
Abstract
A growing body of epidemiologic and experimental data point to chronic bacterial and viral infections as possible risk factors for neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis. Infections of the central nervous system, especially those characterized by a chronic progressive course, may produce multiple damage in infected and neighbouring cells. The activation of inflammatory processes and host immune responses cause chronic damage resulting in alterations of neuronal function and viability, but different pathogens can also directly trigger neurotoxic pathways. Indeed, viral and microbial agents have been reported to produce molecular hallmarks of neurodegeneration, such as the production and deposit of misfolded protein aggregates, oxidative stress, deficient autophagic processes, synaptopathies and neuronal death. These effects may act in synergy with other recognized risk factors, such as aging, concomitant metabolic diseases and the host’s specific genetic signature. This review will focus on the contribution given to neurodegeneration by herpes simplex type-1, human immunodeficiency and influenza viruses, and by Chlamydia pneumoniae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD) and amyotrophic lateral sclerosis (ALS), are devastating pathologies characterized by progressive degeneration and loss of specific subsets of neurons that lead to a decline in brain functions such as cognition and locomotor control. Although these diseases have very different clinical manifestations, depending partly on which region of the brain is affected, they share some common features and pathological hallmarks, including the formation and deposition of aberrant protein conformers, synaptic dysfunctions, oxidative stress, deficient autophagic processes, and inflammation.
The causative agents of these highly complex diseases, which are often the result of several combined genetic and environmental factors, are still unknown and the molecular basis underlying their pathogenesis has yet to be fully clarified. Starting from the pioneer study by Bowery [1] showing the neurodegenerative effects produced by tetanus toxin in rats, a significant emerging body of literature suggests the possibility that CNS infections may play a cofactorial role in inducing neurodegenerative diseases [2]. Serological studies suggest a potential involvement of enteroviruses (EV) and human herpesviruses (HHV) in the aetiology of ALS, a fatal neurodegenerative disease selectively affecting motor neurons [3]. A viral origin of PD has also been proposed on the basis of the close similarity between the clinical symptoms of PD and those of Japanese encephalitic virus [4]. Ever since the earliest papers describing the neurological symptoms associated with influenza [5, 6], the influenza virus has repeatedly been suggested as an aetiological agent for PD [7]. Several findings also support the involvement of infectious agents (in particular herpes simplex virus type-1 [HSV-1] and Chlamydia pneumoniae) in the pathogenesis of AD, a multifactorial disorder characterized by severe memory impairment and cognitive decline that affects hippocampal and basal cortex neurons [8–15]. Among the human herpesviruses, herpes simplex virus, Epstein Barr Virus, varicella zoster virus, cytomegalovirus, human herpesvirus-6 and, more recently, herpesvirus-7 have all been associated with multiple sclerosis (MS), an inflammatory disease leading to demyelization of nerve cell axons in the spinal cord and brain [16–23].
Some of these correlations are not yet supported by conclusive experimental and clinical evidence, but a growing body of data supports the hypothesis that chronic damage induced by different infectious agents may concur to produce neurodegeneration. Obviously, the long-term effects of persistent or lifelong repeated infections may differ in different hosts, according to their general health, pharmacological treatments, genetic background, concurrent diseases, etc. Even considering the different outcomes due to these conditions, several findings indicate that pathogen-related long-term damage may underlie several neuronal dysfunctions typical of aging [2].
In this regard, it is worth noting that the CNS may be particularly vulnerable to infectious agents during aging on account of alterations to the blood–brain barrier (BBB), as well as age-related increased oxidative stress and impaired energy production [2]. Aged neurons are certainly more vulnerable to many insults, including the toxicity of viral or prion proteins, due to increased oxidative stress and impaired neurotrophic factor signalling pathways [24, 25].
Although activation of the host immune response in an attempt to eradicate the pathogen [26] may significantly contribute to produce neuronal damage, different pathogens and/or their products may directly induce long-term degenerative effects, such as the deposit of misfolded protein aggregates, increased levels of oxidative stress, deficient autophagic processes, synaptopathies and neuronal death. This review will focus on the degenerative effects of chronic or persistent infections caused by herpes simplex virus, human immunodeficiency virus, influenza virus and C. pneumoniae.
Route of Entry of Infectious Agents into the CNS
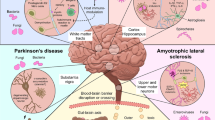
Viruses, bacteria, protozoa and unconventional pathogens such as prion proteins have the ability to invade the CNS and cause acute infections which in some cases may be fatal or which may progress to become chronic illnesses [26, 27]. Here, we focus on selected human viruses and bacteria to describe various mechanisms of neuroinvasion (Fig. 1).

Infectious agents can reach the central nervous system by either crossing the blood–brain barrier (hematogen route) or being transported by axons of cranial nerve neurons (for further details, see text)
In immunocompetent subjects with a fully functional BBB very few pathogens are able to infect the brain through the blood. Indeed, the BBB and cerebral spinal fluid (CSF) barrier prevent the unselective diffusion of vascular and cellular components. The BBB, in particular, is composed mainly of non-fenestrated endothelial cells interconnected by tight junctions in contact with astrocyte processes. Besides limiting the movement of cells and molecules to the brain parenchyma and neurons, these cells also provide a physical and cellular barrier to the perivascular space by producing a basement membrane consisting of laminin. However, some pathogens are able to disrupt the BBB and to cross directly into the CSF through the porous capillaries of the choroid plexus, thus passing into the brain and provoking severe haemorrhagic encephalitis. When the BBB is compromised, as in many diseases, different viruses can enter the brain through the bloodstream.
Other possible routes of entry into the CNS are: (1) infection of cells belonging to the monocyte–macrophage/microglia lineage that are able to cross the BBB; (2) intra- and trans-neuronal transfer from peripheral neurons.
-
1.
Viruses of the lentivirus family, which includes HIV, cross the BBB through the “Trojan Horse” mechanism [28, 29], driven by infected leukocytes, which allow the virus to escape the immune system and to move from the bloodstream to the brain. Inflammation enhances this invasion of the CNS, as inflammatory molecules released during the systemic infection activate infected leukocytes, which in turn attach to and invade the postcapillary venule wall surrounding endothelial and parenchymal basement membranes, thus crossing the BBB. Leukocytes are further activated by interactions between chemokine receptors expressed on their membranes and chemokines circulating in the brain. For example, increased expression of chemokine ligand 2 (CCL2) by endothelial cells and astrocytes following contact with HIV-infected cells [30], together with virus-induced alterations of endothelial adhesion molecules and junctional proteins [31], amplifies disruption of the BBB and viral entry. Moreover, dysregulation of BBB, likely caused by a combination of viral and host factors (e.g., secreted viral proteins, inflammatory mediators, antiviral therapies, drug or alcohol abuse, aging), has been proposed as a critical component of HIV-induced neuropathology [32]. Virus-bearing monocytes then infect perivascular macrophages and microglia, but not neurons, which are, however, negatively affected by the release of inflammatory cytokines and/or viral proteins by neighbouring cells.
-
2.
The peripheral nerve endings located in the skin and the mucosa mediate the other mechanism of entry of viruses that infect the CNS. Depending on the differential expression of viral receptors, a particular neurotropic virus will target a specific type of peripheral nerve ending, including those of sensory, motor and olfactory neurons. Viruses infecting these sites exploit cellular components such as the axonal microtubules and microtubule-based motor proteins [33] to move, through retrograde axonal transport, from the cell periphery to the neuronal cell body that contains the synthetic machinery exploited for viral replication, as well as to return through anterograde transport to the cell periphery and spread into target cells [34].
HSV-1, for example, can infect oral and nasal mucosa and then travels through retrograde axonal transport to the trigeminal ganglion or the olfactory bulb, respectively, where it establishes a latent infection or may rapidly enter the CNS [35]. It is worth noting that HSV-1 infection has been reported to suppress the induction of neuronal apoptosis in the olfactory neuroepithelium and trigeminal and dorsal root ganglia in order to facilitate neuroinvasion of the brain [36, 37]. Periodic reactivations from latency are followed by axonal transport of newly produced HSV-1 virions either back to the site of primary infection, where they cause new skin vesicles or mucosal ulcers, or onward to the CNS, where they can cause a productive, but usually mild infection, which may later become latent, as described for rodents [38, 39]. In particular, newly produced virions may target the limbic system, which includes the hippocampus, thalamus and amygdala [40]. Indeed, in HSV encephalitis, the major site of damage is the limbic system [41] that is presumably reached through the olfactory bulb, since HSV-1 can infect cells in the nasal endothelium [42, 43].
The olfactory system provides a bridge between the peripheral environment and the brain and several other neurotropic viruses, including rabies, influenza A virus and parainfluenza virus are known to infect the CNS by transmission through the olfactory pathway. The A/WSN/33 strain of influenza virus has been shown to enter the CNS via the olfactory epithelium [44]. However, it has also been hypothesized that it can enter the CNS via other cranial nerves, including the vagus, and trigeminal nerves [45–48]. These nerves have processes that innervate visceral organs and tissues that are thought to be initially targeted by intranasal viral infection, including the olfactory epithelium (olfactory nerve, CN I), orofacial mucosa (trigeminal nerve, CN V) and digestive system (vagus nerve, CN X). Accordingly, the virus has been found in the regions innervated by these nerves following infection of animal models via intranasal routes. The virus has also been detected (by immunohistochemical staining of viral nuclear protein, NP) in the visceral ganglia [48, 49]. However, evidence that the A/WSN/33 strain of influenza has an affinity for the substantia nigra [50], a neuronal population with no direct anatomical connection to the cranial nerve system, suggests that other routes of invasion may be involved. In particular, it is possible to hypothesize that it can move into the brain through the ependymal cells lining the ventricles and shedding into the CSF, where it can freely spread throughout the neuraxis; through the extravasation from capillaries that penetrate into the brain; or through direct invasion of the CNS after BBB disruptions.
After entering the CNS, viruses promote cell-to-cell dissemination through different mechanisms, i.e. release into the synaptic cleft or via fusion events with neighbouring neurons. They may also reinfect peripheral tissue. Alphaviruses use the anterograde transport system to move from the cell body to the axonal terminal where they are released by exocytosis into the synaptic cleft [35]. However, during anterograde transport, the virus can also exit through axonal varicosities before reaching the termini and infect neighbouring cells [51, 52].
Invasion of the CNS by pathogens may result in acute infection, possibly followed by latency and reactivations, or in persistent infection causing chronic damage that accumulates with time. The occurrence of each outcome may depend on several factors, including the type of pathogen, the kind of host immune/inflammatory response elicited, the CNS region affected, the general conditions of the host and the presence of concomitant diseases.
Epidemiological Evidence
HSV-1 and Alzheimer's Disease
Chronic and persistent exposure to HSV-1 (see Fig. 2) has been proposed as a potential risk factor for AD. HSV-1 is a ubiquitous neurotropic virus that affects between 56 and 85 % of the world population, with country-to-country variations, and more than one third of the population has recurrent clinical HSV-1 infections and manifestations. Interestingly, epidemiological studies have reported the presence of the HSV-1 genome in post-mortem brain specimens from numerous AD patients, particularly those who carry the type 4 allele of the gene that encodes apolipoprotein E (APOE4), another potential risk factor for AD [14, 56]. Moreover, genes related to HSV-1 reactivation have been detected in the brain of patients with familial AD, associated with β-amyloid deposits [13], and HSV-1 DNA has been found in amyloid plaques from the temporal and frontal cortices of AD sufferers [58]. Recently, a large prospective population-based study also showed that the risk of AD is increased in elderly subjects with positive titers of anti-HSV-1 IgM antibodies, which are markers of primary or reactivated HSV-1 infections, while it is not associated with anti-HSV-1 IgG antibodies, which are markers of a life-long infection [59]. Finally, genome-wide association (GWA) studies have correlated individual brain susceptibility to HSV-1 infection with a genetic risk of AD [60, 61]. In particular, analysis of data from GWA studies of several thousand European AD patients and controls [60] identified a set of AD-linked gene variants that may increase the brain's susceptibility to viral infections [61]. These include: nectin-2, also known as herpes virus entry-mediator-B or poliovirus receptor-related protein-2, which mediates the entry of HSV into host cells; apolipoprotein E (APOE), particularly its ε4 allele, which besides being a well-estabilished genetic risk factor for AD, has also been shown to influence susceptibility to viral infections and spreading into neuronal cells; translocase of the outer mitochondrial membrane 40 homolog (TOMM40), whose variations might influence mitochondrial damage induced by HSV DNAase such as UL12.5, and other genes.

HSV is a double-stranded DNA virus, structurally composed of a linear genome packaged into an icosahedral capsid enclosed by tegument proteins and surrounded by a lipid bilayer membrane with embedded proteins and glycoproteins (envelope). Primary infection in humans usually occurs in the orofacial mucosa during childhood. There the virus replicates within the epithelial cells and undergoes its typical lytic life cycle ending with the production of infectious virions and lysis of the host cell. HSV entry into the host cell requires sequential interaction between specific viral membrane glycoproteins (gB, gC, gD, gH and gL) and cellular receptors [heparan sulphate proteoglycans (HSPG), nectin-1 and 2, herpesvirus entry mediator (HVEM) or 3-O sulphated heparan sulphate (3-OS HS)]. On entry, the nucleocapsid is transported to the nuclear membrane and the viral DNA is released into the nucleus for transcription of viral genes and replication. The HSV genome consists of two long structures of unique sequences (designated long (UL) and short (US)), that encode over 80 distinct genes, and it is transcribed by the RNA polymerase II of the infected host. Immediate-early genes are the first to be expressed following infection and they encode proteins that regulate the subsequent expression of early and late viral genes. Early gene expression then allows the synthesis of enzymes involved in DNA replication and the production of certain envelope glycoproteins. Expression of late genes occurs last; this group of genes predominantly encodes proteins that form the virus particle. This latter is then released from host cells by budding. HSV-1 is able to establish a lifelong latent infection in sensory neurons, particularly in cellular bodies of those feeding the site of primary infection [53]. This latency is characterized by the presence of a functional viral genome without production of the infectious virus. During this time, the latency-associated transcripts (LATs) are the only prominent transcripts [54] whose role in generating functional peptides or proteins is still a matter of debate [55]. Reactivation from latency can be triggered by several external stimuli (stress, immunosuppression, etc.) that activate viral gene expression. Newly produced virions are transported to the sites of primary infection where they cause recurrent herpetic lesions in some people. Interestingly, APOE4 is a risk factor for these recurrent herpetic lesions [56, 57]
These variants form a genetic signature that may determine individual brain susceptibility to HSV-1 infection during aging or susceptibility to pathogen-driven damages, particularly those leading to neurodegeneration.
