification methods), we euthanize mice with ruffled coats, those that have lost the ability to ambulate, and those scratching or biting their infected tissues. We also remove any animals that have lost more than 20 % of their day 0 body weight. For mice infected directly into the CNS via the lateral ventricle, weight loss is the sole metric for removing unhealthy animals. Measurements of physiological and biochemical parameters including heart rate, respiratory rate, and stress hormones are also used to assess animal health (Hawkins et al. 2011). Finally, behavioral signs including aggression and withdrawal can determine an animal’s psychological condition and inform a decision leading to euthanasia (Hawkins et al. 2011). In some cases, these criteria are used to formulate a disease score to quantitate clinical sickness and remove animals from the study (Hawkins et al. 2011). Disease presentation can vary markedly depending on the characteristics of the respective HSV infection, so investigators should thoroughly understand each model to avoid inappropriately euthanizing animals or misinterpreting disease severity.
Experimental variables that affect the course of animal infection
In addition to the animal species selected for establishing an experimental HSV model, other factors can significantly alter the course of virus infection and the ensuing immune response. The HSV serotype and specific viral strain dictate the target cells infected, ability to invade the CNS, and the extent of productive infection in a given cell type, tissue, and host. HSV-1 and HSV-2 share approximately 83 % homology in their coding sequences, but their epidemiology and disease characteristics are certainly not identical (Whitley and Roizman 2001). Although most orofacial infections are typically attributed to HSV-1 and genital infections to HSV-2, HSV-1 infection of the genital tract has been increasingly observed (Roberts et al. 2003). Manifestations of the potentially fatal disease HSE have shown a serotype and human host age disparity, with HSV-1 accounting for most adult cases and HSV-2 responsible for most instances involving neonates and children (Whitley 2006). Other variables that influence HSV disease severity include host genetics, route of inoculation, and dose of virus used. How each of these variables affects disease outcome will be illustrated with specific examples relating to pathogenic outcome.
HSV strains display a spectrum of neurovirulent outcomes
An extensive number of clinical isolates and laboratory-adapted HSV-1 and HSV-2 strains are available for use in experimental studies. These strains can vary significantly in their ability to cause disease and infect the nervous system. Thus, viruses must be selected carefully for use in different types of experimental designs (Sprecher and Becker 1987). Mutational analyses and generation of HSV-1/2 intertypic recombinants has led to the elucidation of viral determinants affecting strain pathogenicity and neurovirulence. A selection of these gene products includes thymidine kinase (UL23), ribonucleotide reductase (UL39/40), and ICP34.5 (RL1), which are involved in viral DNA synthesis and evasion of the immune response (Cameron et al. 1988; Cassady et al. 1998; Field and Wildy 1978; Orvedahl et al. 2007; Ushijima et al. 2007). Table 2 lists HSV-1 and HSV-2 strains commonly used for experimental infections as well as the source of their original isolation and relative virulence in adult mice (Brown et al. 1973; Duff and Rapp 1971; Ejercito et al. 1968; Gudnadottir et al. 1964; Irvine and Kimura 1967; Rawls et al. 1968; Wander et al. 1980).
Mouse strain-dependent spectrum of resistance
Mice represent an important starting point for assessing a number of experimental parameters of HSV disease. Mice support productive HSV replication, are relatively inexpensive, and have been manipulated extensively to generate a multitude of genetically modified strains. The molecular biology reagents available for work with mice facilitate a sophisticated analysis of immunological responses following infection. Inbred strains of mice, however, display a spectrum of susceptibility to HSV-1 infection, pathogenesis, and disease (Kastrukoff et al. 1986; Lopez 1975). Table 3 summaries the seminal studies that have highlighted these significant mouse strain-dependent resistance phenotypes. Following a systemic intraperitoneal infection with HSV-1 2391 (a strain known to be virulent in BALB/c mice), the following susceptibility and resistance trends were observed: C57BL/6 and C57BL/10 mice showed the greatest resistance to infection (median lethal dose [LD50] > 106 plaque-forming units [PFU]); C3H/He and BALB/c strains demonstrated intermediate susceptibility; and AKR and SWR mice demonstrated the greatest level of susceptibility (LD50 = 101 PFU) (Lopez 1975). In support of this trend, C57BL/6 mice showed restricted access of HSV-1 throughout the CNS, whereas C3H mice and (to an even greater extent) BALB/c mice exhibited elevated viral spread throughout the cerebellum and cerebrum following a lip infection (Kastrukoff et al. 1982). Importantly, while the ability of HSV-1 to access selected regions of the CNS differed among these groups of mice, the frequency of latent infection of the trigeminal ganglia was similar in all three groups of mice, suggesting that the resistant mice restrict viral spread from the PNS to the CNS more efficiently than do the susceptible strains (Kastrukoff et al. 1982). Involvement of the innate immune response was suggested by the correlation between resistance and an early burst of type I interferon (IFN) production (Engler et al. 1981; Engler et al. 1982; Kirchner et al. 1983; Zawatzky et al. 1982b). This mouse strain resistance spectrum must be considered when interpreting the results of pathogenicity studies performed by different investigators because different parameters relative to immune system function may be engaged based on the extent and nature of disease severity. Although it remains possible that factors other than early production of type I IFN are involved in determining mouse resistance, the restricted spread of the virus from the site of inoculation to the nervous system positively correlated with protection against severe disease (Kastrukoff et al. 1982; Kastrukoff et al. 1986; Kastrukoff et al. 2012).
Mouse age-dependent spectrum of resistance
Innate and adaptive immune responses are qualitatively different in neonates compared to adults, making the young generally more susceptible to developing life-threatening complications from a number of different infectious diseases. Animal models of HSV disease have also demonstrated an increased vulnerability in younger animals. Intranasal, intraperitoneal, ocular, and subcutaneous HSV-1 infections in neonatal mice have demonstrated reduced early type I IFN production and elevated viral replication as compared to older mice (Ben-Hur et al. 1983; Bukowski and Welsh 1986; Johnson 1964; Kintner and Brandt 1995; Kopp et al. 2013; McKendall 1980; Zawatzky et al. 1982a). Neonatal immunity is biased toward a tolerogenic T helper type 2 (TH2) response in order to prevent reactivity toward maternal antigens (Martin et al. 2011). Underdeveloped TH1 immune responses have been shown to enhance vulnerability to intracellular pathogens (Burl et al. 2011; Hoffmann et al. 2005; Marodi 2006). Indeed, young laboratory animals have been shown to be more susceptible to a variety of microorganisms including Theiler’s virus, HSV, and Salmonella (Blackstock and Murphy 2004; Rhee et al. 2005; Steiner et al. 1984). Studies using human cells have corroborated the trend observed in young experimental animals, with the inflammatory potential of cells isolated at selected times throughout the first 1.5 years of life gradually increasing in response to TLR ligation with the peak response reached at adulthood. Younger mice may be more susceptible to virus infections (including HSV) for the same reasons. Studies on neonatal immunity have suggested that specific components of the innate immune response are essential for protection against infectious agents in early life. In contrast, these aspects of the innate immune response may be less critical for control of infection later in life when other features of immunity have begun to develop and redundancy has been shown to exist within the mature immune system.
Routes of inoculation
Depending on the goals of a given infection model, investigators may use one of many different known sites for virus inoculation to initiate the primary disease syndrome (Fig. 3). This provides a great deal of versatility to the experimentalist but introduces complexity when studies are compared across the literature when multiple inoculation sites are used to study virologic and/or immunologic events. As will become clear in the following discussion, certain inoculation approaches are well suited to establish HSV latency (such as the mouse corneal scarification model) and a number of infection routes are preferentially used for reactivation studies (including intravaginal infection of guinea pigs). In some circumstances, systemic disease has been caused by intraperitoneal or intravenous injections. In contrast, peripheral infections have been initiated by ocular, vaginal, skin, and nasal/oral tissue inoculation, whereas encephalitis has been induced by directly injecting HSV into the brain (Barr et al. 2007; Eloranta et al. 1996). The subsequent discussion will address the typical kinetics and characteristics of the host’s response for each of the inoculation routes used to establish HSV infection.

