
Abstract
The synthesis of α/β-chimeras comprises peptide bond formation from α- to β-, from β- to β-, and from β- to α-amino acid residues. The fine-tuned solid phase synthesis of –GXXG– chimera peptides containing the simplest achiral α-amino acid glycine and two cyclic SAAs of different ring size [X denoting cyclic β-Sugar Amino Acids (β-SAA)] is reported, variants containing Fmoc–RibAFU(ip)–OH a furanoid-, and Fmoc–GlcAPU(Me)–OH a pyranoid-type structural “Lego-element”. Systematic search for the best coupling strategy with both H–β-SAA–OHs is described, including the comparison of the different coupling reagents and conditions. Selecting the optimal reagent (from commonly used PyBOP, HATU and HOBt) was assisted by time-resolved 1H-NMR: formation and stability of the Fmoc protected active esters were compared. We found that PyBOP is the best choice for successfully coupling both H–β-SAA–OH prototypes. The present comparative results open a reasonable route for building efficiently various –β-SAA– containing homo- and heterooligomers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chimeric synthetic polypeptides containing β-amino acids have achieved increasing attention in foldamer chemistry and drug design, due to their prolonged resistance against proteases and their ability of forming designed nanostructures of reduced internal dynamics (Horne and Gellman 2008; Pilsl and Reiser 2011; Guichard and Huc 2011; Kiss and Fülöp 2014; Cabrele et al. 2014; Mándity and Fülöp 2015; Pohl et al. 2013). Cyclic β-amino acid residues—as conformationally restricted building blocks—are of special interest, as they have the same number of backbone torsional angles (ϕ and ψ) as α-peptides. These oligo- and polypeptides composed of cyclic β-amino acids have backbone folding properties primarily determined by local configuration. Configuration driven, fine-tuned conformational properties enhance their receptor-binding ability and macromolecule formation potential (Beke et al. 2004).
The cis- and trans-stereoisomers of 2-aminocyclobutanecarboxylic acid (ACBC), 2-aminocyclopentanecarboxylic acid (ACPC) and 2-aminocyclohexanecarboxylic acid (ACHC) derivatives are now widely applied to construct chimeras (Giuliano et al. 2013; Torres et al. 2009, 2010; Gorrea et al. 2012; Herradón and Seebach 1989; Kessler et al. 1995; Kiss et al. 2017). However, all three of the above compounds are highly hydrophobic and their hydrophilic cyclic analogues are barely known and thus used. During the last 20 years, both five- and six-membered cyclic Sugar Amino Acids (H–SAA–OHs) appeared sporadically as building blocks (Risseeuw et al. 2013; Pandey et al. 2011; Giri et al. 2012; Long et al. 1999; Simone et al. 2005; Sharma et al. 2008, 2011; Andreini et al. 2009, Chakraborty et al. 1998, von Roedern and Kessler 1994, Feher-Voelger et al. 2014) (Fig. 1). H–SAA–OHs were considered hard to synthesize and costly to manufacture on the gram scale required. Recently, we have accomplished several consecutive steps, starting from d-glucose. The syntheses are fine-tuned, optimized and ready for large-scale and cost-efficient total synthesis; the Fmoc-derivatives are now available on the gram scale (Nagy et al. 2017; Goldschmidt Gőz et al. 2018).