C. pneumoniae and AD
C. pneumoniae (see Fig. 3) infection was first linked to AD on the basis of evidence that a high percentage (90 %) of AD brains were found to be PCR-positive for this pathogen, particularly in the cerebral regions most affected by AD [8]. In particular, this microorganism, which is able to infect microglia, astrocytes, perivascular macrophages and monocytes [8, 62], was isolated from the tissue as metabolically active and propagated in cells. Other studies failed to detect C. pneumoniae in archival tissue of AD patients [63–65], but it has to be underlined that two of these studies were performed on tissue that was paraffin embedded, which may have affected the identification of the organism using the specific PCR technique [63, 65]. Other authors have demonstrated the presence of C. pneumoniae in AD patient brains through PCR analysis of frozen tissue [66]. More recently, Little et al. demonstrated that intranasal inoculation of C. pneumoniae in mice induced AD-like hallmarks in brains [67]. Moreover, C. pneumoniae antibodies have been identified in AD brains, colocalizing with plaques and tangles in vulnerable brain regions [68].

Chlamydiae are Gram-negative bacteria. They are obligate intracellular parasites because their multiplication depends on the host cell for energy and various nutrients. Chlamidiae have evolved a unique biphasic developmental cycle in which they alternate two distinct morphological forms: the elementary body (EB) and the reticulate body (RB). EBs are small tight bodies and represent the metabolically inactive form of bacteria, which can resist environmental stress and survive outside a host for a limited time. Infection begins with the attachment of the EB to the surface of susceptible host cells, followed by its internalization by endocytosis and the formation of phagosomes (Chlamydia inclusions) that are heavily modified by chlamydial proteins which prevent their fusion with lysosomes. Shortly after uptake, EBs differentiate into the metabolically active form of RB and begin to replicate within the phagosomes. RBs replicate by binary fission that, after 24–72 h, becomes asynchronous, with some RBs converting back to EBs. Finally, EBs are released from infected cells, often after causing the death of the host cells, and can infect new cells, either in the same organism or in a new host. C. pneunoniae was classified as the third species of Chlamidia and was associated in humans with acute infections of the lower respiratory tract. It is recognized as a common cause of mild pneumonia in children and young adults. It infects both epithelial cells and macrophages within the lungs and may be disseminated to sites outside of the lungs by infected monocytes and macrophages. Infection may also persist or, alternatively, the bacterium may be present in asyntomatic patients. Recently, a large volume of research showed evidence that C. pneumoniae may contribute directly and indirectly (immuno-mediated) to atherosclerosis. Indeed, it was one of the few infectious agents that have been found within and isolated from cells of human atherosclerotic plaques
HIV and Dementia
Almost 60 million people worldwide have been infected by HIV (see Fig. 4), a virus known for its devastating effects on the immune system, that results in acquired immune deficiency syndrome (AIDS), characterized by increased risk of several opportunistic infections and diseases. Although effective treatments for AIDS have prolonged the survival of infected patients, they have also resulted in a growing number of patients with neurological consequences of HIV infection [69, 70]. The virus can also cause severe neurological disorders, known as HIV-associated neurocognitive disorders (HAND), that are characterized by cognitive, motor and behavioral abnormalities and comprise: asymptomatic neurocognitive impairment (ANI), HIV-associated mild neurocognitive disorder (MND) and HIV-associated dementia (HAD). These pathologies are the result of the invasion of HIV and its replication within the CNS and consequent virus-induced degeneration of synapses and neurons in different brain regions, as well as associated neuroinflammation and immune activation of macrophages, microglia and astrocytes. Interestingly, APOEε4 has been proposed as a genetic factor for the development of a severe form of HAD, as it is for AD [71, 72]. Although the use of antiretroviral drugs, initially as monotherapy (single agents such as zidovudine) and then as combination therapy (highly active antiretroviral therapy or HAART), has changed the clinical management of HIV- and HAND-positive patients by suppressing the systemic viral load and consequently decreasing the mortality rates, the incidences of both opportunistic infections in AIDS patients and the most severe form of HAD, the prevalence of neurocognitive impairment remains high [70, 73–75]. One possible explanation is that HAART does not readily cross the BBB, making the CNS a safe haven for infection and permitting ongoing degenerative changes even when viral titers are low in the periphery. However, other factors have been suggested to be involved in modulating HAND pathologies, including the effects of aging on the vulnerability of the brain, the persistence of HIV replication in brain macrophages, the evolution of highly neurovirulent HIV strains affecting the CNS, and even long-term CNS toxicity due to HAART [reviewed in 76].

HIV-1 is an enveloped icosahedral retrovirus, belonging to the Lentivirus subgroup of Retroviridae family. Its genome is constituted by two identical copies of non-complementary positive single-stranded RNA, enclosed by a capsid composed of several copies of the viral protein p24. The single-stranded RNA is tightly bound to nucleocapsid proteins and enzymes needed for viral replication and assembly such as reverse transcriptase, proteases, ribonuclease and integrase. A matrix composed of the viral protein p17 surrounds the nucleocapsid and this is, in turn, surrounded by the viral envelope containing the surface glycoproteins gp120 and gp41 as protruding spikes. The HIV replication cycle begins with adsorption of the viral particles to CD4 molecules (a member of the immunoglobulin superfamily) on the surface of susceptible cells. The subsequent interaction with a co-receptor belonging to the family of chemokine receptors (CXCR4 or CCR5) plays a major role in membrane fusion and entry. Shortly after entry, subviral particles are partially uncoated in the cytoplasm and initiate the reverse transcription of viral RNA. The newly produced DNA is then transported into the nucleus and integrated into the host DNA by the virus-encoded integrase. The integrated HIV DNA is called provirus. The provirus may remain inactive for a long time, producing few or no new viral particles. The coordinated interaction of the HIV-encoded Tat protein and cellular transcription factors with the RNA polymerase II transcription apparatus starts the production of viral genomic RNA and messenger RNA, which is then spliced into smaller pieces, exported from the nucleus into the cytoplasm and translated into the proteins. In addition to viral structural proteins and enzymes, the HIV genome encodes the regulatory proteins (Rev and Tat, which is secreted by HIV-infected cells) and several accesory proteins: Vif, Vpu, Vpr, Vpx and Nef, playing an important role in the viral replication, disease pathogenesis and immune evasion. At the end of viral replication cycle, envelope polyproteins are transported to the plasma membrane where viral progeny begins assembly and budding from the infected cells. Then, subsequent proteolysis by viral protease generates mature particles
Influenza Virus and Parkinson's Disease
The influenza virus (see Fig. 5) has been implicated as both a direct and an indirect cause of PD, on the basis of both clinical descriptions and epidemiological studies. However, the link with PD is somewhat controversial. Much of the association of Parkinsonism with influenza and many other viruses stems from an outbreak of encephalitis lethargica (EL) (von Economo's disease) and the postencephalitic Parkinsonism that occurred subsequent to the 1918 influenza pandemic caused by a type A H1N1 influenza virus [78, 79, reviewed in 80]. Although the hypothesis that EL was a complication of influenza is supported by several data, it is still a matter of debate. One piece of evidence against the role of influenza as an agent of PD is the absence of viral RNA recovered from the brains of postencephalitic PD patients [81, 82], the absence of any known mutations that would make the 1918 influenza virus neurotropic, and questions regarding the timing of the 1918 pandemic waves and the outbreak of EL. Despite the apparent strength of the direct evidence against the influenza hypothesis, there is a strong epidemiological tie, based mostly on the increased incidence of PD in the wake of the 1918 H1N1 influenza A pandemic [83–85]. It has even been shown that persons born during the 1918 influenza pandemic had a 2–3-fold higher risk of Parkinson's disease than those born prior to 1888 or after 1924 [86, 87]. Poskanzer and Schwab [84] also showed an increase in PD onset based on an external event occurring around 1920. Recently, a large observational study carried out by Toovey et al. [88] using a large database from the UK, identified 3,976 cases of PD and 18,336 of Parkinson symptoms (PS) between 1994 and 2007 and concluded that influenza infections are associated with transient neurological sequelae such as tremor or gait disturbances. Interestingly, the risk of developing PS increases with the number of influenza attacks, suggesting that influenza-associated neuronal injury may be a cumulative process. The relative risk of developing neurological PS was highest within the first few weeks after a diagnosed and recorded influenza infection.

Influenza A viruses are enveloped, negative strand RNA viruses belonging to the Orthomyxoviridae family. Their genome consists of eight single-stranded RNA segments encoding 11 or 12 proteins: the receptor-binding haemagglutinin (HA); the sialic acid-destroying enzyme neuraminidase (NA), the ion channel M2, the matrix protein M1; the nucleoprotein (NP); the polymerase acidic protein (PA), polymerase basic proteins 1 and 2 (PB1, PB2) and the pro-apoptotic protein polymerase basic 1 (PB1)-F2; the nuclear export protein (NEP; also known as NS2) and the host antiviral response antagonist non-structural protein 1 (NS1); the newly identified N40 protein, which is expressed from the PB1 segment and has an unknown function [77]. Within the virion, each of the eight RNA segments forms a viral ribonucleoprotein (RNP) complex: in particular, viral RNA is wrapped around NP, and this structure is in turn bound to the viral polymerase complex, to constitute the viral nucleocapsid. In the initial stages of influenza A virus replication, the viral HA binds to host cell receptors that contain terminal α-2,6-linked or α-2,3-linked sialic acid (α-2,6-SA or α-2,3-SA) moieties, and the virus enters the cell by receptor-mediated endocytosis. Cleavage of HA by cellular proteases is required to expose the HA peptide that is responsible for the fusion between the viral envelope and the endosomal membrane. Acidification of the late endocytic vesicles allows the viral HA to undergo a conformational rearrangement that produces a fusogenic protein. The H+ ions in the acidic endosome are pumped, via the viral M2 ion channel, into the virus structure allowing the virus uncoating and the release of RNP complexes into the cytoplasm. The viral RNA is then imported in an ATP-dependent manner into the cell nucleus for transcription of genomic and messenger RNAs which are transported to the cytosol for translation. Viral HA, NA and M2 are synthesized in the Endoplasmic Reticulum, transported by the trans-Golgi secretory pathway and the mature proteins are inserted in the plasma membrane. New viral RNA is encased in the nucleocapsidic proteins and, together with matrix protein, is transported to cell surface where HA and NA will be incorporated. Progeny virions are then released from cells by budding
Infectious Agents and Neurodegenerative Processes
Protein Misfolding in the Brain
Numerous neurodegenerative disorders share common features, including protein aggregation and the formation of inclusion bodies or aggregate deposits in selected brain regions. These deposits usually consist of insoluble fibrillar aggregates containing misfolded protein with β-sheet conformation. Although the distribution and composition of these protein aggregates are different in each neurodegenerative disease, they show similar morphological, structural and staining characteristics [89] and represent typical disease hallmarks. Two types of protein deposit characterize AD: (1) amyloid plaques, comprising mainly 40- to 42-residue peptides named β-amyloid peptides (Aβs), are deposited extracellularly in the brain parenchyma and around the cerebral vessel wall [90]; (2) neurofribillary tangles containing paired helical filaments, composed mainly of hyperphosphorylated tau protein, accumulate in the cytoplasm of degenerating neurons [91]. The aggregates that characterize PD consist mainly of a protein named α-synuclein (α-SYN), which accumulates in Lewy bodies in the cytoplasm of substantia nigra neurons [92]. In HD patients, intranuclear deposits of a polyglutamine-rich version of huntingtin protein are found in the striatum [93]. SLA is characterized by aggregates composed mainly of superoxide dismutase in cell bodies and axons of motor neurons [94]. Finally, brains with various forms of transmissible spongiform encephalopathies (TSE) are characterized by an accumulation of preotease-resistant aggregates of prion protein (PrP) [95].
Historically, lesions that contain aggregates were considered to be pathogenic. In effect, abnormal aggregates are found in the brain region most affected by each disease. Additionally, mutations in the genes coding for misfolded proteins cause familial forms of these diseases, characterized by an earlier onset and a more severe phenotype than the sporadic forms. Recently, several findings have suggested that the small intermediates, i.e. soluble oligomers, within the complex multistep process by which misfolded proteins assemble into inclusion bodies are the most toxic forms of these aberrant proteins. They appear to be able to affect normal cell activities, whereas aggregates may represent a cellular attempt to wall off potentially toxic material [96] or might function as a reservoir of the bioactive oligomers.
Amyloid peptides (Aβs), found in extraneuronal plaques in AD patients as well as in intraneuronal compartments, result from the amyloidogenic proteolytic cleavage of Amyloid Precursor Protein (APP) by the sequential action of β- and γ-secretase enzymes, and a significant body of evidence suggests that Aβ accumulations in AD are the result of an imbalance between Aβ production and Aβ clearance [97–99]. Mutations in genes encoding APP and presenilin, a component of the γ-secretase complex, have been associated with the familial form of AD, whose pathogenesis is undoubtedly linked to their involvement in Aβ overproduction. However, the causes of the latter in sporadic forms of the disease have yet to be fully clarified. Interestingly, some pathogens have been associated with or involved in Aβ accumulation.