Anatomy of natural human herpes simplex virus (HSV) infection and the corresponding animal models used to mimic human disease. Sites of primary human HSV-1 and HSV-2 infection are depicted in maroon, and their corresponding animal model sites (in blue) are highlighted by the hashed lines. HSV establishes latency in sensory dorsal root ganglia (DRG) of the peripheral nervous system (PNS) and within neurons of the central nervous system (CNS) (simplified in orange) (color figure online)
Cutaneous and subcutaneous HSV infections
HSV is unable to penetrate intact skin; therefore, productive infection requires compromised skin integrity or mucus membrane access. Teague and Goodpasture initially described a cutaneous infection model in 1923 in which the flank of guinea pigs and rabbits was depilated and the epidermis was irritated with a coal-tar layer for a few days (Teague and Goodpasture 1923). The animals were subsequently infected with HSV and local vesicular lesions were observed within 72 h. Notably, a few days after the primary infection, a series of secondary lesions developed in a band formation running parallel to the spinal column. These lesions contained infectious virus and were attributed to viral spread to nearby skin sites innervated by the same nerve root in a “zosteriform” pattern. Simmons and Nash later adapted this approach to the mouse model, highlighting the restrictive role of T cells in controlling the zosteriform spread and studying the processes of establishment and maintenance of latency (Simmons and Nash 1984; Simmons et al. 1992). Carbone and colleagues have used this model to study antigen uptake at the site of infection by dermal dendritic cells, their migration to the draining lymph nodes, and their cooperation with lymph node–resident dendritic cells, which results in antigen presentation to naive T lymphocytes (Allan et al. 2006). Subcutaneous injection of HSV-1 into the rear footpads represents another approach to initiate peripheral virus infection. This method results in a reproducible infection and is useful for studying the generation of anti-HSV-1 T cell responses. The virus establishes a latent infection in the lumbosacral dorsal root ganglia and induces a peak of HSV-1 gB-specific CD8+ T cells in the draining popliteal lymph nodes on day 5 (Bonneau and Jennings 1989; McNally et al. 1999). Signs of disease include loss of fur, necrosis and edema of the foot, and paralysis of the hind limb (Sprecher and Becker 1987). This model can involve encephalitis when neurovirulent viruses are used in young, immunodeficient, or otherwise susceptible animals.
HSV infections of the head
Lip infections
Although this approach is not widely used in the field, inoculation of HSV-1 into the oral tissues very closely mimics one of the routes of primary human HSV exposure. Oral infection is established by injecting the virus into the lip or tooth pulp or by compromising the integrity of the lip mucoepithelium and applying the virus topically. This route of inoculation represents a simple and quick method to establish a reproducible primary infection and results in minimal animal discomfort. After initial replication in local mucosal epithelial cells and keratinocytes, HSV-1 travels via retrograde axonal transport to the trigeminal ganglia where it either establishes a latent infection or spreads centripetally to the brainstem via the trigeminal nerve root entry zone (Barnett et al. 1995; Kastrukoff et al. 1982; Kastrukoff et al. 2010; Kastrukoff et al. 1981; Labetoulle et al. 2000). Erythematous labial ulcers arise 1 to 3 days after infection and may resolve if the animal survives the primary infection (Fig. 4). Additionally, HSV-1 can travel down the trigeminal nerve in an anterograde fashion to infect ocular tissues and cause blepharitis, conjunctivitis, or keratitis (Labetoulle et al. 2000). Kastrukoff and coworkers used this model to reveal mouse strain differences in susceptibility to HSV-1 crossover from the trigeminal nerve to the CNS and to assess the role of T lymphocytes and antibodies in preventing CNS infection (Kastrukoff et al. 1982, 1986). They also demonstrated multifocal CNS demyelination and antigen-positive lesions whose size and frequency were dependent on mouse strain (Kastrukoff et al. 1992, 2012). We have previously used this model to investigate the role of TLR2 in restricting virus spread to the CNS and in generating an antigen-specific CD8+ T cell response. Four-week-old wild-type C57BL/6J mice were significantly more resistant to lethal brainstem encephalitis than were TLR2-deficient animals owing to restricted spread of HSV-1 from the PNS to the CNS at the level of the trigeminal ganglia (manuscript in preparation).

Model of mucocutaneous herpes simplex virus (HSV) infection of the mouse lip. The lower lip of mice is scratched with a needle, and HSV-1 is painted onto the tissue. On day 4 of infection, a portion of the mice exhibit normal tissue structure (left panel), whereas a subset of mice display varying degrees of lip pathology (middle and right panels). Lesions present with redness and swelling and can resolve in a percentage of animals
Intranasal infections
Intranasal inoculations with HSV-1 or HSV-2 have been widely used to induce encephalitis and to examine selected correlates of protection. Infection of young or otherwise susceptible mice through inhalation of the viral inoculum leads to ocular swelling, weight loss, shaking movements, and rapid mortality from cerebral edema (Boivin et al. 2002; Shimeld et al. 1987). Numerous studies have confirmed that HSV-1 invades elements of both the olfactory and trigeminal nerves, with both routes representing significant tracts of virus spread to the CNS (Armien et al. 2009; Barnett et al. 1994). Lokensgard and colleagues have shown that HSV-1 reaches the brain via the olfactory route earlier than through the trigeminal ganglia-to-brainstem pathway. Early neutrophil infiltration was evident in both the olfactory bulb and brainstem, whereas a lymphocytic infiltrate predominated at later time points. Moreover, activated resident cells including astrocytes and microglia were readily observed throughout the course of the chronic infection (Armien et al. 2009; Marques et al. 2008). A series of studies using this route of infection has shown the utility of administering anti-inflammatory therapeutics including methylprednisolone and TLR9 antagonists to mice with HSV-1 CNS infections. Depending on how long after infection the immune-modulating drugs were given, the severity of HSE could be reduced by measuring clinical disease visually, through cranial magnetic resonance imaging and by comparing survival rates (Boivin et al. 2012; Meyding-Lamadé et al. 2003). Although intranasal infection is useful for studying multiple aspects of viral encephalitis, this model can also lead to viral infection of the lungs and create undesired systemic responses. Another caveat is the reproducibility of the infection, which can vary between animals depending on their breathing intensities and the experimentalist’s skill level.