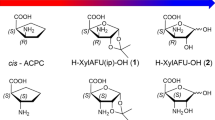
Molecular structure of the two H–β-SAA–OH prototypes 1 and 2, with their carbocyclic analogues ACPC and ACHC
Homo- and heterooligomers containing H–β-SAA–OHs were synthesized by us and others using different coupling reagents (BOP, HATU and HOBt) and amino protection (Fmoc or Boc groups) either in solution or on solid phase (Gruner et al. 2002; Csordás et al. 2016; Chandrasekhar et al. 2004; Suhara et al. 2002). For example, the pentofuranoid Fmoc–RibAFU(ip)–OH (1) was combined with β-homoglycine to form β-chimera peptides using HATU/collidine reagents on solid phase (Gruner et al. 2002). The coupling of the same H–β-SAA–OH with α-glycine was similarly tested with HATU/DIEA (Csordás et al. 2016), but the coupling efficacy of the sugar moiety needs to be proved. Homooligomers of different lengths were prepared from the derivatives of the C-3 epimer H–XylAFU(ip)–OH with EDCI/HOBt in solution (Chandrasekhar et al. 2004). The hexopyranoid N-tert-butyloxycarbonyl-3,4,6-tri-O-acetyl-β-d-glucosamine-1-carboxylic acid was also used to form related types of homooligomers (Suhara et al. 2002). The syntheses were performed using BOP/DIEA reagents and Boc/Bn protection in solution. Although several coupling reactions and conditions were probed, no systematic comparison of the different coupling reagents, strategies and techniques were performed yet. The comprehensive analysis presented here with respect to the coupling conditions and efficacy of two different H–β-SAA–OH prototypes representing different ring sizes: 5-membered ring, 3,4-trans Fmoc–RibAFU(ip)–OH (1) (Nagy et al. 2017) and 6-membered ring, 4,5-trans Fmoc–GlcAPU(Me)–OH (2) (Goldschmidt Gőz et al. 2018) residues were introduced into appropriate –Gly–β-SAA–β-SAA–Gly– heterooligomers. Our aims were to probe the key steps of peptide bond formation between α- and β-, β- and β-, and β- and α-amino acids using various active ester formation methods. Both the active ester types and the right coupling conditions (T, p, solvent type, reaction time, etc.) were optimized. Here, we present a systematic study on the formation and stability fine-tuned by 1H-NMR of the active esters from the furanoid and pyranoid carbohydrate moieties.
Results and discussion
Our comprehensive study was carried out using four different, though commonly used peptide coupling reagents, namely: (i) the HOBt/EDCI used by Chandrasekhar et al. for H–β-SAA–OH in solution (Chandrasekhar et al. 2004); (ii) the HOBt/DIC system, the most prevalent choice of the field (Valeur and Bradley 2009); (iii) the PyBOP/DIEA reagent pair developed for coupling “difficult sequences” without the considerable risk of racemization (Albericio and Carpino 1997); (iv) the HATU/DIEA also applied for “difficult sequences” with the risk of racemization (Valeur and Bradley 2009; Montalbetti and Falque 2005), and thus, limiting the overall coupling times to less than 3 h (Scheme 1).

Active ester formation from a furanoid-type N-Fmoc protected β-sugar amino acid such as Fmoc–RibAFU(ip)–OH (1). Reagents and conditions: a HATU/DIEA, b HOBt/EDCI/DIEA, c HOBt/DIC, and d PyBOP/DIEA
Formation and stability of β-sugar amino acid active esters
During SPPS, the active ester is in situ made to enhance the efficacy of the amide bond formation. It is presumed that the active ester formation occurs fast and quantitatively (conversion > 99%) (Albericio and Carpino 1997). For α-l-amino acid residues, conditions for active ester formation were optimized to complete it within a few minutes (Coste et al. 1990). As cyclic β-sugar amino acids are more compact with a –COOH group sterically hindered, coupling conditions (Fig. 1) were probed and described here for selected H–β-SAA–OHs, using alternative coupling reagents. For a quantitative characterization, time-resolved 1H-NMR spectra were recorded, selected resonance frequencies were assigned, and the time needed of the active ester formation (tf) and hydrolysis (th) was established from the integral-time diagrams (see Supporting Information). The solution of the furanoid (1) or pyranoid (2) prototypes was mixed with the appropriate coupling reagent (PyBOP, HATU or HOBt in an equimolar ratio) in [D7]DMF at T = 25 °C. At t = 0 min either DIEA (2 eqv.), DIC (1 eqv.), or EDCI (1 eqv.) was added to initiate the active ester formation and 1H-NMR spectra were recorded after 10, 20, 30, 60 min, and so on for hours (and day) (Fig. 2). For the Fmoc–RibAFU(ip)–OH (1) and PyBOP/DIEA system, resonance frequencies of the free PyBOP (HB: 8.10 ppm and HD: 8.27 ppm) and that of the Fmoc–RibAFU(ip)–OH (H1: 6.00 ppm) were used in conjunction with those of the active ester 5 (HB′: 7.78 ppm, H1′: 5.93 ppm, Fig. 2a). For Fmoc–GlcAPU(Me)–OH (2) with HOBt/DIC, resonance frequencies of the free HOBt (HA: 7.73 ppm and HD: 7.98 ppm) and that of Fmoc–GlcAPU(Me)–OH (H1: 5.03 ppm) was monitored with those of the active ester 4 (HA′: 8.22 ppm, HD′: 8.36 ppm, and H1′: 5.25 ppm, Fig. 2b).