HSV-1 and Protein's Aggregates
The possible links between HSV-1 and Aβ are supported by experimental findings. Firstly, amyloid peptide is characterized by some degree of sequence homology with the HSV-1 glycoprotein B, and the viral protein has been suggested by some to act as a seed for Aβ deposits in amyloid plaques [100]. Moreover, new HSV-1 particles produced in the PNS have been proposed to recruit cell membranes containing APP, possibly during packaging in the Golgi apparatus [101], and to release APP during transport into the brain, thus contributing in some way to the formation of amyloid deposits. HSV-1 has also been shown to bind APP directly within the axonal transport into neurons [102, 103]. Secondly, Wozniak et al. [104] reported the accumulation of Aβ peptides in neurons and mouse brains infected with HSV-1, and then [58] the presence of the viral genome within amyloid plaques in AD brains. Other studies suggest that HSV-1 infection can interfere with APP processing: Shipley et al. [105] showed that HSV-1 infection of neuroblastoma cells induced the formation of a 55-kDa C-terminal fragment of APP; Wozniak et al. [104] found that BACE1 (β-secretase) and nicastrin (an essential component of the γ-secretase complex) immunolabeling is increased in the brains of HSV-1-infected mice. We recently reported that HSV-1 produces marked changes in neuronal excitability and intracellular Ca2+ signalling that cause APP phosphorylation and intracellular Aβ accumulation in rat cortical neurons [106]. We also demonstrated that HSV-1 triggers amyloidogenic cleavages of APP that are mediated in part by the action of β-secretase, γ-secretase and caspase-3-like enzymes, and that these result in the formation and intracellular accumulation of different APP fragments, including Aβ in both monomeric and oligomeric forms, with established potential for neurotoxicity [107] (Fig. 6). Finally, Cheng and colleagues recently demonstrated that intracellular HSV-1 interacts with APP, and that this interplay enhances viral transport and disrupts APP transport and distribution [108]. As for Aβ production, HSV-1 has been shown to cause the hyperphosphorylation of the microtubule-associated tau protein, thus impairing its intracellular transport functions. Zambrano et al. [109] first linked HSV-1 to tau hyperphosphorylation, showing evidence of altered microtubule dynamics and neurite damage occurring in HSV-1-infected cultures of murine cortical neurons. Subsequently, HSV-1 infection in neuroblastoma and glioblastoma cells was shown to induce the phosphorylation of tau at a number of sites that were shown to be phosphorylated in AD, also demonstrating a consistent increase in the amount of the relevant enzymes, i.e. glycogen synthase kinase 3β and protein kinase A [110]. More recently, Lerchundi and colleagues [111] showed evidence that HSV-1 induces the caspase-3-mediated cleavage of tau protein at its specific site (aspartic acid 421). This event has been associated with an increased kinetics of tau aggregation, observed in neurodegenerative pathologies. Overall, these findings support the idea that HSV-1 could contribute to neurodegeneration that characterizes age-associated pathologies such as AD.

Effects of HSV-1 on APP phosphorylation and intracellular accumulation of amyloid-β protein (Aβ42) in cultured cortical neurons of E18 rats. a–c Time-dependent APP phosphorylation at threonine 668 induced by HSV-1; phosphorylated APP (pAPP) staining (white) in mock-infected cells (a), 1-h after cell challenge with HSV-1 (b) and 18 h post-infection (p.i.) (c). Neuronal infection is documented by HSV-1 labeling (red). d–f: no significant Aβ42 labeling (green) is observed in mock-infected cortical neurons. g–i Most HSV-1-infected neurons (red) exhibit Aβ42 immunoreactivity. j–l: high magnification image of a HSV-1-infected neuron showing marked Aβ42 staining. In f, i and l, the cell nuclei were stained with DAPI (blue). Calibration bars: 20 μm
HIV and Protein's Aggregates
There is a growing body of evidence indicating accelerated deposition of amyloid plaques also in HIV-infected brains. However, how HIV viral infection increases Aβ accumulation is poorly understood. It has been suggested that viral infection leads to increased production and impaired degradation of Aβ, mediated by upregulation of proinflammatory cytokine and inhibition of Aβ degrading enzyme, respectively [112–116]. Macrophages and microglia, both of which are productively infected by HIV in brains, play a pivotal role in Aβ degradation through the expression and execution of two endopeptidases, neprilysin (NEP) and insulin-degrading enzyme. It has been reported that HIV viral protein Tat-derived peptide inhibits NEP activity in vitro, and recombinant Tat added directly to brain cultures resulted in a 125 % increase in soluble Aβ oligomers [113, 116]. Recently, it was shown that monomeric Aβ degradation by primary cultured macrophages and microglia was significantly impaired by HIV infection due to an impairment in NEP endopeptidase activity, probably caused by the diminished transport of NEP to the cell surface and intracellular accumulation at the endoplasmic reticulum and in lysosomes [117]. The authors suggest that the malfunctioning of NEP in infected macrophages may contribute to the acceleration of β amyloidosis in HIV-infected brains, and propose modulation of macrophages as a potential preventive strategy for Aβ-related cognitive disorders in HIV-affected patients. Increased Aβ amyloidosis appears to be accelerated in brains of patients undergoing HAART [118]. The mechanisms through which HAART contributes to Aβ deposition are largely unknown. HAART is also reported to induce metabolic dysregulation, resulting in a hyperlipidemia, alterations in body fat distribution, diabetes, insuline resistance and coronary artery disease. Recently, Giunta and colleagues [119] provided evidence that antiretroviral treatment, especially when as combination, impairs microglial phagocytosis of β-amyloid and increases its production by neurons. HIV has also recently been shown to increase the level of hyperphosphorylated tau, through upregulation of its kinase CHK5, in a subset of HIV patients with the inflammatory condition known as HIV encephalitis [120]. A similar pattern of neurodegeneration, which includes increased brain levels of CDK5 and p35, alterations in tau phosphorylation and dendritic degeneration, was observed in transgenic mice engineered to express the HIV protein gp120. A previous paper by Giunta et al. [121] reported an increased amyloid-β deposition and tau hyperphosphodrylation in a mouse model of HIV-1 Tat-induced AD-like pathology generated by crossing PSAPP (APPswe and PSEN1dE9) [122] and HIV-1 Tat transgenic mice [123] compared with PSAPP mice, indicating that the viral protein Tat significantly promotes an AD-like pathology in PSAPP/Tat mice.
C. pneumoniae and Aβ Aggregates
Intranasal inoculation of C. pneumoniae (isolated from post-mortem brains of AD patients) has been shown to induce the formation of amyloid plaques in the brains of wild-type mice that increase in number as the disease progresses [67], together with intraneuronal Aβ42 accumulation. However, the bacterium effects on plaque formation have been contested by a subsequent similar work showing that a different strain of Chlamydia was unable to induce Aβ plaque formation in the brains of infected mice [124]. More recently, Chlamydia antigens have been detected alongside amyloid deposits and tau tangles in post-mortem AD brain tissue [68]. Further research is required to demonstrate definitely how both intracellular and extracellular amyloid aggregates and C. pneumoniae are interrelated.
Exposure of mammalian neuronal and glial cells and organotypic cultures to Borrelia burgdorferi spirochetes has been shown to produce amyloid deposits and tau hyperphosphorylation, indicating that bacteria and/or their degradation products may enhance the cascade of events leading to AD [125]. In this regard, it is noteworthy that spirochetes frequently co-infect with other bacteria and viruses, i.e. C. pneumoniae and HSV-1, suggesting that concurrent infections with several pathogens may also occur in AD. Finally, recent data reporting that Aβ peptides show an anti-microbial activity [126] and might act as a defence molecule of innate immunity which in some way support the hypothesis of the involvement of infectious agents in AD pathogenesis and aetiology: the accumulation of Aβ and plaque deposits may derive from an over-production of Aβ peptides directed against pathogenic neuroinvasions.
Influenza Virus and α-Synuclein Aggregates
The PD hallmark Lewy bodies are composed mainly of aggregated α-SYN. This protein aggregation has been shown to depend on the phosphorylation of a number of serine residues in α-SYN, particularly serine 129 [127]. Jang et al. [128] demonstrated that the highly infectious, neurotropic A/Vietnam/1203/04 (H5N1) influenza virus progresses from the peripheral nervous system into the CNS, where it can induce PD symptoms and the activation of microglia, a significant increase in phosphorylation and aggregation of α-SYN, which likely results in the observed substantia nigra pars compacta (SNpc) dopaminergic neuron degeneration 60 days after resolution of the infection.
Alteration of Autophagic Processes
Autophagy is a highly conserved mechanism for the degradation and recycling of superfluous or damaged proteins as well as entire organelles that occurs mainly in cells under metabolic stress in order to provide an energy supply. This pathway is therefore essential for promoting health and longevity by regulating cellular survival, development, differentiation and homeostasis, as well as for defending cells from infectious intruders [129]. Different forms of autophagy (macroautophagy, microautophagy and chaperone-mediated autophagy) have been described as regulators of cell fate [130] and their differences depend on their physiological functions and the mode of cargo delivery to lysosomes. In particular, macroautophagy, the major regulated catabolic process of long-lived protein and organelle degradation, takes place within double-membrane vesicles, termed autophagosomes that deliver cytoplasmic cargo to lysosomes, whose hydrolases ensure the complete degradation of unwanted material. The formation of autophagosomes is directly catalyzed by LC3, a ubiquitin-like molecule, and is regulated by specific proteins of the yeast Atg family [131]. In particular, Beclin-1, the mammalian orthologue of yeast Atg6, has a central role in autophagy by interacting with several cofactors to regulate the biogenesis and maturation of autophagosomes as well as their fusion to lysosomes, thereby promoting autophagy.
The dysregulation of autophagy may contribute to a number of diseases, including aging, cancer, infectious diseases and neurodegenerative disorders [132–136]. In the latter case, defects in autophagy have been associated with the presence of aberrant proteins in the brain both in experimental models and in human patients. Mice engineered to suppress basal autophagy show neurodegenerative hallmarks, such as accumulation of inclusion bodies and intracellular protein aggregates in the brain [137, 138]. Increased numbers of autophagosomes have been found in AD, TSE, PD and HD brains where they may reflect the activation of autophagy as a physiological response to the disease or as a consequence of autophagosomal maturation defects, as in the case of AD [139–141]. Indeed, autophagy plays a key role in the clearance of aggregated proteins (oligomers and fibrils) associated with several neurodegenerative diseases, as they are poor substrates for proteasomal degradation. Acquired defects in autophagosome formation may result from the sequestration of autophagy proteins in the aggregates formed by mutant or misfolded proteins, as well as from defects in Beclin-1 and other autophagic proteins or from impaired delivery of autophagosomes to lysosomes due to mutation or alterations that affect the dynein motor machinery. Autophagy also targets RNA and DNA viruses and microbes, including herpes simplex virus, for sequestration and elimination [142–144], and this effect is particularly useful for long-lived cells such as neurons, in which it may represent a non-cytolytic mechanism of viral clearance. Moreover, autophagy may also exert a protective function during infection with neurotropic viruses, by promoting the survival of infected neurons and minimizing neuronal dysfunction. In addition, it is possible that autophagy may contribute to class II presentation of viral antigens by microglia or astrocytes during CNS infection. However, some infectious agents may overcome or take advantage of autophagy by replicating within autophagic vacuoles [145–148] or by inhibiting this process.
HSV-1 and Autophagic Pathways
HSV-1 encodes the ICP34.5 protein—infected cell polypeptide 34.5—that can disrupt the autophagic process [144, 149], thus protecting itself against destruction. This protein has also been shown to reverse the protein kinase R (PKR) defence mechanism, which is activated by the presence of virus-derived double-stranded RNA (dsRNA) and is aimed at shutting off cellular protein synthesis through the phosphorylation of elongation initiation factor 2α (eIF2α) [150]. Such a shutting-off would inhibit the synthesis of viral proteins, initiate apoptosis [151] and enhance autophagy. ICP34.5 mediates HSV1-induced eIF2α dephosphorylation [152], thus allowing viral protein synthesis. However, it uses two different mechanisms to antagonize host autophagy and PKR function. The N-terminal domain of ICP34.5, which is not required for the reversal of PKR-mediated eIF2α phosphorylation, binds to Beclin-1 and inhibits its autophagic function in autophagosome biogenesis [153], while the C-terminal domain of ICP34.5 (the GADD34 domain) recruits a host phosphatase, PP1α, to reverse PKR-mediated eIF2α phosphorylation and host cell translational shut-off [154]. These two mechanisms cooperate to block autophagy and may exacerbate the defective autophagy processes that characterize aging brains. Moreover, HSV-1-induced PKR activation in neuroblastoma cells and peripheral nervous tissue from infected mice has been suggested to increase the amyloidogenic APP processing and Aβ production by promoting BACE1 translation via eIF2α phosphorylation [155]. Interestingly, a recent paper from Santana et al. [156] shows that HSV-1 infection in neuroblastoma cells induces intracellular Aβ accumulation in autophagosomes, whose fusion with lysosomes appears to be aborted. As described above, HSV-1 has been proposed as a risk factor for AD, and APP, Aβ and the enzymes responsible for Aβ formation are present in autophagosomes in AD brains [157], which may constitute a reservoir of these pathogenic proteins. In this context, viral action via the ICP34.5 gene may prevent not only degradation of HSV-1 in the brain but also degradation of the aberrant cell proteins, including Aβ and hyperphosphorylated tau, thus possibly contributing to the formation of amyloid aggregates and neurofibrillary tangles.