Ocular infections
HSV-1 is one of the most common causes of infectious human blindness in the developed world; therefore, viral pathogenicity of ocular tissue is an area of intensive research interest. Rabbits and mice are the most commonly used animals for these studies. The viral inoculum is painted onto normal or scarified corneas to facilitate viral uptake and disease progression is measured by examining corneal opacity and perforation. Hill and colleagues pioneered the use of the rabbit model to investigate the neural spread of HSV-1 to the trigeminal ganglia, superior cervical ganglia, trigeminal nerve, and trigeminal nerve root entry zone from ocular tissues in rabbits (Shimomura et al. 1985). Parameters dictating the levels of viral ocular shedding, which can occur spontaneously in the rabbit model, were also examined (Berman and Hill 1985). After initial replication in the periphery, HSV-1 infects the ophthalmic branch of the trigeminal nerve and can be detected in the trigeminal ganglia within 2 to 3 days of infection (Decman et al. 2005). In response to ganglionic invasion, macrophages and T cells infiltrate the ganglia and produce nitric oxide and TNF-α to help contain viral replication and promote the establishment of latency. In this regard, Hendricks and colleagues have examined the activation and migration patterns of HSV-1-specific CD8+ T lymphocytes to the eye and trigeminal ganglia and their involvement in the maintenance of latency and protection against reactivation (Dennis et al. 1995; Knickelbein et al. 2008). Alternatively, the virus can spread to the CNS if these repressive processes are not generated prior to crossover from the PNS. Ocular infection has also been used effectively to study mechanisms responsible for ocular immunopathology and resolution including chemokine and cytokine production, activation of TH1-biased CD4+ T cells, and generation of protective regulatory T cell responses. The severe disease herpetic stromal keratitis is a consequence of both virus-mediated destruction of the epithelium as well as neutrophil- and CD4+ T lymphocyte-mediated stromal tissue cytotoxicity (Hendricks et al. 1992; Ksander and Hendricks 1987; Russell et al. 1984). Elevated levels of interleukin (IL)-6, IL-10, IL-12, and IFN-γ have been observed within a few days of infection in corneal tissues and their sustained production has been implicated in disease severity (Banerjee et al. 2005; Biswas et al. 2005; Daheshia et al. 1998; Kanangat et al. 1996; Sarangi et al. 2008; Sehrawat et al. 2008; Stumpf et al. 2002). The transgenic rabbit model of herpetic conjunctivitis has been utilized to study the activation of CD8+ T cells specific for human HLA-presented HSV-1 antigens. This model has been preferred over the transgenic mouse model because production of neutralizing antibodies can help control HSV-1 replication within the mouse eye but may prevent complete investigation of the T cell arm of immunity (Chentoufi et al. 2010). The ocular infection model leads to significant discomfort in the animals and requires frequent administration of analgesics.
Inoculation of HSV directly into the CNS
HSV can be injected directly into the CNS in order to bypass peripheral immune events and reduce the confounding variables of age and genetic determinants of disease susceptibility. This approach has been useful for determining a gene’s role specifically within the brain as well as the contribution of resident brain cells with respect to neuroinflammation (Kopp et al. 2009; Vilela et al. 2011; Wang et al. 2011). HSV-1 can be introduced directly into specific regions of the brain including the olfactory bulb, hippocampus, lateral ventricle, and confluences of the sinuses (Boggian et al. 2000; Conrady et al. 2013). Once the virus is inoculated into the CNS, resident cells initiate robust inflammatory responses to limit viral replication and attract peripheral blood cells for additional support in controlling viral spread.
The cellular responses of individual brain cell populations can be assessed after the cells are removed from the host, purified, and infected in vitro. Astrocytes have been shown to sustain productive HSV-1 infection and respond by upregulating TLR2 and TLR4 expression and by activating TLR-dependent signaling pathways (Ecob-Johnston and Whetsell 1979; Villalba et al. 2012). Oligodendrocytes have also been shown to support HSV-1 replication in organotypic cultures of mouse spinal cords (Ecob-Johnston and Whetsell 1979). Specifically, oligodendrocytes responded in a host genetic strain-dependent manner, with resistant C57BL/6J mouse cells yielding less infectious HSV-1 compared with cells from the more susceptible mouse strains BALB/cByJ and A/J (Kastrukoff et al. 1987; Thomas et al. 1991). These observations have been corroborated using primary human oligodendrocytes and human cell lines, which exhibited increasing HSV-1 titers throughout the course of infection (Bello-Morales et al. 2005; Kastrukoff and Kim 2002). In contrast, the virus successfully enters microglia but is efficiently eliminated before progeny virions are produced (Lokensgard et al. 2001). To restrict HSV-1 replication, microglia initiate a robust inflammatory response characterized by elevated IL-1β, IL-6, TNF-α, and nitric oxide production that is partially TLR-dependent (Aravalli et al. 2005). A number of research investigators have examined the contribution of microglia-mediated inflammation toward controlling HSV-1 replication and exacerbating encephalitis through activation of other brain cells and recruitment of inflammatory cells from the circulation (Esaki et al. 2010; Schachtele et al. 2012; Wang et al. 2011). The interaction of HSV with the neuronal population is more complex. Some neuronal subpopulations are not susceptible to HSV infection, whereas other subpopulations support productive HSV infection. Some neuronal subsets exhibit necrotic or apoptotic endpoints following infection, whereas other neuronal subsets are differentially responsive to HSV gene expression and the subsequent cellular responses. In the latter subsets, cellular responses guide the virus into the latent state and control the genomic activation and gene expression pathways leading to viral reactivation and recurrent peripheral disease. The interplay between neuronal and glial cell types in response to HSV infection and bystander inflammation is complex, and the intracranial model has helped to tease apart some of the intricate cellular processes and responses.
Genital herpes models
Female mice and guinea pigs can be infected vaginally with HSV-1 or HSV-2 to closely mimic human genital HSV disease. Mice must be pretreated with the hormone medroxyprogesterone to synchronize estrous cycles and promote virus uptake. As this treatment thins the uterine lining, interpretation of viral pathogenicity is altered significantly (Baeten et al. 2007). Nonetheless, the virus is introduced directly into the vagina whereby it infects epithelial cells and causes ulcers to appear as early as day 2 after infection (Shin and Iwasaki 2013). Additionally, HSV can spread to the vulva, urethra, bladder, rectum, and perineal tissues (Parr and Parr 2003). Vaginal lavage is used to measure local viral shedding and inflammation, allowing removal of uninfected animals from the study (Lund et al. 2006; Skoberne et al. 2013). After initial infection, the virus accesses the sacral ganglia to establish latency. A fraction of infected animals progress to fatal encephalitis, especially if a large dose of a neurovirulent virus strain is used. The genital herpes model is frequently chosen to investigate the role of innate immune responses, vaccine efficacy, and correlates of immunity directed against recurrent disease, which occurs spontaneously in guinea pigs (Bernstein et al. 1991; Lund et al. 2006; Svensson et al. 2007; Wagner and Bloom 1997). The genital HSV model has also been used recently to examine the mechanisms by which genital HSV disease may enhance susceptibility to HIV transmission (Rollenhagen et al. 2014).
Conclusions
Multiple experimental animal models are available to study parameters of acute, latent, and recurrent HSV-1 and HSV-2 disease. The outcome of these asymptomatic or pathogenic interactions is dictated by numerous factors including host age, genetics, virus strain, inoculum size, and route of infection (Tables 1, 2, and 3). Because different studies have utilized divergent HSV infection methods, care must be taken when integrating experimental results. We hope that research using these models will help in developing a mechanistic understanding to prevent the establishment of latent HSV infection, irreversibly block viral reactivation, and/or eliminate the HSV genome from latently infected cells. Current studies focused on bolstering protective immune responses, preventing reactivation events, and reducing disease severity will undoubtedly reduce the morbidity associated with this ubiquitous human pathogen. Efficient use of the available animal model systems will likely play a critical role in this interdisciplinary and translational research initiative.