Characteristic 1H-NMR resonance frequency changes (T = 25 °C) during the active ester formation used to monitor and decipher kinetics: a Fmoc–RibAFU(ip)–OH (1) with PyBOP/DIEA, b Fmoc–GlcAPU(Me)–OH (2) with HOBt/DIC
When the active ester formation with HOBt/EDCI/DIEA is very slow (tf = 960 min), the reaction is complete; however, when it is fast (tf = 20 min), conversion is poor (20%, Fmoc–GlcAPU(Me)–OH, 2), and in addition, the product decomposes upon formation, the degree of the decomposition was monitored by the formation of HOBt side-product, the 1H-NMR signals of which appeared in the spectra: HA: 7.78 ppm, HB: 7.60 ppm, HD: 7.93 ppm. The active ester formation is slow (tf = ~ 60–120 min) when HOBt/DIC is used and the conversion also remains low (50%, Fmoc–RibAFU(ip)–OH, 1); moreover, the starting uronic acid (1) is fully regained. Additional experiments revealed that a stable active ester cannot be formed with HOBt directly as it contains equimolar H2O, which, in principle, is enough for the complete hydrolysis of the nascent ester. Furthermore, HOBt decomposes if crystal water is removed by drying with molecular sieves etc. Thus, stabilized forms of HOBt had to be used. Reaction of Fmoc–GlcAPU(Me)–OH (2) with HATU/DIEA is slow (tf = 240 min) presenting an additional problem as racemization can occur after 3 h. We found the active ester formation of Fmoc–RibAFU(ip)–OH (1) with HATU/DIEA or PyBOP/DIEA is fast (tf = 20 min) and the product remains stable for more than 24 h. Similarly, Fmoc–GlcAPU(Me)–OH (2) reacts with PyBOP/DIEA quickly (tf = 10 min) forming an active ester intact and stable for over 6 h. Thus, PyBOP/DIEA seems to be the only solution good enough to use and to match conditions required for standard SPPS coupling.
Comparing the four coupling reagent pairs and the time needed for the active ester formation, significant differences were found (Table 1). The mechanisms of active ester formation can explain these differences. We found that the rate-determining steps of the ester formation are different using the HATU/DIEA or PyBOP/DIEA pairs and the HOBt/EDCI/DIEA or HOBt/DIC pairs. When PyBOP/DIEA (or HATU/DIEA) (Scheme 2a) was used, the deprotonation of the carboxylic acid (2) by DIEA occurred quickly. The rate-determining step is the next when the carboxylate anion as a nucleophile attacks on the electrophilic center, namely on the P atom of the phosphonium moiety of PyBOP (or at the C atom of the amidinium moiety of HATU).

Differences between the rate-determining steps, highlighted by green frames, for the two types of coupling agents: a PyBOP/DIEA; b HOBt/DIC
However, using HOBt/DIC (or HOBt/EDCI), a weaker base (DIC or EDCI) deprotonates the carboxylic acid (1). This ion-pair formation is likely slow enough to become the rate-determining reaction step (Scheme 2b).
Thus, the active ester formation was found to be faster with PyBOP/DIEA and HATU/DIEA (Table 1) compared to HOBt/EDCI/DIEA and HOBt/DIC, in line with their mechanism explained above. Both for the furanoid Fmoc–RibAFU(ip)–OH (1) and for the pyranoid prototype, Fmoc–GlcAPU(Me)–OH (2), active ester formation with PyBOP is fast: tf = ~ 20 min and tf = ~ 10 min, respectively. On the contrary, the reaction with HATU is considerably slower (tf = ~ 240 min); therefore, in that case, pre-activation is necessary to avoid racemization during the coupling (3 h).
Due to their differing electronic structure, nucleophile attacks may occur differently at P and C centers: the P atom can form a trigonal bipyramid structure, clearly more favorable for a nucleophile attack (Fig. 3), than that of C atom.