HIV and Autophagosome Maturation
Like HSV-1, HIV has been shown to block autophagosome maturation, probably to prevent its own degradation. The autophagic step affected is autophagosome fusion to lysosomes. In particular, the viral protein Nef has been shown to inhibit autophagosome fusion with lysosomes by interacting with Beclin-1 and disabling its correct complex formation with the autophagic proteins that promote this fusion [158]. Moreover, HIV also appears able to inhibit neuronal autophagy. In particular, a recent study showed that products of microglia infected by the simian immunodeficiency virus (SIV, the HIV-analog that infects monkeys) inhibit neuronal autophagy, resulting in decreased neuronal survival, and that two major mediators of HIV-induced neurotoxicity, tumour necrosis factor-alpha and glutamate, had similar autophagy-reducing effects in neurons. Interestingly, the induction of autophagy in neurons through rapamycin treatment conferred significant protection to neurons in SIV-infected brains [159].
Influenza Virus and Authophagic Pathways
Influenza virus has also been reported to affect autophagy, causing autophagy activation in early phases of the infection in order to promote viral replication [160], while later it induces autophagosome accumulation in the cytoplasm of infected cells. Again, this latter event seems to be due to virus-induced defects in autophagosome maturation, achieved possibly via viral matrix protein 2, a proton-channel pumping unit, which binds ATG6/Beclin 1 [158, 161] and blocks the fusion between lysosomes and autophagosomes. The functional consequence of this block is a higher susceptibility of influenza A virus-infected cells to apoptosis. Our group [160] has recently demonstrated that pepstatin A, an inhibitor of cathepsin D that is involved in both autophagy and apoptosis [162], is able to decrease viral replication by interfering with this pathway. Interestingly, as previously reported [163], the influenza virus is able to induce apoptotic cell death in neuronal cells, even though they are characterized by a high expression of the antiapoptotic protein Bcl-2 and relatively low permissiveness to influenza virus replication, and it is possible that in this context a blocking of the autophagic process may enhance virus-induced apoptosis and thus neurodegeneration.
The reported findings suggest that autophagy is an important process that needs to be preserved in neurons exposed to insults that can lead to neurodegeneration, including viral infections.
Oxidative Stress and Neurodegeneration
Several experimental findings point to oxidative stress as a key element in the pathogenesis of neurodegenerative diseases. The brain is particularly vulnerable to oxidative damage on account of its high oxygen levels, the presence of polyunsaturated fatty acids, transition metals in ionic form and low amounts of antioxidants [164]. In the elderly, where the redox state is unbalanced because of high levels of pro-oxidant species, the presence of metals such as iron ions in an oxygen-rich environment can induce further production of reactive oxygen species (ROS). The analysis at autopsy of brains from AD patients shows high levels of ROS and a drastic decrease in the intraneuronal content of glutathione (GSH), mainly in the hippocampus and cortex. These features also characterize the substantia nigra of PD patients, and are found in spinal fluid of patients with ALS [reviewed in 165]. This is associated with a high rate of cell membrane lipid peroxidation and nitration or oxidation of proteins and nucleic acids [166]. In particular, several of these oxidative events seem to be target-specific, such as for tyrosine nitration in α-SIN and in tau protein that are found in PD and AD, respectively [167, 168].
Viral Infections and Oxidative Stress
Viral infections are frequently associated with host-cell redox changes characteristic of oxidative stress [169–173].
HSV-1
HSV-1 infection induces an alteration of the intracellular redox state towards a pro-oxidant state through the depletion of GSH (the main endogenous antioxidant), the production of ROS, the induction of mitochondrial DNA damage, and endoplasmic reticulum (ER) stress [173–176]. Furthermore, HSV-1 infection in murine neuronal cells increases ROS levels and lipid peroxidation [177]. In agreement with these observations, high levels of lipid peroxidation products and protein nitrosylation were found in those brain areas where replicating or latent HSV-1 were detected after infection in primary sites [178, 179]. Overall, these data indicate the occurrence of oxidative damage in the brain following virus infection.
Alterations in the intracellular redox state towards a pro-oxidant state have been associated with formation of the typical histological alterations of brain tissue that occur in neurodegenerative diseases, including Aβ peptide generation and deposition [180–182]. HSV-1 was found to trigger multiple amyloidogenic APP processing mediated mainly by virus-induced upregulation of β- and γ-secretases. Interestingly, this event was prevented by antioxidant agents [107], suggesting that HSV-1-induced oxidative stress in neuronal cells may trigger β- and γ-secretase activation and, consequently, APP processing and Aβ formation. Some kinases involved in tau and APP phosphorylation [183–185] belong to the stress-activated protein kinase family, known to be activated during oxidative stress. A strong activation of these kinases has been observed both in post-mortem brains from AD patients [186, 187] and during in vitro HSV-1 infection [188, 189]. HSV-1 has been shown to induce AD-specific tau and APP phosphorylation, as well as the upregulation of the kinases involved in this event, such as GSK3β and PKA [106, 109, 110]. These effects may be further enhanced in vivo as consequence of oxidative stress response produced by microglial cells during HSV-1 infection [190].
HIV
Pioneer studies [191–193] showed evidence of oxidative stress markers in the cells and body fluids of HIV-infected patients. Garaci et al. [194] demonstrated that in vitro HIV infection significantly decreases the GSH content of human macrophages and suggested that this effect might be related to the preferential use of cysteine (the rate-limiting amino acid for GSH synthesis) for the synthesis of viral proteins. Interestingly, the brains of patients with HIV dementia show oxidative damage markers, including increased amounts of peroxynitrite, 4-hydroxynonenal and protein carbonyls [195]. The viral proteins gp120 and Tat have been shown to increase oxidative stress and to induce several damaging effects in neurons, including destruction of the cytoskeleton. These effects were reversed by antioxidants, suggesting an important role for oxidative damage in the pathogenesis of HIV dementia [196, 197]. Overproduction of superoxide anions and other free-radical species, possibly via the release of proinflammatory molecules by HIV-infected macrophages/microglial cells, has been suggested to play a role in the astroglial apoptotic cell death that characterizes HAD [198]. In this context, gp41, an envelope glycoprotein of HIV, has been shown to trigger inducible NO synthase (iNOS) expression and consequently NO production in human astrocytes and murine cortical brain cells in culture [199, 200], thus possibly contributing to the HAD pathogenesis.
Influenza Virus
Decreases in GSH levels and oxidative stress have also been found in cells and animals infected by influenza virus [172, 201–204]. Virus-induced pro-oxidant conditions play an important role in viral replication, by activating cellular kinases involved in nucleo-cytoplasmic traffick of viral proteins and by promoting the maturation of viral HA [163, 205].
Virus-induced oxidative stress has also been associated with the onset and progression of influenza virus-associated encephalopathy (IE) in murine models [reviewed in 206]. Markers of oxidative stress have been found in serum and CSF of patients with IE and have been proposed as possible predictive biomarkers of the severity of the disease [207]. As mentioned before, influenza virus infection has been related to the appearance of biochemical markers typical of PD [128], a neurodegenerative disease characterized by high levels of oxidative stress in substantia nigra and other brain regions. On the basis of these evidences, it is possible to speculate that virus-induced oxidative stress may play a role in this process. As a matter of fact, oxidative stress has shown to play a major role in the degeneration of the dopaminergic neurons that characterize PD [208]. The vulnerability of these neurons to oxidative stress was demonstrated by the ability of pro-oxidant molecules, such as 6-hydroxydopamine (6-OHDA), paraquat and rotenone to damage substantia nigra cells after stereotaxic or systemic administrations. Thus, chronic age-related oxidative stress [209] may result in the accumulation of misfolded proteins [210], through a direct oxidation of protein disulfide isomerse (PDI), an oxido-reductase involved in folding of glycoprotein. Specifically, the S-nitrosylation of PDI may play a central role in the progression of PD. Indeed, PDI S-nitrosylation disrupts its neuroprotective role of preventing neuronal cell death triggered by ER stress, the accumulation of misfolded proteins, or proteasome inhibition [211].
It is possible to speculate that an increased “oxidative burst” generated when the influenza virus infects aged neuronal cells may contribute to alterations in PDI function, increasing the production of misfolded proteins and contributing to the pathogenesis of PD.
Synaptic Alterations
A large body of evidence suggests that deficits in synaptic transmission and plasticity play a major role in cognitive impairment and memory loss that characterize several neurodegenerative disorders including AD [212, 213]. Synaptic transmission occurs through chemical neurotransmitters that are released from pre-synaptic terminals in response to action potentials and bind specific receptors present on the post-synaptic side. The interaction between neurotransmitters and their post-synaptic receptors generates electrical signals (named postsynaptic potentials) that, when appropriately summed, may give rise again to action potentials thus allowing the propagation of information through the neural networks. Synaptic plasticity reflects the ability of a synaptic contact to change its efficiency (i.e. its signalling strength) depending on the activation pattern it previously experienced, and these plastic changes in information transmission are critical to learning and memory. However, the signal transmission at the synaptic level implies ion fluxes through the plasma membranes of both pre- and post-synaptic neurons that may have profound impact on neuronal functions and viability independently of electrical signal generation. A representative example is given by glutamate-dependent excitotoxicity, consisting of hyper-activation of calcium-permeable glutamate receptors (i.e. the N-methyl-d-aspartate receptors [NMDAR]) that produce intracellular calcium overload triggering pro-apototic pathways and leading to neuronal death [214, 215].
Virus and Synaptic Alterations
Viruses have been reported to significantly affect the synaptic function.
HIV
HIV-1 infection induces a progressive loss of synaptic connections in the CNS that appears in the early phases of the HAD pathology and correlates with the progression of the disease [216, 217]. In vivo studies demonstrated that some viral proteins are directly involved in these effects: mice engineered to express gp120 in astrocytes under an astrocyte-specific promoter show reduction of presynaptic terminals and dendrites in the CNS [218], whereas mice selectively expressing gp160 (the gp120 and gp141 heterodimers) in neurons exhibit synaptic dysfunctions [219]. Reportedly, gp120 induces synaptic damage through indirect and direct mechanisms involving NMDAR activation. Its indirect action is primarily mediated by soluble factors released from glial cells upon gp120 treatment [220–225]. In particular, gp120 activation of macrophages and microglia induces glutamate-related hyper-activation of NMDAR, with consequent ER stress and Ca2+ release [226]. Direct effects of gp120 on NMDA-evoked calcium influx involve modifications in the spatial location and density of NMDAR, possibly mediated by alterations of the biophysical properties of neuronal membranes induced by the viral protein that stabilizes the structure of lipid microdomains containing the receptor. This effect, together with gp-120-induced PKA- and PKC-dependent phosphorylation of NMDAR, results in a perturbation of the surface expression and spatial location of NMDARs [227].
NMDARs are also activated by the viral protein Tat [228, 229], resulting in Ca2+ rise [226, 230, 231] that in turn activates neuronal nitric oxide synthase (nNOS) leading to cell death [197]. Tat also affects neuronal dendritic structures causing proteasome-mediated degradation of microtubule-associated protein 2 (MAP2) and the collapse of cytoskeletal filaments [232]. Kim et al. [233] hypothesized that these effects, rather than the neuronal death, could account for the impaired synaptic plasticity observed in neural networks exposed to Tat [234]. Consistently, they found that the impaired network function and the decreased neuronal survival produced by Tat, both effects induced by LRP-dependent activation of NMDARs, result from distinct mechanisms that are mediated by the ubiquitin-proteasoma pathway and nNOS activation, respectively.
Influenza Virus
Influenza virus A nucleoprotein NP has also been shown to affect synaptic functions: rat hippocampal neurons exposed to recombinant NP, fused to 11–amino acid peptide transduction domain (PTD) of TAT, showed disturbances in postsynaptic functions, documented by reduced frequency and amplitude of the miniature excitatory postsynaptic currents. The authors hypothesized that the viral NP that localized in dendritic spinelike processes interferes with the expression or anchoring of postsynaptic glutamate receptors thereby disturbing synaptic functions [235].
HSV-1
Besides being specific target of viral proteins, synapses also represent a route through which viruses may travel between neurons, as this is the case of HSV-1 [236]. Consistently, nectin proteins, among which nectin-1 and nectin-2 that are used by HSV as virus entry and cell–cell spread mediator [237–240] have been involved in synapse formation [241]. We found that HSV-1 binding to neuronal membrane markedly affected the electrophysiological properties of rat cortical neurons and enhanced their excitability [106]. This effect consisted of persistent Na+ channel activation and K+ current inhibition leading to membrane depolarization and increased neuronal firing. Voltage-gated Ca2+ channel were consequently activated thus triggering intracellular Ca2+ signals raising the basal intracellular Ca2+ levels. Calcium signals potently promoted APP phosphorylation and processing with consequent intracellular and extracellular accumulation of several neurotoxic fragments including Aβ oligomers [106, 107]. These virus-induced APP fragments might induce synaptic dysfunction resembling that underlying the cognitive deficits observed in AD.
Apoptosis
Neuronal cell death by apoptosis, a highly organised process characterized by chromatin condensation, shrinkage of the nucleus and cytoplasm, DNA fragmentation and disintegration of the cell into small apoptotic bodies that are destined to be phagocytized [242], underlies the symptoms of many neurodegenerative diseases. Apoptotic death of hippocampal and cortical neurons results in AD symptoms such as memory loss and cognitive decline; the death of dopaminergic neurons and the consequent apoptosis of midbrain neurons that use dopamine as a neurotransmitter lead to the characteristic tremors of PD; the loss of striatal neurons that control body movements characterizes HD; apoptosis of motor neurons in the spinal cord is responsible for the manifestations of ALS.