Perhaps the most important guiding principle when selecting an appropriate animal model for the study of any human disease, whether it be to recapitulate HSV-induced disease or any other infectious, oncogenic, inflammatory, metabolic, or genetically inherited disease, is that the pathogenesis and disease observed in a given animal model closely resemble the human disease in all aspects from the initial phase of disease to the final endpoint. Clearly, it is known from many decades of studying a vast number of diseases that this is rarely possible in most, if not all model systems, due to basic differences in anatomy, physiology, immune system, nervous system, and other organ-specific differences. Nevertheless, the knowledge gained from a particular physiologic process in a given animal species has provided a basis to leverage this understanding to more closely examine the pathogenesis of a number of infectious disease pathogens. With respect to HSV infections, the understanding of the genes and gene products involved in the murine immune response and in the definition of their role(s) in defense against viral infection can be applied to human infections, and vice versa. The ever-growing availability of genetically modified mice that can be utilized to define the role of various immune response genes within the context of host defense against numerous pathogens has defined the role of pattern recognition receptors of the innate immune response (Davey et al. 2010; Kurt-Jones et al. 2004; Lund et al. 2006), and it is now clear that the genetic susceptibility to HSV infections in humans can be similarly linked to such receptors (Casrouge et al. 2006; Herman et al. 2012; Perez de Diego et al. 2010; Sancho-Shimizu et al. 2011; Zhang et al. 2007). Furthermore, the seminal studies in mice by the Hendricks group defining the role of the adaptive immune response, particularly the class I MHC-restricted CD8+ T cell population (Khanna et al. 2003; Liu et al. 2001; Liu et al. 2000; Liu et al. 1996), have now been confirmed to have their counterpart in human infections by either HSV-1 (Chentoufi et al. 2008; Theil et al. 2003; Verjans et al. 1998; Verjans et al. 2007) or HSV-2 (Laing et al. 2010; Peng et al. 2012; Tigges et al. 1992). A similar wealth of knowledge is derived from the anatomy and neurobiology of the rat nervous system, which has well-established correlations to the human nervous system and can be applied directly to human infections. This knowledge serves as the basis to define protective and inflammatory cell populations and signal pathways involved in response to viral infection of the brain. Nevertheless, the desire to have every aspect of human disease be accurately reflected within the context of a single animal model remains difficult. As most disease processes involve multiple cells, tissues, and/or organs, modeling every aspect of a particular disease process from beginning to end is rendered difficult. The multi-faceted differences between human and animal physiology often lead to variances in pathogenic outcome between humans and the given animal model. Therefore, the primary focus in the selection and design of a model system is to answer highly specific and focused questions.
References
Ahmed M, Lock M, Miller CG, Fraser NW (2002) Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol 76:717–729. doi:10.1128/JVI. 76.2.717-729.2002
Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR (2006) Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25:153–162. doi:10.1016/j.immuni.2006.04.017
Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR (2005) Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol 175:4189–4193. doi:10.4049/jimmunol.175.7.4189
Armien AG, Hu S, Little MR, Robinson N, Lokensgard JR, Low WC, Cheeran MC (2009) Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis. Brain Pathol 20:738–750. doi:10.1111/j.1750-3639.2009.00354.x
Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ (2013) Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J Virol 87:11332–11345. doi:10.1128/JVI. 01700-13
Avitabile E, Forghieri C, Campadelli-Fiume G (2009) Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J Virol 83:10752–10760. doi:10.1128/JVI. 01287-09
Awasthi S, Balliet JW, Flynn JA, Lubinski JM, Shaw CE, DiStefano DJ, Cai M, Brown M, Smith JF, Kowalski R, Swoyer R, Galli J, Copeland V, Rios S, Davidson RC, Salnikova M, Kingsley S, Bryan J, Casimiro DR, Friedman HM (2014) Protection provided by a herpes simplex virus 2 (HSV-2) glycoprotein C and D subunit antigen vaccine against genital HSV-2 infection in HSV-1-seropositive guinea pigs. J Virol 88:2000–2010. doi:10.1128/JVI. 03163-13
Baeten JM, Benki S, Chohan V, Lavreys L, McClelland RS, Mandaliya K, Ndinya-Achola JO, Jaoko W, Overbaugh J (2007) Hormonal contraceptive use, herpes simplex virus infection, and risk of HIV-1 acquisition among Kenyan women. AIDS 21:1771–1777. doi:10.1097/QAD.0b013e328270388a
Banerjee K, Biswas PS, Rouse BT (2005) Elucidating the protective and pathologic T cell species in the virus-induced corneal immunoinflammatory condition herpetic stromal keratitis. J Leukoc Biol 77:24–32. doi:10.1189/jlb.0904486
Barker KR, Sarafino-Wani R, Khanom A, Griffiths PD, Jacobs MG, Webster DP (2014) Encephalitis in an immunocompetent man. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology 59:1–3. doi:10.1016/j.jcv.2013.05.014
Barnett EM, Jacobsen G, Evans G, Cassell M, Perlman S (1994) Herpes simplex encephalitis in the temporal cortex and limbic system after trigeminal nerve inoculation. J Infect Dis 169:782–786. doi:10.1093/infdis/169.4.782
Barnett EM, Evans GD, Sun N, Perlman S, Cassell MD (1995) Anterograde tracing of trigeminal afferent pathways from the murine tooth pulp to cortex using herpes simplex virus type 1. J Neurosci 15:2972–2984
Barr DP, Belz GT, Reading PC, Wojtasiak M, Whitney PG, Heath WR, Carbone FR, Brooks AG (2007) A role for plasmacytoid dendritic cells in the rapid IL-18-dependent activation of NK cells following HSV-1 infection. Eur J Immunol 37:1334–1342. doi:10.1002/eji.200636362
Bello-Morales R, Fedetz M, Alcina A, Tabarés E, López-Guerrero JA (2005) High susceptibility of a human oligodendroglial cell line to herpes simplex type 1 infection. J Neurovirol 11:190–198. doi:10.1080/13550280590924179
Ben-Hur T, Hadar J, Shtram Y, Gilden DH, Becker Y (1983) Neurovirulence of herpes simplex virus type 1 depends on age in mice and thymidine kinase expression. Arch Virol 78:303–308. doi:10.1007/BF01311326
Berman EJ, Hill JM (1985) Spontaneous ocular shedding of HSV-1 in latently infected rabbits. Invest Ophthalmol Vis Sci 26:587–590
Bernstein DI, Harrison CJ, Jenski LJ, Myers MG, Stanberry LR (1991) Cell-mediated immunologic responses and recurrent genital herpes in the guinea pig. Effects of glycoprotein immunotherapy. J Immunol 146:3571–3577
Biswas PS, Banerjee K, Kim B, Kinchington PR, Rouse BT (2005) Role of inflammatory cytokine-induced cyclooxygenase 2 in the ocular immunopathologic disease herpetic stromal keratitis. J Virol 79:10589–10600. doi:10.1128/JVI. 79.16.10589-10600.2005
Blackstock R, Murphy JW (2004) Age-related resistance of C57BL/6 mice to Cryptococcus neoformans is dependent on maturation of NKT cells. Infect Immun 72:5175–5180. doi:10.1128/IAI. 72.9.5175-5180.2004