Several neurotropic viruses induce apoptosis in neural cells and these effects can contribute to the pathogenesis of the virus-induced disease. Other viruses prevent apoptotic death of host cells in order to estabilish a persistent infection. On the other hand, programmed cell death may also be used by the host as a non-inflammatory response aimed at removing the virus. Thus, the fate of infected cells depends on a complex network of interactions between a virus and its host cell, which has yet to be fully delucitated.
HSV-1
During strong and acute infection, such as occurs in HSE, HSV-1 induces neuronal cell death, although apoptosis does not occur during later sequelae even when inflammation is still present [243], and is likely the cause of long-term entorhinal cortex and hippocampal cell loss along with memory deficits in mice [244]. Additionally, encephalitic HSV-1 has been reported to induce apoptosis in hippocampal neurons through the activation of JNK pathways [245]. On the other hand, the presence of HSV-1 DNA in many healthy brains [11] suggests that the virus does not necessarily provoke cell death in cerebral tissue. In this regard, viral latency-associated transcript (LAT) has been reported to inhibit apoptosis [246], particularly by blocking the two major mammalian apoptotic pathways, i.e. the extrinsic apoptotic pathway and the intrinsic pathway [247–249], to promote neurite sprouting in neuroblastoma cells and to protect C1300 and N2A cells from killing by CD8 T lymphocytes in vitro [250, 251].
Based on these findings and others showing that LAT play a key role in the HSV-1 latency-reactivation cycle [252–254], the authors proposed that by preserving latently infected neurons from apoptotic cell death, it supports HSV-1 reactivation rates and spread into neuronal tissue. Viral protein expressed during productive infection with HSV-1 can also induce or inhibit apoptosis in a cell type-dependent manner following the infection of cultured cells [255–259]. In particular, experiments performed with genetically modified viruses showed that the apoptotic pathway induced by HSV-1 in neuronal cells within the first hour of infection could be blocked at multiple steps of the viral replicative cycle. The viral proteins synthesized between 3 and 6 h post-infection [260] and also some viral glycoproteins such as glycoproteins D and J, which are involved in the virus entry, have a role in this suppression of apoptosis in HSV-1-infected neuronal cells [261]. Moreover, the R1 subunit of viral ribonucleotide reductase inhibits apoptosis in hippocampal neurons and in differentiated PC12 cells through upregulation of the anti-apoptotic protein Bag-1 expression and activation of the ERK and Ras/MEK/MAPK pathways [262, 263].
In agreement with these findings, we also were unable to detect apoptotic markers in HSV-1-infected rat cortical neurons up to 24 h p.i.. However, when we challenged primary rat cortical neurons with the supernatants of neuroblastoma cells infected with HSV-1 in the presence or absence of β- and γ-secretase inhibitors, we found that infected-culture supernatants triggered apoptosis in these cells, even when the cultures were exposed to UV light, which inactivates the virus they contain. The infected supernatants obtained in the presence of β- and γ-secretase inhibitors exhibited much lower neurotoxicity. These findings suggest that supernatants of HSV-1-infected cells are highly neurotoxic for primary neurons, and that this effect is related to the presence of virus-induced APP fragments released into the extracellular medium rather than to the presence of “active” viral particles [107]. Nevertheless, they indicate an apoptotic pathway indirectly activated by the virus and mediated mainly by virus-induced Aβ peptides. Carter [264] addresses the hypothesis that activation of the immune system by other pathogens, i.e. those implicated in AD (e.g. Helicobacter pylori, C. pneumoniae and others [265]) might disturb the fragile balance between HSV-1-and host neuronal cells, allowing viral destruction, but causing neuronal loss. Activation of the immune system thus appears to be a potent inducer of neuronal death via inflammatory mediators [266].
C. pneumoniae
C. pneumoniae has been reported to inhibit the apoptotic process following infection in different celly types, including neutrophils [267], monocytes and epithelial cells [268–271], microglial cells [124] and neuronal cells [272]. When the latter were infected with C. pneumoniae, they appeared resistant to staurosporine-induced apoptosis. In particular, C. pneumoniae infection downregulated pro-apoptotic cytoplasmic proteins such as cytochrome c released from mithocondria and activated caspase-3/7, which are normally upregulated following staurosporine treatment. However, considering that they detected C. pneumoniae in AD brains [8, 273, 274], the authors hypothesized that, as for other cell types [275], this bacterium could both inhibit apoptosis and promote neuronal death by necrosis. This could also account for the inflammatory process activated by the pathogen. The ability of C. pneumoniae to inhibit the apoptotic process could result in chronic or prolonged infection in the CNS that, by promoting amyloidogenesis and neuroinflammation, may contribute to the neuropathogenesis of AD.
HIV
HIV infection reportedly induces apoptosis and neuronal loss in the CNS [276–279], but the mechanisms underlying these events have yet to be clarified. Multiple viral proteins, including gp120 and tat, have been involved in virus-induced neuronal loss. Cell death similar to that described in the brains of HAD patients has been detected in cerebral regions of transgenic mice expressing gp120, suggesting that the HIV glycoprotein is able to induce apoptosis alone [218]. A large body of evidence supports this notion, showing that gp120 causes degeneration and death of several types of neurons maintained in culture. However, it is still unknown whether gp120 is able to induce neuronal apoptosis directly or indirectly. Some findings support the hypothesis that gp120 infects infiltrating macrophages and lymphocytes, causing the release of pro-inflammatory and neurotoxic cytokines, such as IL-6, thus damaging sensory neurons and producing neuropathies [280]. Other data support the hypothesis that gp120 may interact directly with neurons, which indeed express both the gp120 co-receptors CXCR4 and CCR5 [281, 282]. In particular, gp-120-induced apoptosis has been demonstrated to be mediated mainly following its interaction with CXCR4 [283, 284], as demonstrated both in vivo and in vitro studies. For example, treatment of cultured human or rat neurons with gp120 leads to neuronal apoptosis [285–288] and intracerebroventricular injection of HIV-1 gp120 in rats produces apoptotic neuronal death in vivo [289–291].
Tat, a transactivating nuclear regulatory protein that is critical for viral replication, is released by infected macrophages and microglia and can be taken up by neighbouring cells, including neurons, where it can express its pro-apoptotic potential [292]. This action is inhibited by neurotrophic factors, including brain-derived neurotrophic factor (BDNF) and nerve growth factor [292], which activate the transcription factor NF-kB and upregulate the expression of the anti-apoptotic protein Bcl-2. Interestingly, long-term incubation of Tat in cultured PC12 cells causes decreased expression and activity of the transcription factor CREB, which in turn plays a key role in neuronal survival through the upregulation of BDNF [293]. Thus neurotoxic HIV proteins are able to activate pro-apoptotic cascades and to inhibit pro-survival pathways. However, together with other neurotoxic viral proteins such as Vpr, gp140 and Nef [294–296], other endogenous factors released from infected cells, especially macrophages, contribute to HIV-induced neuronal apoptosis, including excitatory amino acids, NO, MMPs and proinflammatory cytokines such as TNF-a and SDF-1 [297–299]. Moreover, in HIV-1-infected patients that are also drug abusers, the activation of opioid receptors, widely expressed by astrocytes and astrocyte precursors, can induce apoptosis, thus combining with the viral effects to accelerate the progression of HAD.
Influenza Virus
The influenza virus is also known to induce apoptosis in infected cells through other mechanisms. Among these, we previously demonstrated that in neurons, influenza virus activates the mitochondrial (intrinsic) apoptotic pathway through p38MAPK-mediated phosphorylation of Bcl-2 [163]. The interaction between Bcl-2 and this kinase diminishes the ability of Bcl-2 to prevent the cell undergoing virally induced apoptosis, but it also reduces the ability of the virus to replicate effectively. The immediate result is programmed death of infected cells and the release of a relatively low number of infective virions. These data let us hypothesize that despite the low virus replication of neuronal cells [172] the apoptotic signals activated by the virus, may contribute to the onset of neurodegeneration.
It has previously been reported that the avian H5N1 type of influenza A virus can be detected in neurons and astrocytes of human brains at autopsy [300, 301]. Recently, Ng et al. [302] demonstrated that the H5N1 virus can infect human astrocytic and neuronal cells, resulting in the induction of direct cellular damage and pro-inflammatory cytokine cascades. Indeed, increased expression of IL-6 and/or TNF-α mRNA was detected in both astrocytic and neuronal cells (human glioblastoma and neuroblastoma cells) at 6 and 24 h p.i.. TNF-a treatment induced apoptosis, as well as pro-inflammatory cytokine, chemokine and inflammatory responses in differentiated cells.
The recent immunohistochemical study in PD performed by Rhon and Catlin [303] revealed that the presence of influenza virus within the SNpc in post-mortem PD brain sections was associated with apoptotic oligodendrocytes labelled by the Beclin-1 caspase-cleavage product antibody (BeclinCCP) in the white matter of the SN of PD and DLB (dementia with Lewy body) patients. Many of the oligodendrocytes labelled with the BeclinCCP antibody displayed hallmark features of apoptosis, including fragmentation of processes and shrunken cell bodies. This degeneration of oligodendrocytes, which are known to play a key role in the myelination of axons in the CNS, may contribute to the extrapyramidal symptoms associated with PD.
Apoptosis has also been identified in the brains of influenza encephalopathy and encephalitis patients [304]. Microglial cells were markedly increased in TUNEL-positive influenza encephalopathy and encephalitis brains compared with TUNEL-negative brains. Immunoreactivity for active-caspase 3, demonstrated by immunohistochemistry, and the overexpression of a caspase-cleaved fragment of poly(ADP-ribose) polymerase, indicated that activation of caspase 3 is involved in the apoptotic pathway in the brains of influenza encephalopathy patients.
Conclusions
Advances in microbiological research have led to in-depth understanding of the structure and replication mechanisms of several pathogens, as well as of their interactions with host cells. However, a growing body of evidence suggests that when an infectious agent reaches sites different from those of its primary replication it may produce mild infections that eventually cause additional and unexpected effects. This is the case for persistent CNS infections caused by continuous pathogen replications (e.g. HIV and C. pneumoniae infections), repeated infections (e.g. influenza virus) or latent infections followed by life-long reactivations (as in the case of HSV-1). Repeated cycles of pathogen replication within the CNS produce functional and molecular hallmarks of neurodegeneration, including protein misfolding, deposition of misfolded protein aggregates, alterations of autophagic pathways, oxidative stress, neuronal functional alterations and apoptotic cell death (Fig. 7). These effects accumulate over time, thus contributing to neurodegeneration. The pathogen-induced effects add to and are possibly amplified by several factors such as metabolic disorders, genetic alterations and other environmental risk factors, involved in the pathogenesis of neurodegenerative diseases. As a result, the pathogen-induced damage amplifies and accelerates the neurodegenerative process, whose signs are usually manifested during aging.

Molecular hallmarks of neurodegeneration induced by infectious agents in neurons of the central nervous system (for further details, see text)
The data reviewed in our paper suggest that more detailed understanding of the molecular mechanisms underlying pathogen-mediated neuronal damage may pave the way to the identification of new preventive and/or therapeutic strategies aimed at counteracting the progression of these devastating pathologies.
References
Bowery NG, Bagetta G, Nisticó G et al (1992) Intrahippocampal tetanus toxin produces generalized convulsions and neurodegeneration in rats: antagonism by NMDA receptor blockers. Epilepsy Res Suppl 9:249–256
Mattson MP (2004) Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev 3:105–120
Cermelli C, Vinceti M, Beretti F et al (2003) Risk of sporadic amyotrophic lateral sclerosis associated with seropositivity for herpesvirus and echovirus-7. Eur J Epidemiol 18:123–127
Ogata A, Tashiro K, Nukuzuma S et al (1997) A rat model of Parkinson's disease induced by Japanese encephalitis virus. J Neurovirol 3:141–147
Menninger KA (1919) Psychoses associated with influenza. Arch Neurol Psychiatr 2:291–337
Menninger KA (1926) Influenza and schizophrenia. An analysis of post-influenzal “dementia precox” as of 1918 and five years later. Am J Psychiatr 5:469–529
Takahashi M, Yamada T (1999) Viral etiology for Parkinson's disease—a possible role of influenza A virus infection. Jpn J Infect Dis 52:89–98
Balin BJ, Gerard HC, Arking EJ et al (1998) Identification and localization of Chlamydia pneumoniae in the Alzheimer's brain. Med Microbiol Immunol (Berl) 187:23–42
Balin BJ, Appelt DM (2001) Role of infection in Alzheimer's disease. J Am Osteopath Assoc 101:S1–S6
Dobson CB, Itzhaki RF (1999) Herpes simplex virus type 1 and Alzheimer's disease. Neurobiol Aging 20:457–465
Jamieson GA, Maitland NJ, Craske J et al (1991) Detection of herpes simplex virus type 1 DNA sequences in normal and Alzheimer's disease brains using polymerase chain reaction. Biochem Soc Trans 19:122S
Itabashi S, Arai H, Matsui T et al (1997) Herpes simplex virus and risk of Alzheimer's disease. Lancet 349:1102
Mori I, Kimura Y, Naiki H et al (2004) Reactivation of HSV-1 in the brain of patients with familial Alzheimer's disease. J Med Virol 73:605–611
Wozniak MA, Shipley SJ, Combrinck M et al (2005) Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer's disease patients. J Med Virol 75:300–306
Wozniak MA, Frost AL, Preston CM, Itzhaki RF (2011) Antivirals reduce the formation of key Alzheimer's disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One 6:e25152
Ben Fredj N, Rotola A, Nefzi F et al (2011) Identification of human herpesviruses 1 to 8 in Tunisian multiple sclerosis patients and healthy blood donors. J Neurovirol. doi:10.1007/s13365-011-0056-z
Giovannoni G, Cutter GR, Lunemann J et al (2006) Infectious causes of multiple sclerosis. Lancet Neurol 5:887–894
Giovannoni G, Ebers G (2007) Multiple sclerosis: the environment and causation. Curr Opin Neurol 20:261–268
Kurtzke JF (1993) Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev 6:382–427
Mancuso R, Delbue S, Borghi E et al (2007) Increased prevalence of Varicella zoster virus DNA in cerebrospinal fluid from patients with multiple sclerosis. J Med Virol 79:192–199
Salvetti M, Giovannoni G, Aloisi F (2009) Epstein-Barr virus and multiple sclerosis. Curr Opin Neurol 22:201–206
Serafini B, Rosicarelli B, Franciotta D et al (2007) Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med 204:2899–2912
Zivadinov R, Nasuelli D, Tommasi MA et al (2006) Positivity of cytomegalovirus antibodies predicts a better clinical and radiological outcome in multiple sclerosis patients. Neurol Res 28:262–269
Ballatori N, Krance SM, Notenboom S et al (2009) Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem 390:191–214
Govoni S, Amadio M, Battaini F, Pascale A (2010) Senescence of the brain: focus on cognitive kinases. Curr Pharm Des 16:660–671
McGavern DB, Kang SS (2011) Illuminating viral infections in the nervous system. Nat Rev Immunol 11:318–329
Kristensson K (2011) Microbes' roadmap to neurons. Nat Rev Neurosci 12:345–357
Haase AT (1986) Pathogenesis of lentivirus infections. Nature 322:130–136
Peluso R, Haase A, Stowring L et al (1985) A Trojan horse mechanisms for the spread of visna virus in monocytes. Virology 147:231–236
Muratori C, Mangino G, Affabris E, Federico M (2010) Astrocytes contacting HIV-1-infected macrophages increase the release of CCL2 in response to the HIV-1-dependent enhancement of membrane-associated TNFα in macrophages. Glia 58:1893–1904
Roberts TK, Buckner CM, Berman JW (2010) Leukocyte transmigration across the blood–brain barrier: perspectives on neuroAIDS. Front Biosci 15:478–536
Strazza M, Pirrone V, Wigdahl B, Nonnemacher MR (2011) Breaking down the barrier: the effects of HIV-1 on the blood–brain barrier. Brain Res 1399:96–115
Hirokawa N, Noda Y (2008) Intracellular trafficking and kinesin superfamily proteins, KIFs: structure, function and dynamics. Physiol Rev 88:1089–1118
Hsieh MJ, White PJ, Pouton CW (2010) Interaction of viruses with host cell molecular motors. Curr Opin Biotechnol 21:633–639
Salinas S, Schiavo G, Kremer EJ (2010) A hitchhiker's guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol 8:645–655
Mori I, Nishiyama Y, Yokochi T, Kimura Y (2004) Virus-induced neuronal apoptosis as pathological and protective responses of the host. Rev Med Virol 14:209–216
Mori I, Nishiyama Y, Yokochi T, Kimura Y (2005) Olfactory transmission of neurotropic viruses. J Neurovirol 11:129–137
Kastrukoff L, Hamada T, Schumacher U et al (1982) Central nervous system infection and immune response in mice inoculated into the lip with herpes simplex virus type 1. J Neuroimmunol 2:295–305
Lewandowski G, Zimmerman MN, Denk LL et al (2002) Herpes simplex type 1 infects and establishes latency in the brain and trigeminal ganglia during primary infection of the lip in cotton rats and mice. Arch Virol 147:167–179
Bach LM (1963) Regional physiology of the central nervous system. Prog Neurol Psychiatry 18:46–106
Liedtke W, Opalka B, Zimmermann CW, Lignitz E (1993) Age distribution of latent herpes simplex virus 1 and varicella-zoster virus genome in human nervous tissue. J Neurol Sci 116:6–11
Boggian I, Buzzacaro E, Calistri A et al (2000) Asymptomatic herpes simplex type 1 virus infection of the mouse brain. J Neurovirol 6:303–313
Mori I, Goshima F, Ito H, Koide N, Yoshida T, Yokochi T, Kimura Y, Nishiyama Y (2005) The vomeronasal chemosensory system as a route of neuroinvasion by herpes simplex virus. Virology 334:51–58
Aronsson F, Robertson B, Ljunggren HG, Kristensson K (2003) Invasion and persistence of the neuroadapted influenza virus A/WSN/33 in the mouse olfactory system. Viral Immunol 16:415–423
Iwasaki T, Itamura S, Nishimura H et al (2004) Productive infection in the murine central nervous system with avian influenza virus A (H5N1) after intranasal inoculation. Acta Neuropathol (Berl) 108:485–492
Matsuda K, Park CH, Sunden Y et al (2004) The vagus nerve is one route of transneural invasion for intranasally inoculated influenza a virus in mice. Vet Pathol 41:101–107
Reinacher M, Bonin J, Narayan O, Scholtissek C (1983) Pathogenesis of neurovirulent influenza A virus infection in mice. Route of entry of virus into brain determines infection of different populations of cells. Lab Investig 49:686–692
Shinya K, Shimada A, Ito T et al (2000) Avian influenza virus intranasally inoculated infects the central nervous system of mice through the general visceral afferent nerve. Arch Virol 145:187–195
Rimmelzwaan GF, van Riel D, Baars M et al (2006) Influenza A virus (H5N1) infection in cats causes systemic disease with potential novel routes of virus spread within and between hosts. Am J Pathol 168:176–183
Takahashi M, Yamada T, Nakajima S et al (1995) The substantia nigra is a major target for neurovirulent influenza A virus. J Exp Med 181:2161–2169
De Regge N, Nauwynck HJ, Geenen K et al (2006) α-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J Cell Biol 174:267–275
Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL (2008) Transport and egress of herpes simplex virus in neurons. Rev Med Virol 18:35–51
Baringer JR, Swoveland P (1973) Recovery of herpes-simplex virus from human trigeminal ganglions. N Engl J Med 288:648–650
Stevens JG, Wagner EK, Devi-Rao GB et al (1987) RNA complementary to a herpes virus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059
Henderson G, Jaber T, Carpenter D et al (2009) Identification of herpes simplex virus type 1 proteins encoded within the first 1.5 kb of the latency-associated transcript. J Neurovirol 15:439–448
Itzhaki RF, Lin WR, Shang D et al (1997) Herpes simplex virus type 1 in brain and risk of Alzheimer's disease. Lancet 349:241–244
Koelle DM, Magaret A, Warren T et al (2010) APOE genotype is associated with oral herpetic lesions but not genital or oral herpes simplex virus shedding. Sex Transm Infect 86:202–206
Wozniak MA, Mee AP, Itzhaki RF (2009) Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol 217:131–138
Letenneur L, Peres K, Fleuri H et al (2008) Sieropositive to herpes virus antibodies and risk of Alzheimer's disease: a population-based cohort study. Plos One 3:e3637
Lambert JC, Heath S, Even G et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41:1094–1099
Porcellini E, Carbone I, Ianni M, Licastro F (2010) Alzheimer's disease gene signature says: beware of brain viral infections. Immun Ageing 7:16–20
MacIntyre A, Hammond CJ, Little CS et al (2002) Chlamydia pneumoniae infection alters the junctional complex proteins of human brain microvascular endothelial cells. FEMS Microbiol Lett 217:167–172
D'Andrea MR, Nagele RG, Wang HY, Lee DH (2002) Consistent immunohistochemical detection of intracellular beta-amyloid42 in pyramidal neurons of Alzheimer's disease entorhinal cortex. Neurosci Lett 333:163–166
Nochlin D, Shaw CM, Campbell LA, Kuo CC (1999) Failure to detect Chlamydia pneumoniae in brain tissues of Alzheimer's disease. Neurology 53:1888
Ring RH, Lyons JM (2000) Failure to detect Chlamydia pneumoniae in the late-onset Alzheimer's brain. J Clin Microbiol 38:2591–2594
Mahony JB, Chong S, Coombes BK et al (2000) Analytical sensitivity, reproducibility of results, and clinical performance of five PCR assays for detecting Chlamydia pneumoniae DNA in peripheral blood mononuclear cells. J Clin Microbiol 38:2622–2627
Little CS, Hammond CJ, MacIntyre A et al (2004) Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging 25:419–429
Hammond CJ, Hallock LR, Howanski RJ et al (2010) Immunohistological detection of Chlamydia pneumoniae in the Alzheimer's disease brain. BMC Neurosci 11:121–132
Gartner S (2000) HIV infection and dementia. Science 287:602–604
McArthur JC, McDermott MP, McClernon D et al (2004) Attenuated central nervous system infection in advanced HIV/AIDS with combination antiretroviraltherapy. Arch Neurol 61:1687–1696
Corder EH, Robertson K, Lannfelt L et al (1998) HIV-infected subjects with the E4 allele for APOE have excess dementia and peripheral neuropathy. Nat Med 4:1182–1184
Letendre SL, Ellis RJ, Ances BM et al (2010) Neurologic complications of HIV disease and their treatment. Top HIV Med 18:45–55
Heaton RK, Clifford DB, Franklin DR Jr et al (2010) HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology 75:2087–2096
Nath A, Schiess N, Venkatesan A et al (2008) Evolution of HIV dementia with HIV infection. Int Rev Psychiatry 20:25–31
Sacktor N, McDermott MP, Marder K et al (2002) HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol 8:136–142
Gannon P, Khana MZ, Kolsona DL (2011) Current understanding of HIV-associated neurocognitive disorders pathogenesis. Curr Opin Neurol 24:275–283
Wise HM, Foeglein A, Sun J et al (2009) A complicated message: identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J Virol 83:8021–8031
Jordon E (1927) Epidemic influenza: a survey. American Medical Association, Chicago
Oxford JS (2000) Influenza A pandemics of the 20th century with special reference to 1918: virology, pathology and epidemiology. Rev Med Virol 10:119–133
Taubenberger JK (2006) The origin and virulence of the 1918 “Spanish” influenza virus. Proc Am Philos Soc 150:86–112
Lo KC, Geddes JF, Daniels RS, Oxford JS (2003) Lack of detection of influenza genes in archived formalin-fixed, paraffin wax-embedded brain samples of encephalitis lethargica patients from 1916 to 1920. Virchows Arch 442:591–596
Ruest A, Michaud S, Deslandes S, Frost E (2003) Comparison of the Directigen flu A + B test, the QuickVue influenza test, and clinical case definition to viral culture and reverse transcription-PCR for rapid diagnosis of influenza virus infection. J Clin Microbiol 41:3487–4393
Maurizi CP (1985) Whywas the 1918 influenza pandemic so lethal? The possible role of a neurovirulent neuraminidase. Med Hypotheses 16:1–5
Poskanzer DC, Schwab RS (1963) Cohort analysis of Parkinson's syndrome. Evidence for a single etiology related to subclinical infection about 1920. J Chronic Dis 16:961–973
Ravenholt RT, Foege WH (1982) 1918 influenza, encephalitis lethargica, parkinsonism. Lancet 2:860–864
Martyn CN, Osmond C (1995) Parkinson's disease and the environment in early life. J Neurol Sci 132:201–206
Martyn CN (1997) Infection in childhood and neurological diseases in adult life. Br Med Bull 53:24–39
Toovey S, Jick SS, Meier CR (2011) Parkinson's disease or Parkinson symptoms following seasonal influenza. Influenza Other Respir Viruses 5:328–333
Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4:49–60
Glenner GG, Wong CW (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890
Grundke-Iqbal I, Iqbal K, Tung YC et al (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83:4913–4917
Spillantini MG, Schmidt ML, Lee VM et al (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
DiFiglia M, Sapp E, Chase KO et al (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993
Bruijn LI, Houseweart MK, Kato S et al (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854
Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science 218:1309–1311
Arrasate M, Mitra S, Schweitzer ES et al (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256:184–185
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood brain barrier disorder in Alzheimer's disease. Acta Neuropathol 118:103–113
Cribbs DH, Azizeh BY, Cotman CW, LaFerla FM (2000) Fibril formation and neurotoxicity by a herpes simplex virus glycoprotein B fragment with homology to the Alzheimer's A beta peptide. Biochemistry 39:5988–5994
Bearer EL (2004) Perspectives on herpes-APP interactions. Aging Cell 3:81–84
Satpute-Krishnan P, De Giorgis JA, Bearer EL (2003) Fast anterograde transport of herpes simplex virus: role for the amyloid precursor protein of Alzheimer disease. Aging Cell 2:305–318
Satpute-Krishnan P, De Giorgis JA, Conley MP et al (2006) A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc Natl Acad Sci U S A 103:16532–16537
Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB (2007) Herpes simplex virus infection causes cellular-amyloid accumulation and secretase upregulation. Neurosci Lett 429:95–100
Shipley SJ, Parkin ET, Ithzaki RF, Dobson CB (2005) Herpes simplex virus interferes with amyloid precursor protein processing. BMC Microbiol 5:48
Piacentini R, Civitelli L, Ripoli C et al (2011) HSV-1 promotes Ca2+-mediated APP phosphorylation and Aβ accumulation in rat cortical neurons. Neurobiol Aging 32:2323.e13–2323.e26
De Chiara G, Marcocci ME, Civitelli L et al (2010) APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS One 5:e13989
Cheng SB, Ferland P, Webster P, Bearer EL (2011) Herpes simplex virus dances with amyloid precursor protein while exiting the cell. PLoS One 6:e17966
Zambrano A, Solis L, Salvadores N et al (2008) Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1. J Alzheimers Dis 14:259–269
Wozniak MA, Frost AL, Itzhaki RF (2009) Alzheimer's disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimers Dis 16:341–350
Lerchundi R, Neira R, Valdivia S et al (2011) Tau cleavage at D421 by caspase-3 is induced in neurons and astrocytes infected with herpes simplex virus type 1. J Alzheimers Dis 23:513–520
Blasko I, Veerhuis R, Stampfer-Kountchev M et al (2000) Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol Dis 7:682–689
Daily A, Nath A, Hersh LB (2006) Tat peptides inhibit neprilysin. J Neurovirol 12:153–160
Yamamoto M, Kiyota T, Horiba M et al (2007) Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol 170:680–692
Pulliam L (2009) HIV regulation of amyloid beta production. J Neuroimmune Pharmacol 4:213–217
Rempel HC, Pulliam L (2005) HIV-1 Tat inhibits neprilysin and elevates amyloid beta. AIDS 19:127–135
Lan X, Xu J, Kiyota T et al (2011) HIV-1 reduces Abeta-degrading enzymatic activities in primary human mononuclear phagocytes. J Immunol 186:6925–6932
Xu J, Ikezu T (2009) The comorbidity of HIV-associated neurocognitive disorders and Alzheimer's disease: a foreseeable medical challenge in post-HAART era. J Neuroimmune Pharmacol 4:200–212
Giunta B, Ehrhart J, Obregon DF et al (2011) Antiretroviral medications disrupt microglial phagocytosis of β-amyloid and increase its production by neurons: implications for HIV-associated neurocognitive disorders. Mol Brain 4:23
Patrick C, Crews L, Desplats P et al (2011) Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am J Pathol 178:1646–1661
Giunta B, Hou H, Zhu Y et al (2009) HIV-1 Tat contributes to Alzheimer's disease-like pathology in PSAPP mice. Int J Clin Exp Pathol 2:433–443
Holcomb L, Gordon MN, McGowan E et al (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4:97–100
Kim BO, Liu Y, Ruan Y et al (2003) Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol 162:1693–1707
Boelen E, Steinbusch HW, van der Ven AJ (2007) Chlamydia pneumoniae infection of brain cells: an in vitro study. Neurobiol Aging 28:524–532
Miklossy J, Kis A, Radenovic A et al (2006) Beta-amyloid deposition and Alzheimer's type changes induced by Borrelia spirochetes. Neurobiol Aging 27:228–236
Soscia SJ, Kirby JE, Washicosky KJ et al (2010) The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505
Anderson JP, Walker DE, Goldstein JM et al (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281:29739–29752
Jang H, Boltz D, Sturm-Ramirez K et al (2009) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 106:14063–14068
Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469:323–335
Todde V, Veenhuis M, van der Klei IJ (2009) Autophagy: principles and significance in health and disease. Biochim Biophys Acta 1792:3–13
Sir D, Ou JH (2010) Autophagy in viral replication and pathogenesis. Mol Cells 29:1–7
Cuervo AM, Bergamini E, Brunk UT et al (2005) Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1:13140
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995
Nixon RA (2007) Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120:4081–4091
White E, Karp C, Strohecker AM et al (2010) Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol 22:212–217
Hara T, Nakamura K, Matsui M et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Komatsu M, Waguri S, Chiba T et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Martinez-Vicente M, Cuervo AM (2007) Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 6:352–361
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ (2007) Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6:304–312
Williams A, Jahreiss L, Sarkar S et al (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76:89–101
Orvedahl A, Levine B (2008) Autophagy and viral neurovirulence. Cell Microbiol 10:1747–1756
Seay MD, Dinesh-Kumar SP (2005) Life after death: are autophagy genes involved in cell death and survival during plant innate immune responses? Autophagy 1:185–186
Talloczy Z, Virgin HW 4th, Levine B (2006) PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29
Colombo MI (2005) Pathogens and autophagy: subverting to survive. Cell Death Differ 12(Suppl 2):1481–1483
Jackson WT, Giddings TH, Taylor MP et al (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3:e156
Taylor MP, Kirkegaard K (2007) Modification of cellular autophagy protein LC3 by poliovirus. J Virol 81:12543–12553
Wileman T (2006) Aggresomes and autophagy generate sites for virus replication. Science 312:875–878
Talloczy Z, Jiang W, Virgin HWT et al (2002) Regulation of starvation and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 99:190–195
Cassady KA, Gross M, Roizman B (1998) The second site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol 72:7005–7011
Chang RC, Suen KC, Ma CH et al (2002) Involvement of double-stranded RNAdependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J Neurochem 83:1215–1225
Chou J, Chen JJ, GrossM RB (1995) Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5-mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A 92:10516–10520
Orvedahl A, Alexander D, Tallóczy Z et al (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35
He B, Gross M, Roizman B (1997) The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848
Ill-Raga G, Palomer E, Wozniak MA et al (2011) Activation of PKR causes amyloid ß-peptide accumulation via de-repression of BACE1 expression. PLoS One 6:e21456
Santana S, Recuero M, Bullido MJ et al (2012) Herpes simplex virus type I induces the accumulation of intracellular β-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol Aging 33:430.e19–430.e33
Yu WH, Kumar A, Peterhoff C et al (2004) Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol 36:2531–2540
Gannagé M, Rämer PC, Münz C (2010) Targeting Beclin 1 for viral subversion of macroautophagy. Autophagy 6:166–167
Alirezaei M, Kiosses WB, Fox HS (2008) Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy 4:963–966
Matarrese P, Nencioni L, Checconi P et al (2011) Pepstatin A alters host cell autophagic machinery and leads to a decrease in influenza A virus production. J Cell Physiol 226:3368–3377
Gannagé M, Dormann D, Albrecht R et al (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380
Zheng X, Chu F, Mirkin BL et al (2008) Role of the proteolytic hierarchy between cathepsin L, cathepsin D and caspase-3 in regulation of cellular susceptibility to apoptosis and autophagy. Biochim Biophys Acta 1783:2294–2300
Nencioni L, De Chiara G, Sgarbanti R et al (2009) Bcl-2 expression and p38MAPK activity in cells infected with influenza A virus: impact on virally induced apoptosis and viral replication. J Biol Chem 284:16004–16015
Butterfield DA, Reed T, Newman SF, Sultana R (2007) Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med 43:658–677
Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10(Suppl):S18–S25
Sultana R, Butterfield DA (2009) Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. J Bioenerg Biomembr 41:441–446
Giasson BI, Duda JE, Murray IV et al (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290:985–989
Horiguchi T, Uryu K, Giasson BI et al (2003) Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol 163:1021–1031
Ciriolo MR, Palamara AT, Incerpi S et al (1997) Loss of GSH, oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J Biol Chem 272:2700–2708
Gong G, Waris G, Tanveer R, Siddiqui A (2001) Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci U S A 98:9599–9604
Kaul P, Biagioli MC, Singh I, Turner RB (2000) Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J Infect Dis 181:1885–1890
Nencioni L, Iuvara A, Aquilano K et al (2003) Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J 17:758–760
Peterhans E (1997) Oxidants and antioxidants in viral diseases: disease mechanisms and metabolic regulation. J Nutr 127:S962–S965
Nucci C, Palamara AT, Ciriolo MR et al (2000) Imbalance in corneal redox state during herpes simplex virus 1-induced keratitis in rabbits. Effectiveness of exogenous glutathione supply. Exp Eye Res 70:215–220
Palamara AT, Perno CF, Ciriolo MR et al (1995) Evidence for antiviral activity of glutathione: in vitro inhibition of herpes simplex virus type 1 replication. Antiviral Res 27:237–253
Valyi-Nagy T, Dermody TS (2005) Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol Histopathol 20:957–967
Kavouras JH, Prandovszky E, Valyi-Nagy K et al (2007) Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J Neurovirol 13:416–425
Fujii S, Akaike T, Maeda H (1999) Role of nitric oxide in pathogenesis of herpes simplex virus encephalitis in rats. Virology 256:203–212
Valyi-Nagy T, Olson SJ, Valyi-Nagy K et al (2000) Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology 278:309–321
Tamagno E, Bardini P, Obbili A et al (2002) Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis 10:279–288
Tamagno E, Parola M, Bardini P et al (2005) Beta-siteAPP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediatedby stress-activated protein kinases pathways. J Neurochem 92:628–636
Tamagno E, Guglielmotto M, Aragno M et al (2008) Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem 104:683–695
Ferrer I (2004) Stress kinases involved in tau phosphorylation in Alzheimer's disease, tauopathies and APP transgenic mice. Neurotox Res 6:469–475
Lee MS, Kao SC, Lemere CA et al (2003) APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol 163:83–95
Yoshida H, Hastie CJ, McLauchlan H et al (2004) Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J Neurochem 90:352–358
Zhu X, Raina AK, Rottkamp CA et al (2001) Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem 76:435–441
Zhu X, Rottkamp CA, Hartzler A et al (2001) Activation of MKK6, an upstream activator of p38, in Alzheimer's disease. J Neurochem 79:311–318
Zachos G, Koffa M, Preston CM et al (2001) Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J Virol 75:2710–2728
Hargett D, McLean T, Bachenheimer SL (2005) Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J Virol 79:8348–8360
Schachtele SJ, Hu S, Little MR, Lokensgard JR (2010) Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J Neuroinflammation 7:35
Buhl R, Jaffe HA, Holroyd KJ et al (1989) Systemic glutathione deficiency in symptom-free HIV-seropositive individuals. Lancet 2:1294–1298
Eck HP, Gmunder H, Hartmann M et al (1989) Low concentrations of acid-soluble thiol (cysteine) in the blood plasma of HIV-1-infected patients. Biol Chem Hoppe Seyler 370:101–108
Elbim C, Pillet S, Prevost MH et al (1999) Redox and activation status of monocytes from human immunodeficiency virus-infected patients: relationship with viral load. J Virol 73:4561–4566
Garaci E, Palamara AT, Ciriolo MR et al (1997) Intracellular GSH content and HIV replication in human macrophages. J Leukoc Biol 62:54–59
Turchan J, Pocernich CB, Gairola C et al (2003) Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology 28:307–314
Butler TR, Smith KJ, Self RL et al (2011) Neurodegenerative effects of recombinant HIV-1 Tat(1–86) are associated with inhibition of microtubule formation and oxidative stress-related reductions in microtubule-associated protein-2(a, b). Neurochem Res 36:819–828
Kruman II, NathA MMP (1998) HIV-1 proteinTat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol 154:276–288
Mollace V, Salvemini D, Riley DP et al (2002) The contribution of oxidative stress in apoptosis of human-cultured astroglial cells induced by supernatants of HIV-1-infected macrophages. J Leukoc Biol 71:65–72
Adamson DC, Kopnisky KL, Dawson TM, Dawson VL (1999) Mechanisms and structural determinants of HIV-1 coat protein gp41-induced neurotoxicity. J Neurosci 19:64–71
Hori K, Burd PR, Furuke K et al (1999) Human immunodeficiency virus-1-infected macrophages inducible nitric oxide synthase and nitric oxide (NO) production astrocytes: astrocytic NO as a possible mediator of neuronaldamage in acquired immunodeficiency syndrome. J Immunol 93:1843–1850
Cai J, Chen Y, Seth S et al (2003) Inhibition of influenza infection by glutathione. Free Radic Biol Med 34:928–936
Hennet T, Peterhans E, Stocker R (1992) Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J Gen Virol 73:39–46
Mileva M, Tancheva L, Bakalova R et al (2000) Effect of vitamin E on lipid peroxidation and liver monooxigenase activity in experimental influenza virus infection. Toxicol Lett 114:39–45
Nencioni L, Sgarbanti R, De Chiara G et al (2007) Influenza virus and redox mediated cell signaling: a complex network of virus/host interaction. New Microbiol 30:367–375
Sgarbanti R, Nencioni L, Amatore D et al (2011) Redox regulation of the influenza hemagglutinin maturation process: a new cell-mediated strategy for anti-influenza therapy. Antioxid Redox Signal 15:593–606
Yao D, Kuwajima M, Kido H (2003) Pathologic mechanisms of influenza encephalitis with an abnormal expression of inflammatory cytokines and accumulation of mini-plasmin. J Med Investig 50:1–8
Yamanaka G, Kawashima H, Suganami Y et al (2006) Diagnostic and predictive value of CSF d-ROM level in influenza virus-associated encephalopathy. J Neurol Sci 243:71–75
Martin HL, Teismann P (2009) Glutathione-a review on its role and significance in Parkinson's disease. FASEB J 23:3263–3272
Dröge W, Schipper HM (2007) Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 6:361–370
Nuss JE, Choksi KB, DeFord JH, Papaconstantinou J (2008) Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem Biophys Res Commun 365:355–361
Uehara T, Nakamura T, Yao D et al (2006) S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 44:513–517
van Spronsen M, Hoogenraad CC (2010) Synapse pathology in psychiatric and neurologic disease. Curr Neurol Neurosci Rep 10:207–214