Boggian I, Buzzacaro E, Calistri A, Calvi P, Cavaggioni A, Mucignat-Caretta C, Palu G (2000) Asymptomatic herpes simplex type 1 virus infection of the mouse brain. J Neurovirol 6:303–313. doi:10.3109/13550280009030756
Boivin G, Coulombe Z, Rivest S (2002) Intranasal herpes simplex virus type 2 inoculation causes a profound thymidine kinase dependent cerebral inflammatory response in the mouse hindbrain. Eur J Neurosci 16:29–43. doi:10.1046/j.1460-9568.2002.02057.x
Boivin N, Menasria R, Piret J, Boivin G (2012) Modulation of TLR9 response in a mouse model of herpes simplex virus encephalitis. Antiviral Res 96:414–421. doi:10.1016/j.antiviral.2012.09.022
Bonneau RH, Jennings SR (1989) Modulation of acute and latent herpes simplex virus infection in C57BL/6 mice by adoptive transfer of immune lymphocytes with cytolytic activity. J Virol 63:1480–1484
Brown SM, Ritchie DA, Subak-Sharpe JH (1973) Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J Gen Virol 18:329–346. doi:10.1099/0022-1317-18-3-329
Bukowski JF, Welsh RM (1986) The role of natural killer cells and interferon in resistance to acute infection of mice with herpes simplex virus type 1. J Immunol 136:3481–3485
Burl S, Townend J, Njie-Jobe J, Cox M, Adetifa UJ, Touray E, Philbin VJ, Mancuso C, Kampmann B, Whittle H, Jaye A, Flanagan KL, Levy O (2011) Age-dependent maturation of Toll-like receptor-mediated cytokine responses in Gambian infants. PLoS One 6:e18185. doi:10.1371/journal.pone.0018185
Cameron JM, McDougall I, Marsden HS, Preston VG, Ryan DM, Subak-Sharpe JH (1988) Ribonucleotide reductase encoded by herpes simplex virus is a determinant of the pathogenicity of the virus in mice and a valid antiviral target. J Gen Virol 69(Pt 10):2607–2612. doi:10.1099/0022-1317-69-10-2607
Campadelli-Fiume G (2007) Menotti L (2007) Entry of alphaherpesviruses into the cell. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (eds) Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, Chapter 7
Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Sénéchal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Héron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL (2006) Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312. doi:10.1126/science.1128346
Cassady KA, Gross M, Roizman B (1998) The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol 72:7005–7011
Chentoufi AA, Zhang X, Lamberth K, Dasgupta G, Bettahi I, Nguyen A, Wu M, Zhu X, Mohebbi A, Buus S, Wechsler SL, Nesburn AB, BenMohamed L (2008) HLA-A*0201-restricted CD8+ cytotoxic T lymphocyte epitopes identified from herpes simplex virus glycoprotein D. J Immunol 180:426–437
Chentoufi AA, Dasgupta G, Christensen ND, Hu J, Choudhury ZS, Azeem A, Jester JV, Nesburn AB, Wechsler SL, BenMohamed L (2010) A novel HLA (HLA-A*0201) transgenic rabbit model for preclinical evaluation of human CD8+ T cell epitope-based vaccines against ocular herpes. J Immunol 184:2561–2571. doi:10.4049/jimmunol.0902322
Chung E, Sen J (2012) The ongoing pursuit of a prophylactic HSV vaccine. Rev Med Virol 22:285–300. doi:10.1002/rmv.1709
Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ (2012) Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol 5:173–183. doi:10.1038/mi.2011.63
Conrady CD, Zheng M, van Rooijen N, Drevets DA, Royer D, Alleman A, Carr DJ (2013) Microglia and a functional type I IFN pathway are required to counter HSV-1-driven brain lateral ventricle enlargement and encephalitis. J Immunol 190:2807–2817. doi:10.4049/jimmunol.1203265
Daheshia M, Kanangat S, Rouse BT (1998) Production of key molecules by ocular neutrophils early after herpetic infection of the cornea. Exp Eye Res 67:619–624. doi:10.1006/exer.1998.0565
Davey GM, Wojtasiak M, Proietto AI, Carbone FR, Heath WR, Bedoui S (2010) Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J Immunol 184:2243–2246. doi:10.4049/jimmunol.0903013
Davison AJ, McGeoch DJ (1986) Evolutionary comparisons of the S segments in the genomes of herpes simplex virus type 1 and varicella-zoster virus. J Gen Virol 67(Pt 4):597–611
Decman V, Freeman ML, Kinchington PR, Hendricks RL (2005) Immune control of HSV-1 latency. Viral Immunol 18:466–473. doi:10.1089/vim.2005.18.466
Dennis RF, Siemasko KF, Tang Q, Hendricks RL, Finnegan A (1995) Involvement of LFA-1 and ICAM-1 in the herpetic disease resulting from HSV-1 corneal infection. Curr Eye Res 14:55–62. doi:10.3109/02713689508999914
Dervillez X, Qureshi H, Chentoufi AA, Khan AA, Kritzer E, Yu DC, Diaz OR, Gottimukkala C, Kalantari M, Villacres MC, Scarfone VM, McKinney DM, Sidney J, Sette A, Nesburn AB, Wechsler SL, BenMohamed L (2013) Asymptomatic HLA-A*02:01-restricted epitopes from herpes simplex virus glycoprotein B preferentially recall polyfunctional CD8+ T cells from seropositive asymptomatic individuals and protect HLA transgenic mice against ocular herpes. J Immunol 191:5124–5138. doi:10.4049/jimmunol.1301415
Dropulic LK, Cohen JI (2012) The challenge of developing a herpes simplex virus 2 vaccine. Expert Rev Vaccines 11:429–1440. doi:10.1586/erv.12.129
Duff R, Rapp F (1971) Properties of hamster embryo fibroblasts transformed in vitro after exposure to ultraviolet-irradiated herpes simplex virus type 2. J Virol 8:469–477
Ecob-Johnston MS, Whetsell WO Jr (1979) Host-cell response to herpes virus infection in central and peripheral nervous tissue in vitro. J Gen Virol 44:747–757. doi:10.1099/0022-1317-44-3-747
Ejercito PM, Kieff ED, Roizman B (1968) Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi:10.1099/0022-1317-2-3-357
Eloranta ML, Sandberg K, Alm GV (1996) The interferon-alpha/beta responses of mice to herpes simplex virus studied at the blood and tissue level in vitro and in vivo. Scand J Immunol 43:356–360
Engler H, Zawatzky R, Goldbach A, Schroder CH, Weyand C, Hammerling GJ, Kirchner H (1981) Experimental infection of inbred mice with herpes simplex virus II. Interferon production and activation of natural killer cells in the peritoneal exudate. J Gen Virol 55:25–30. doi:10.1099/0022-1317-55-1-25
Engler H, Zawatzky R, Kirchner H, Armerding D, Engler H, Zawatzky R, Kirchner H, Armerding D (1982) Experimental infection of inbred mice with herpes simplex virus. IV. Comparison of interferon production and natural killer cell activity in susceptible and resistant adult mice. Arch Virol 74(74):239–247. doi:10.1007/BF01314157
Esaki S, Goshima F, Katsumi S, Watanabe D, Ozaki N, Murakami S, Nishiyama Y (2010) Apoptosis induction after herpes simplex virus infection differs according to cell type in vivo. Arch Virol 155:1235–1245. doi:10.1007/s00705-010-0712-2
Field HJ, Wildy P (1978) The pathogenicity of thymidine kinase-deficient mutants of herpes simplex virus in mice. J Hyg (Lond) 81:267–277
Forrester JV, Xu H, Kuffova L, Dick AD, McMenamin PG (2010) Dendritic cell physiology and function in the eye. Immunol Rev 234:282–304. doi:10.1111/j.0105-2896.2009.00873.x
Greco A, Arata L, Soler E, Gaume X, Couté Y, Hacot S, Callé A, Monier K, Epstein AL, Sanchez JC, Bouvet P, Diaz JJ (2012) Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J Virol 86:1449–1457. doi:10.1128/JVI. 06194-11
Gudnadottir M, Helgadottir H, Bjarnason O, Jonsdottir K (1964) Virus isolated from the brain of a patient with multiple sclerosis. Exp Neurol 9:85–95. doi:10.1016/0014-4886(64)90008-1
Guo YJ, Zhao L, Li XF, Mei YW, Zhang SL, Tao JY, Zhou Y, Dong JH (2010) Effect of Corilagin on anti-inflammation in HSV-1 encephalitis and HSV-1 infected microglias. Eur J Pharmacol 635:79–86. doi:10.1016/j.ejphar.2010.02.049
Hankenson FC, Ruskoski N, van Saun M, Ying GS, Oh J, Fraser NW (2013) Weight loss and reduced body temperature determine humane endpoints in a mouse model of ocular herpesvirus infection. J Am Assoc Lab Anim Sci 52:277–285
Hawkins P, Morton DB, Burman O, Dennison N, Honess P, Jennings M, Lane S, Middleton V, Roughan JV, Wells S, Westwood K, UK Joint Working Group on Refinement BVAAWF/FRAME/RSPCA/UFAW (2011) A guide to defining and implementing protocols for the welfare assessment of laboratory animals: eleventh report of the BVAAWF/FRAME/RSPCA/UFAW Joint Working Group on Refinement. Lab Anim 45:1–13. doi:10.1258/la.2010.010031
Hendricks RL, Tumpey TM, Finnegan A (1992) IFN-gamma and IL-2 are protective in the skin but pathologic in the corneas of HSV-1-infected mice. J Immunol 149:3023–3028
Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Perez de Diego R, Abhyankar A, Israelsson E, Guo Y, Cardon A, Rozenberg F, Lebon P, Tardieu M, Heropolitanska-Pliszka E, Chaussabel D, White MA, Abel L, Zhang SY, Casanova JL (2012) Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 209:1567–1582. doi:10.1084/jem.20111316
Hoffmann F, Albert MH, Arenz S, Bidlingmaier C, Berkowicz N, Sedlaczek S, Till H, Pawlita I, Renner ED, Weiss M, Belohradsky BH (2005) Intracellular T-cell cytokine levels are age-dependent in healthy children and adults. Eur Cytokine Netw 16:283–288
Hsiung GD, Mayo DR, Lucia HL, Landry ML (1984) Genital herpes: pathogenesis and chemotherapy in the guinea pig model. Rev Infect Dis 6:33–50, http://www.jstor.org/stable/4453263
Hu J, Peng X, Schell TD, Budgeon LR, Cladel NM, Christensen ND (2006) An HLA-A2.1-transgenic rabbit model to study immunity to papillomavirus infection. J Immunol 177:8037–8045. doi:10.4049/jimmunol.177.11.8037
Irvine AR, Kimura SJ (1967) Experimental stromal herpes simplex keratitis in rabbits. Arch Ophthalmol 78:654–663. doi:10.1001/archopht.1967.00980030656018
Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi:10.1038/nature08476
Iyer AV, Pahar B, Chouljenko VN, Walker JD, Stanfield B, Kousoulas KG (2013) Single dose of glycoprotein K (gK)-deleted HSV-1 live-attenuated virus protects mice against lethal vaginal challenge with HSV-1 and HSV-2 and induces lasting T cell memory immune responses. Virol J 10:317. doi:10.1186/1743-422X-10-317
Johnson RT (1964) The pathogenesis of herpes virus encephalitis. II. A cellular basis for the development of resistance with age. J Exp Med 120:359–374. doi:10.1084/jem.120.3.359
Kanangat S, Thomas J, Gangappa S, Babu JS, Rouse BT (1996) Herpes simplex virus type 1-mediated up-regulation of IL-12 (p40) mRNA expression. Implications in immunopathogenesis and protection. J Immunol 156:1110–1116
Kastrukoff LF, Kim SU (2002) Oligodendrocytes from human donors differ in resistance to herpes simplex virus 1 (HSV-1). Glia 38:87–92. doi:10.1002/glia.10043
Kastrukoff L, Long C, Doherty PC, Wroblewska Z, Koprowski H (1981) Isolation of virus from brain after immunosuppression of mice with latent herpes simplex. Nature 291:432–433. doi:10.1038/291432a0
Kastrukoff L, Hamada T, Schumacher U, Long C, Doherty PC, Koprowski H (1982) Central nervous system infection and immune response in mice inoculated into the lip with herpes simplex virus type 1. J Neuroimmunol 2:295–305. doi:10.1016/0165-5728(82)90062-5
Kastrukoff LF, Lau AS, Puterman ML (1986) Genetics of natural resistance to herpes simplex virus type 1 latent infection of the peripheral nervous system in mice. J Gen Virol 67(Pt 4):613–621. doi:10.1099/0022-1317-67-4-613
Kastrukoff LF, Lau AS, Kim SU (1987) Multifocal CNS demyelination following peripheral inoculation with herpes simplex virus type 1. Ann Neurol 22:52–59. doi:10.1002/ana.410220113
Kastrukoff LF, Lau AS, Leung GY, Walker DG, Thomas EE (1992) Herpes simplex virus type I (HSV I)-induced multifocal central nervous system (CNS) demyelination in mice. J Neuropathol Exp Neurol 51:432–439
Kastrukoff LF, Lau AS, Takei F, Smyth MJ, Jones CM, Clarke SR, Carbone FR (2010) Redundancy in the immune system restricts the spread of HSV-1 in the central nervous system (CNS) of C57BL/6 mice. Virology 400:248–258. doi:10.1016/j.virol.2010.02.013
Kastrukoff LF, Lau AS, Thomas EE (2012) The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS). Herpesviridae 3:4. doi:10.1186/2042-4280-3-4
Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL (2003) Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18:593–603. doi:10.1016/S1074-7613(03)00112-2
Kintner RL, Brandt CR (1995) The effect of viral inoculum level and host age on disease incidence, disease severity, and mortality in a murine model of ocular HSV-1 infection. Curr Eye Res 14:145–152. doi:10.3109/02713689508999926
Kirchner H, Engler H, Schroder CH, Zawatzky R, Storch E (1983) Herpes simplex virus type 1-induced interferon production and activation of natural killer cells in mice. J Gen Virol 64(Pt 2):437–441. doi:10.1099/0022-1317-64-2-437
Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL (2008) Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322:268–271. doi:10.1126/science.1164164
Kopp SJ, Banisadr G, Glajch K, Maurer UE, Grunewald K, Miller RJ, Osten P, Spear PG (2009) Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci U S A 106:17916–17920. doi:10.1073/pnas.0908892106
Kopp SJ, Karaba AH, Cohen LK, Banisadr G, Miller RJ, Muller WJ (2013) Pathogenesis of neonatal herpes simplex 2 disease in a mouse model is dependent on entry receptor expression and route of inoculation. J Virol 87:474–481. doi:10.1128/JVI. 01849-12
Ksander BR, Hendricks RL (1987) Cell-mediated immune tolerance to HSV-1 antigens associated with reduced susceptibility to HSV-1 corneal lesions. Invest Ophthalmol Vis Sci 28:1986–1993
Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW (2004) Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101:1315–1320. doi:10.1073/pnas.0308057100
Labetoulle M, Kucera P, Ugolini G, Lafay F, Frau E, Offret H, Flamand A (2000) Neuronal propagation of HSV1 from the oral mucosa to the eye. Invest Ophthalmol Vis Sci 41:2600–2606
Laing KJ, Magaret AS, Mueller DE, Zhao L, Johnston C, De Rosa SC, Koelle DM, Wald A, Corey, L (2010) Diversity in CD8(+) T cell function and epitope breadth among persons with genital herpes. J Clin Immunol 30:703–722. doi:10.1007/s10875-010-9441-2
Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC (1998) Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J Virol 72:6119–6130
Lee AJ, Ashkar AA (2012) Herpes simplex virus-2 in the genital mucosa: insights into the mucosal host response and vaccine development. Curr Opin Infect Dis 25:92–99. doi:10.1097/QCO.0b013e32834e9a56
Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA (2009) Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol 83:12164–12171. doi:10.1128/JVI. 01676-09