Südhof TC, Malenka RC (2008) Understanding synapses: past, present, and future. Neuron 60:469–476
Gilman CP, Mattson MP (2002) Do apoptotic mechanisms regulate synaptic plasticity and growth-cone motility? Neuromol Med 2:197–214
Mattson MP (2003) Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromol Med 3:65–94
Everall IP, Heaton RK, Marcotte TD et al (1999) Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. The HIV Neurobehavioral Research Center. Brain Pathol (Zurich, Switzerland) 9:209–217
Spencer DC, Price RW (1992) Human immunodeficiency virus and the central nervous system. Annu Rev Microbiol 46:655–693
Toggas SM, Masliah E, Rockenstein EM et al (1994) Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 367:188–193
Michaud J, Fajardo R, Charron G et al (2001) Neuropathology of NFHgp160 transgenic mice expressing HIV-1 env protein in neurons. J Neuropathol Exp Neurol 60:574–587
Catani MV, Corasaniti MT, Navarra M et al (2000) gp120 induces cell death in human neuroblastoma cells through the CXCR4 and CCR5 chemokine receptors. J Neurochem 74:2373–2379
Corasaniti MT, Melino G, Navarra M et al (1995) Death of cultured human neuroblastoma cells induced by HIV-1 gp120 is prevented by NMDA receptor antagonists and inhibitors of nitric oxide and cyclooxygenase. Neurodegeneration 4:315–321
Lipton SA, Sucher NJ, Kaiser PK, Dreyer EB (1991) Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron 7:111–118
Medina I, Ghose S, Ben-Ari Y (1999) Mobilization of intracellular calcium stores participates in the rise of [Ca2+]i and the toxic actions of the HIV coat protein GP120. Eur J Neurosci 11:1167–1178
Geeraerts T, Deiva K, M'sika I et al (2006) Effects of SDF-1alpha and gp120IIIB on apoptotic pathways in SK-N-SH neuroblastoma cells. Neurosci Lett 399:115–120
Ushijima H, Ando S, Kunisada T (1993) HIV-1 gp120 and NMDA induce protein kinase C translocation differentially in rat primary neuronal cultures. J Acquir Immune Defic Syndr 6:339–343
Haughey NJ, Mattson MP (2002) Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr 31(Suppl 2):S55–S61
Xu H, Bae M, Tovar-Y-Romo LB et al (2011) The human immunodeficiency virus coat protein gp120 promotes forward trafficking and surface clustering of NMDA receptors in membrane microdomains. J Neurosci 31:17074–17090
Self RL, Mulholland PJ, Nath A et al (2004) The human immunodeficiency virus type-1 transcription factor Tat produces elevations in intracellular Ca2+ that require function of an N-methyl-d-aspartate receptor polyamine-sensitive site. Brain Res 995:39–45
Song L, Nath A, Geiger JD et al (2003) Humanimmunodeficiency virus type 1 Tat protein directly activates neuronal N-methyl-d-aspartate receptors at an allosteric zinc-sensitive site. J Neurovirol 9:399–403
Bonavia R, Bajetto A, Barbero S et al (2001) HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun 288:301–308
Perez A, Probert AW, Wang KK, Sharmeen L (2001) Evaluation of HIV-1 Tat induced neurotoxicity in rat cortical cell culture. J Neurovirol 7:1–10
Aprea S, Del Valle L, Mameli G et al (2006) Tubulin-mediated binding of human immunodeficiency virus-1 Tat to the cytoskeleton causes proteasomal-dependent degradation of microtubule-associated protein 2 and neuronal damage. J Neurosci 26:4054–4062
Kim HJ, Martemyanov KA, Thayer SA (2008) Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 28:12604–12613
Li ST, Matsushita M, Moriwaki A et al (2004) HIV-1 Tat inhibits long-term potentiation and attenuates spatial learning. Ann Neurol 55:362–371
Brask J, Chauhan A, Hill RH et al (2005) Effects on synaptic activity in cultured hippocampal neurons by influenza A viral proteins. J Neurovirol 11:395–402
Labetoulle M, Kucera P, Ugolini G et al (2000) Neuronal pathways for the propagation of herpes simplex virus type 1 from one retina to the other in a murine model. J Gen Virol 81:1201–1210
Cocchi F, Menotti L, Dubreuil P et al (2000) Cell-to-cell spread of wild-type herpes simplex virus type 1, but not of syncytial strains, is mediated by the immunoglobulin-like receptors that mediate virion entry, nectin1 (PRR1/HveC/HIgR) and nectin2 (PRR2/HveB). J Virol 74:3909–3917
Geraghty RJ, Krummenacher C, Cohen GH et al (1998) Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620
Sakisaka T, Taniguchi T, Nakanishi H et al (2001) Requirement of interaction of nectin-1 alpha/HveC with afadin for efficient cell–cell spread of herpes simplex virus type 1. J Virol 75:4734–4743
Warner MS, Geraghty RJ, Martinez WM et al (1998) A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology 246:179–189
Mizoguchi A, Nakanishi H, Kimura K et al (2002) Nectin: an adhesion molecule involved in formation of synapses. J Cell Biol 156:555–565
Assunção Guimarães C, Linden R (2004) Programmed cell deaths. Apoptosis and alternative death styles. Eur J Biochem 271:1638–1650
DeBiasi RL, Kleinschmidt-DeMasters BK, Richardson-Burns S, Tyler KL (2002) Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J Infect Dis 186:1547–1557
Armien AG, Hu S, Little MR et al (2009) Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis. Brain Pathol 20:738–750
Perkins D, Gyure KA, Pereira F, Aurelian L (2003) Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J Neurovirol 9:101–111
Perng GC, Jones C, Ciacci-Zanella J et al (2000) Virus induced neuronal apoptosis blocked by the herpes simplex virus latency associated transcript (LAT). Science 287:1500–1503
Henderson G, Perng GC, Nesburn AB et al (2004) The latency-related gene encoded by bovine herpesvirus 1 can suppress caspase 3 and caspase 9 cleavage during productive infection. J Neurovirol 10:64–70
Jin L, Peng W, Perng GC et al (2003) Identification of herpes simplex virus type 1 latency associated transcript sequences that both inhibit apoptosis and enhance the spontaneous reactivation phenotype. J Virol 77:6556–6561
Peng W, Jin L, Henderson G et al (2004) Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J Neurovirol 10:260–265
Jiang X, Chentoufi AA, Hsiang C et al (2011) The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol 85:2325–2332
Li S, Carpenter D, Hsiang C et al (2010) Herpes simplex virus type 1 latency-associated transcript inhibits apoptosis and promotes neurite sprouting in neuroblastoma cells following serum starvation by maintaining protein kinase B (AKT) levels. J Gen Virol 91:858–866
Leib DA, Bogard CL, Kosz-Vnenchak M et al (1989) A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900
Perng GC, Dunkel EC, Geary PA et al (1994) The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055
Trousdale MD, Steiner I, Spivack JG et al (1991) In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-associated transcript variant in a rabbit eye model. J Virol 65:6989–6993
Asano S, Honda T, Goshima F et al (1999) US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J Gen Virol 80:51–56
Aubert M, Blaho JA (1999) The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J Virol 73:2803–2813
Galvan V, Roizman B (1998) Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type dependent manner. Proc Natl Acad Sci U S A 95:3931–3936
Galvan V, Brandimarti R, Roizman B (1999) Herpes simplex virus 1 blocks caspase-3-independent and caspasedependent pathways to cell death. J Virol 73:3219–3226
Leopardi R, Roizman B (1996) The herpes simplex virus major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proc Natl Acad Sci U S A 93:9583–9587
Gautier I, Coppey J, Durieux C (2003) Early apoptosis-related changes triggered by HSV-1 in individual neuronlike cells. Exp Cell Res 289:174–183
Zhou G, Galvan V, Campadelli-Fiume G, Roizman B (2000) Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J Virol 74:11782–11791
Perkins D, Pereira EF, Gober M et al (2002) The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J Virol 76:1435–1449
Perkins D, Pereira EF, Aurelian L (2003) The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1. J Virol 77:1292–1305
Carter CJ (2011) Alzheimer's disease plaques and tangles: cemeteries of a pyrrhic victory of the immune defence network against herpes simplex infection at the expense of complement and inflammation-mediated neuronal destruction. Neurochem Int 58:301–320
Honjo K, van Reekum R, Verhoeff NP (2009) Alzheimer's disease and infection: do infectious agents contribute to progression of Alzheimer's disease? Alzheimers Dement 5:348–360
Conrady CD, Drevets DA, Carr DJ (2010) Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol 220:1–9
van Zandbergen G, Gieffers J, Kothe H et al (2004) Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol 172:1768–1776
Airenne S, Surcel HM, Tuukkanen J (2002) Chlamydia pneumoniae inhibits apoptosis in human epithelial and monocyte cell lines. Scand J Immunol 55:390–398
Carratelli CR, Rizzo A, Catania MR et al (2002) Chlamydia pneumoniae infections prevent the programmed cell death on THP-1 cell line. FEMS Microbiol Lett 215:69–74
Paland N, Rajalingam K, Machuy N et al (2006) NF-kappaB and inhibitor of apoptosis proteins are required for apoptosis resistance of epithelial cells persistently infected with chlamydophila pneumoniae. Cell Microbiol 8:1643–1655
Rajalingam K, Al-Younes H, Muller A et al (2001) Epithelial cells infected with chlamydophila pneumonia. Infect Immun 69:7880–7888
Appelt DM, Roupas MR, Way DS et al (2008) Inhibition of apoptosis in neuronal cells infected with Chlamydophila (Chlamydia) pneumoniae. BMC Neurosci 9:13
Gerard HC, Wildt KL, Whittum-Hudson JA et al (2005) The load of Chlamydia pneumoniae in the alzheimer's brain varies with APOE genotype. Microb Pathog 39:19–26
Gerard HC, Dreses-Werringloer U, Wildt KS et al (2006) Chlamydophila (chlamydia) pneumoniae in the alzheimer's brain. FEMS Immunol Med Microbiol 48:355–366
Nazzal D, Cantero AV, Therville N et al (2006) Chlamydia pneumoniae alters mildly oxidized low-density lipoprotein-induced cell death in human endothelial cells, leading to necrosis rather than apoptosis. J Infect Dis 193:136–145
Everall IP, Luthert PJ, Lantos PL (1991) Neuronal loss in the frontal cortex in HIV infection. Lancet 337:1119–1121
Jones G, Power C (2006) Regulation of neural cell survival by HIV-1 infection. Neurobiol Dis 21:1–17
Ketzler S, Weis S, Haug H, Budka H (1990) Loss of neurons in the frontal cortex in AIDS brains. Acta Neuropathol 80:92–94
Petito CK, Roberts B (1995) Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol 146:1121–1130
Yoshioka M, Bradley WG, Shapshak et al (1995) Role of immune activation and cytokine expression in HIV-1-associated neurologic diseases. Adv Neuroimmunol 5:335–358
Corasaniti MT, Nistico R, Costa A et al (2001) The HIV-1 envelope protein, gp120, causes neuronal apoptosis in the neocortex of the adult rat: a useful experimental model to study neuroaids. Funct Neurol 16:31–38
Oh JW, Drabik K, Kutsch O et al (2001) CXC chemokine receptor 4 expression and function in human astroglioma cells. J Immunol 166:2695–2704
Hesselgesser J, Taub D, Baskar P et al (1998) Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol 8:595–598
Zheng J, Thylin MR, Ghorpade A et al (1999) Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol 98:185–200
Brenneman DE, Westbrook GL, Fitzgerald SP (1988) Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature 335:639–642
Lannuzel A, Barnier JV, Hery C et al (1997) Human immunodeficiency virus type 1 and its coat protein gp120 induce apoptosis and activate JNK and ERK mitogen-activated protein kinases in human neurons. Ann Neurol 42:847–856
Meucci O, Miller RJ (1996) gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGF-beta1. J Neurosci 16:4080–4088
Müller WE, Schröder HC, Ushijima H et al (1992) gp120 of HIV-1 induces apoptosis in rat cortical cell cultures: prevention by memantine. Eur J Pharmacol 226:209–214
Acquas E, Bachis A, Nosheny RL et al (2004) Human immunodeficiency virus type 1 protein gp120 causes neuronal cell death in the rat brain by activating caspases. Neurotox Res 5:605–615
Bagetta G, Corasaniti MT, Berliocchi L et al (1995) HIV-1 gp120 produces DNA fragmentation in the cerebral cortex of rat. Biochem Biophys Res Commun 211:130–136
Bagetta G, Corasaniti MT, Malorni W et al (1996) The HIV-1 gp120 causes ultrastructural changes typical of apoptosis in the rat cerebral cortex. Neuroreport 7:1722–1724
Ramirez SH, Sanchez JF, Dimitri CA et al (2001) Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem 78:874–889
Zauli G, Milani D, Mirandola P et al (2001) HIV-1 Tat protein down-regulates CREB transcription factor expression in PC12 neuronal cells through a phosphatidylinositol 3-kinase/ AKT/cyclic nucleoside phosphodiesterase pathway. FASEB J 15:483–491
Ferrucci A, Nonnemacher MR, Wigdahl B (2011) Human immunodeficiency virus viral protein R as an extracellular protein in neuropathogenesis. Adv Virus Res 81:165–199
Piller SC, Jans P, Gage PW, Jans DA (1998) Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc Natl Acad Sci U S A 95:4595–4600
Patel CA, Mukhtar M, Pomerantz RJ (2000) HIV-1 Vpr induces apoptosis in human neuronal cells. J Virol 74:9717–9726
Kaul M, Lipton SA (1999) Chemokines and activated macrophages in HIV gp120-induced neronal apoptosis. Proc Natl Acad Sci U S A 96:8212–8216
Garden GA (2002) Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 40:240–251
Zhang K, McQuibban GA, Silva C et al (2003) HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci 6:1064–1071
Zhang Z, Zhang J, Huang K et al (2009) Systemic infection of avian influenza A virus H5N1 subtype in humans. Hum Pathol 40:735–739
Gu J, Xie Z, Gao Z, Liu J et al (2007) H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet 370:1137–1145
Ng YP, Lee SM, Cheung TK et al (2010) Avian influenza H5N1 virus induces cytopathy and proinflammatory cytokine responses in human astrocytic and neuronal cell lines. Neuroscience 168:613–623
Rohn TT, Catlin LW (2011) Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson's disease brain. PLoS One 6:e20495
Nakai Y, Itoh M, Mizuguchi M et al (2003) Apoptosis and microglial activation in influenza encephalopathy. Acta Neuropathol 105:233–239
Acknowledgments
This work was supported by grants from the Italian Ministry of Education University and Research (PRIN 2009PM9B33_001 and PON01-01802 to ATP; PRIN 2009PM9B33_002 to CG).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
De Chiara, G., Marcocci, M.E., Sgarbanti, R. et al. Infectious Agents and Neurodegeneration. Mol Neurobiol 46, 614–638 (2012). https://doi.org/10.1007/s12035-012-8320-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8320-7