Liu F (2007) Hong Zhou Z (2007) Comparative virion structures of human herpesviruses. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (eds) Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, Chapter 3
Liu T, Tang Q, Hendricks, RL (1996) Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol 70:264–271
Liu T, Khanna KM, Chen X, Fink DJ, Hendricks, RL (2000) CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med 191:1459–1466. doi:10.1084/jem.191.9.1459
Liu T, Khanna KM, Carriere BN, Hendricks, RL (2001) Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J Virol 75:11178–11184. doi:10.1128/JVI.75.22.11178-11184
Lokensgard JR, Hu S, Sheng W, vanOijen M, Cox D, Cheeran MC, Peterson PK (2001) Robust expression of TNF-alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J Neurovirol 7:208–219
Lopez C (1975) Genetics of natural resistance to herpesvirus infections in mice. Nature 258:152–153. doi:10.1038/258152a0
Lund JM, Linehan MM, Iijima N, Iwasaki A (2006) Cutting edge: plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol 177:7510–7514. doi:10.4049/jimmunol.177.11.7510
Margolis TP, Imai Y, Yang L, Vallas V, Krause PR (2007) Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J Virol 81:1872–1878. doi:10.1128/JVI. 02110-06
Marodi L (2006) Neonatal innate immunity to infectious agents. Infect Immun 74:1999–2006. doi:10.1128/IAI. 74.4.1999-2006.2006
Marques CP, Cheeran MC, Palmquist JM, Hu S, Urban SL, Lokensgard JR (2008) Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J Immunol 181:6417–6426. doi:10.4049/jimmunol.181.9.6417
Martin R, Nauta AJ, Ben Amor K, Knippels LM, Knol J, Garssen J (2011) Early life: gut microbiota and immune development in infancy. Benef Microbes 1:367–382. doi:10.3920/BM2010.0027
McKendall RR (1980) Comparative neurovirulence and latency of HSV1 and HSV2 following footpad inoculation in mice. J Med Virol 5:25–32. doi:10.1002/jmv.1890050104
McNally JM, Dempsey D, Wolcott RM, Chervenak R, Jennings SR (1999) Phenotypic identification of antigen-dependent and antigen-independent CD8 CTL precursors in the draining lymph node during acute cutaneous herpes simplex virus type 1 infection. J Immunol 163:675–681
Meyding-Lamadé UK, Oberlinner C, Rau PR, Seyfer S, Heiland S, Sellner J, Wildemann BT, Lamadé WR (2003) Experimental herpes simplex virus encephalitis: a combination therapy of acyclovir and glucocorticoids reduces long-term magnetic resonance imaging abnormalities. J Neurovirol 9:118–125. doi:10.1080/713831335
Mou F, Forest T, Baines JD (2007) US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81:6459–6470. doi:10.1128/JVI. 00380-07
Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. doi:10.1016/j.chom.2006.12.001
Pan M, Wang X, Liao J, Yin D, Li S, Pan Y, Wang Y, Xie G, Zhang S, Li Y (2012) Prediction and identification of potential immunodominant epitopes in glycoproteins B, C, E, G, and I of herpes simplex virus type 2. Clin Dev Immunol 2012:205313. doi:10.1155/2012/205313
Parr MB, Parr EL (2003) Intravaginal administration of herpes simplex virus type 2 to mice leads to infection of several neural and extraneural sites. J Neurovirol 9:594–602. doi:10.1080/714044481
Peng T, Zhu J, Phasouk K, Koelle DM, Wald A, Corey, L (2012) An effector phenotype of CD8+ T cells at the junction epithelium during clinical quiescence of herpes simplex virus 2 infection. J Virol 86:10587–10596. doi:10.1128/JVI.01237-12
Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, Herman M, Cardon A, Durandy A, Bustamante J, Vallabhapurapu S, Bravo J, Warnatz K, Chaix Y, Cascarrigny F, Lebon P, Rozenberg F, Karin M, Tardieu M, Al-Muhsen S, Jouanguy E, Zhang SY, Abel L, Casanova JL (2010) Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 33:400–411. doi:10.1016/j.immuni.2010.08.014
Piret J, Boivin G (2010) Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother 55:459–472. doi:10.1128/AAC. 00615-10
Rawls WE, Laurel D, Melnick JL, Glicksman JM, Kaufman RH (1968) A search for viruses in smegma, premalignant and early malignant cervical tissues. The isolation of Herpesviruses with distinct antigenic properties. Am J Epidemiol 87:647–655
Rhee SJ, Walker WA, Cherayil BJ (2005) Developmentally regulated intestinal expression of IFN-gamma and its target genes and the age-specific response to enteric Salmonella infection. J Immunol 175:1127–1136. doi:10.4049/jimmunol.175.2.1127
Roberts CM, Pfister JR, Spear SJ (2003) Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 30:797–800. doi:10.1097/01.OLQ.0000092387.58746.C7
Roizman B (1996) The function of herpes simplex virus genes: a primer for genetic engineering of novel vectors. Proc Natl Acad Sci U S A 93:11307–11312. doi:10.1073/pnas.93.21.11307
Rollenhagen C, Lathrop MJ, Macura SL, Doncel GF, Asin SN (2014) Herpes simplex virus type-2 stimulates HIV-1 replication in cervical tissues: implications for HIV-1 transmission and efficacy of anti-HIV-1 microbicides. Mucosal Immunol. doi:10.1038/mi.2014.3
Roth K, Ferreira VH, Kaushic C (2013) HSV-2 vaccine: current state and insights into development of a vaccine that targets genital mucosal protection. Microb Pathog 58:45–54. doi:10.1016/j.micpath.2012.11.001
Russell RG, Nasisse MP, Larsen HS, Rouse BT (1984) Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Invest Ophthalmol Vis Sci 25:938–944
Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, Fabrega S, Cardon A, Maluenda J, Tatematsu M, Mahvelati F, Herman M, Ciancanelli M, Guo Y, AlSum Z, Alkhamis N, Al-Makadma AS, Ghadiri A, Boucherit S, Plancoulaine S, Picard C, Rozenberg F, Tardieu M, Lebon P, Jouanguy E, Rezaei N, Seya T, Matsumoto M, Chaussabel D, Puel A, Zhang SY, Abel L, Al-Muhsen S, Casanova JL (2011) Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest 121:4889–4902. doi:10.1172/JCI59259
Sarangi PP, Sehrawat S, Suvas S, Rouse BT (2008) IL-10 and natural regulatory T cells: two independent anti-inflammatory mechanisms in herpes simplex virus-induced ocular immunopathology. J Immunol 180:6297–6306. doi:10.4049/jimmunol.180.9.6297
Schachtele SJ, Hu S, Little MR, Lokensgard JR (2010) Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J Neuroinflammation 7:35. doi:10.1186/1742-2094-7-35
Schachtele SJ, Hu S, Lokensgard JR (2012) Modulation of experimental herpes encephalitis-associated neurotoxicity through sulforaphane treatment. PLoS One 7:e36216. doi:10.1371/journal.pone.0036216
Sehrawat S, Suvas S, Sarangi PP, Suryawanshi A, Rouse BT (2008) In vitro-generated antigen-specific CD4+ CD25+ Foxp3+ regulatory T cells control the severity of herpes simplex virus-induced ocular immunoinflammatory lesions. J Virol 82:6838–6851. doi:10.1128/JVI. 00697-08
Shimeld C, Dyson H, Lewkowicz-Moss S, Hill TJ, Blyth WA, Easty DL (1987) Spread of HSV-1 to the mouse eye after inoculation in the skin of the snout requires an intact nerve supply to the inoculation site. Curr Eye Res 6:9–12. doi:10.3109/02713688709020061
Shimomura Y, Dudley JB, Gangarosa LP Sr, Hill JM (1985) HSV-1 quantitation from rabbit neural tissues after epinephrine-induced reactivation. Invest Ophthalmol Vis Sci 26:121–125
Shin H, Iwasaki A (2013) Generating protective immunity against genital herpes. Trends Immunol 34:487–494. doi:10.1016/j.it.2013.08.001
Simmons A (2002) Clinical manifestations and treatment considerations of herpes simplex virus infection. J Infect Dis 186(Suppl 1):S71–S77. doi:10.1086/342967
Simmons A, Nash AA (1984) Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J Virol 52:816–821
Simmons A, Nash AA (1985) Role of antibody in primary and recurrent herpes simplex virus infection. J Virol 53:944–948
Simmons A, Nash AA (1987) Effect of B cell suppression on primary infection and reinfection of mice with herpes simplex virus. J Infect Dis 155:649–654. doi:10.1093/infdis/155.4.649
Simmons A, Slobedman B, Speck P, Arthur J, Efstathiou S (1992) Two patterns of persistence of herpes simplex virus DNA sequences in the nervous systems of latently infected mice. J Gen Virol 73(Pt 5):1287–1291. doi:10.1099/0022-1317-73-5-1287
Skoberne M, Cardin R, Lee A, Kazimirova A, Zielinski V, Garvie D, Lundberg A, Larson S, Bravo FJ, Bernstein DI, Flechtner JB, Long D (2013) An adjuvanted herpes simplex virus 2 subunit vaccine elicits a T cell response in mice and is an effective therapeutic vaccine in Guinea pigs. J Virol 87:3930–3942. doi:10.1128/JVI. 02745-12
Smith G (2012) Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi:10.1146/annurev-micro-092611-150051
Sprecher E, Becker Y (1987) Herpes simplex virus type 1 pathogenicity in footpad and ear skin of mice depends on Langerhans cell density, mouse genetics, and virus strain. J Virol 61:2515–2522
Steiner CM, Rozhon EJ, Lipton HL (1984) Relationship between host age and persistence of Theiler's virus in the central nervous system of mice. Infect Immun 43:432–434
Steiner I, Spivack JG, Deshmane SL, Ace CI, Preston CM, Fraser NW (1990) A herpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J Virol 64:1630–1638
Stumpf TH, Case R, Shimeld C, Easty DL, Hill TJ (2002) Primary herpes simplex virus type 1 infection of the eye triggers similar immune responses in the cornea and the skin of the eyelids. J Gen Virol 83:1579–1590
Svensson A, Bellner L, Magnusson M, Eriksson K (2007) Role of IFN-alpha/beta signaling in the prevention of genital herpes virus type 2 infection. J Reprod Immunol 74:114–123. doi:10.1016/j.jri.2006.09.002
Teague O, Goodpasture EW (1923) Experimental herpes zoster. JAMA 81:377. doi:10.1001/jama.1923.02650050031010
Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, Strupp M, Arbusow V, Brandt T (2003) Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol 163:2179–2184. doi:10.1016/S0002-9440(10)63575-4
Thomas EE, Lau AS, Kim SU, Osborne D, Kastrukoff LF (1991) Variation in resistance to herpes simplex virus type 1 of oligodendrocytes derived from inbred strains of mice. J Gen Virol 72(Pt 9):2051–2057. doi:10.1099/0022-1317-72-9-2051
Tigges MA, Koelle D, Hartog K, Sekulovich RE, Corey L, Burke, RL (1992) Human CD8+ herpes simplex virus-specific cytotoxic Tlymphocyte clones recognize diverse virion protein antigens. J Virol 66:1622–1634
Tomazin R, van Schoot NE, Goldsmith K, Jugovic P, Sempe P, Fruh K, Johnson DC (1998) Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol 72:2560–2563
Toth LA (2000) Defining the moribund condition as an experimental endpoint for animal research. ILAR J 41:72–79. doi:10.1093/ilar.41.2.72
Ushijima Y, Luo C, Goshima F, Yamauchi Y, Kimura H, Nishiyama Y (2007) Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect 9:142–149. doi:10.1016/j.micinf.2006.10.019
Valencia F, Veselenak RL, Bourne N (2013) In vivo evaluation of antiviral efficacy against genital herpes using mouse and guinea pig models. Methods Mole Biol 1030:315–326. doi:10.1007/978-1-62703-484-5_24
Verjans GM, Feron EJ, Dings ME, Cornelissen JG, Van der Lelij A, Baarsma GS, Osterhaus, AD (1998) T cells specific for the triggering virus infiltrate the eye in patients with herpes simplex virus-mediated acute retinal necrosis. J Infect Dis 178:27–34. doi:10.1086/515586
Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, Langerak AW, Kinchington PR, Osterhaus, AD (2007) Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A 104:3496–3501. doi:10.1073/pnas.0610847104
Vilela MC, Campos RD, Mansur DS, Rodrigues DH, Lacerda-Queiroz N, Lima GK, Rachid MA, Kroon EG, Campos MA, Teixeira AL (2011) Role of IL-4 in an experimental model of encephalitis induced by intracranial inoculation of herpes simplex virus-1 (HSV-1). Arq Neuropsiquiatr 69:237–241. doi:10.1590/S0004-282X2011000200019
Villalba M, Hott M, Martin C, Aguila B, Valdivia S, Quezada C, Zambrano A, Concha MI, Otth C (2012) Herpes simplex virus type 1 induces simultaneous activation of Toll-like receptors 2 and 4 and expression of the endogenous ligand serum amyloid A in astrocytes. Med Microbiol Immunol 201:371–379. doi:10.1007/s00430-012-0247-0
Wagner EK, Bloom DC (1997) Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev 10:419–443
Wander AH, Centifanto YM, Kaufman HE (1980) Strain specificity of clinical isolates of herpes simplex virus. Arch Ophthalmol 98:1458–1461. doi:10.1001/archopht.1980.01020040310020
Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, Finberg RW, Kurt-Jones EA (2011) Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J Virol 86:2273–2281. doi:10.1128/JVI. 06010-11
Ward PL, Roizman B (1994) Herpes simplex genes: the blueprint of a successful human pathogen. Trends Genet 10:267–274. doi:10.1016/0168-9525(90)90009-U
Whitley RJ (2006) Herpes simplex encephalitis: adolescents and adults. Antiviral Res 71:141–148. doi:10.1016/j.antiviral.2006.04.002
Whitley RJ, Roizman B (2001) Herpes simplex virus infections. Lancet 357:1513–1518. doi:10.1016/S0140-6736(00)04638-9
Zawatzky R, Engler H, Kirchner H (1982a) Experimental infection of inbred mice with herpes simplex virus. III. Comparison between newborn and adult C57BL/6 mice. J Gen Virol 60:25–29. doi:10.1099/0022-1317-60-1-25
Zawatzky R, Kirchner H, DeMaeyer-Guignard J, DeMaeyer E (1982b) An X-linked locus influences the amount of circulating interferon induced in the mouse by herpes simplex virus type 1. J Gen Virol 63:325–332. doi:10.1099/0022-1317-63-2-325
Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Héron B, Vallée L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL (2007) TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527
Zhang X, Castelli FA, Zhu X, Wu M, Maillere B, BenMohamed L (2008) Gender-dependent HLA-DR-restricted epitopes identified from herpes simplex virus type 1 glycoprotein D. Clin Vaccine Immunol 15:1436–1449. doi:10.1128/CVI. 00123-08
Acknowledgments
The authors would like to thank Diana Winthers, Drexel University College of Medicine Academic Publishing Services, for offering editorial services for the manuscript. Dr. Jennings is supported by career developmental funds provided by the Department of Microbiology and Immunology and the Institute for Molecular Medicine and Infectious Disease, Drexel University College of Medicine. Dr. Wigdahl (Principal Investigator) is supported by grants from the Public Health Service, National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS32092 and NS4626) and the National Institute of Drug Abuse (DA19807).
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kollias, C.M., Huneke, R.B., Wigdahl, B. et al. Animal models of herpes simplex virus immunity and pathogenesis. J. Neurovirol. 21, 8–23 (2015). https://doi.org/10.1007/s13365-014-0302-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-014-0302-2