Possibility of nucleophilic attacks at P- and C-reaction centers: unlike the C atom, the P atom can form a trigonal bipyramid structure, which favors the nucleophilic attack
In conclusion, the use of PyBOP/DIEA pair provides the most promising conditions—the active esters are (i) formed quickly, (ii) products are stable for hours—to reliably form amide bonds between α- and β-, β- and β-, and β- and α-amino acid residues both in solution and on a solid support.
Synthesis of model peptides on solid phase
We probed partners of different complexity to form amide bonds. On one hand, using the highly mobile Gly as the representative of α-amino acids, for which no steric hindrance has to be taken into account as Gly has none and no racemization of the α-amino acid partner has to be considered as it is achiral. On the other hand, the bulky and cyclic H–β-SAA–OH residues which have several chiral C atoms, with –OHs differently protected by large protecting groups, indicating considerable steric hindrance. Using SPPS and Fmoc chemistry, the synthesis of two –GXXG– tetrapeptides was completed, and thus, all three amide bond types probed, namely, the α–β, β–β, and β–α. Coupling conditions for both 1 and 2 (–RibAFU(ip)– and –GlcAPU(Me)–) were probed, and –GX–, –XX–, and –XG– amide bond formations were carefully monitored and competed (Tables 2, 3). Fmoc–β-SAA–OH 1 and 2 were synthesized according to our recently improved, multigram scale, environmentally friendly protocol (Scheme 3), starting from d-glucose and methyl α-d-glucopyranoside (Nagy et al. 2017; Goldschmidt Gőz et al. 2018).

“Bird’s eye view” of the total synthesis of the furanoid (Fmoc–RibAFU(ip)–OH (1)) and pyranoid (Fmoc–GlcAPU(Me)–OH (2)) β-Sugar Amino Acid derivatives, the total synthesis of which fine-tuned recently
For difficult sequences, a low capacity resin (< 0.5 mmol/g) is recommended to be used. Therefore, RAM-Tentagel® resin (0.24 mmol/g) for the Fmoc–GlcAPU(Me)–OH (2) and 2-chlorotrityl chloride (2-Cl–Trt–Cl) resin (an 1.6 mmol/g original capacity resin was tuned down to 0.25–0.36 mmol/g with the first amino acid coupled to it) for the Fmoc–RibAFU(ip)–OH (1) was used. Coupling efficacy of the resin was completed by measuring its Fmoc capacity. The first glycine moiety was linked to the resin in the usual manner via Fmoc–Gly–OH (Scheme 4), followed by the subsequent removal of Fmoc-protecting group (see “Experimental section”). Coupling time need of our Fmoc–β-SAA–OHs (1 and 2) was optimized by 1H-NMR as described above (Table 1). Coupling of both 1 and 2 was achieved to the free N-terminus of the first Gly by using all four different coupling reagent pairs, namely: HATU/DIEA, PyBOP/DIEA, HOBt/DIC and HOBt/EDCI/DIEA in DMF. Both the XX and the closing XG couplings were executed with the above four reagent types followed by the removal of the Fmoc protection (Tables 2, 3). Identification of both –GXXG– tetrapeptides, 6 and 7, was done by ESI–MS ([M+H]+ = 503 and [M+H]+ = 870 (see in Supporting Information) once cleaved from the resins. Peptide 6 was cleaved with the mild AcOH:MeOH:DCM 1:1:8 cocktail to preserve the otherwise acid-labile isopropylidene O-protecting group of 1, while 7 with the milder 50% TFA (instead of the standard 95% TFA) to avoid the removal of O-benzyl protecting groups.

Solid-phase synthesis of the –GXXG– α/β-chimera peptides probed with four different coupling conditions. Reagents and conditions: a 1. HATU/DIEA, 2. piperidine (2%), DBU (2%), DMF; b 1. EDCI/HOBt/DIEA, 2. piperidine (2%), DBU (2%), DMF; c 1. DIC/HOBt, 2. piperidine (2%), DBU (2%), DMF; d 1. PyBOP/DIEA, 2. piperidine (2%), DBU (2%), DMF; e DCM:MeOH:AcOH (8:1:1); f TFA (50%), DCM (45%), H2O (2.5%), and TIS (2.5%)
The efficacy of all four probed coupling reagent pairs was jointly evaluated (Tables 2, 3), and we found that coupling with HOBt/EDCI/DIEA is insufficiently moderate: 32% and 58% for the two SAAs, respectively. Despite of the longer reaction time needed (18 h coupling time) due to the slow active ester formation, we concluded that HOBt/EDCI/DIEA is unsuitable for producing chimera peptides with cyclic H–β-SAA–OHs on a solid support, in spite of the fact that it was successfully used for coupling of H–XylAFU(ip)–OH in solution (Chandrasekhar et al. 2004). Similarly to our previous results (Csordás et al. 2016) for HATU/DIEA reagent pair, we also achieved low efficacy: 34% and 46%, respectively. In addition, the use of HATU is problematic from yet another aspect: its coupling times might be too long to avoid racemization (< 3 h).
We found HOBt/DIC for coupling Fmoc–GlcAPU(Me)–OH (2) effective (92%), especially to form the GX amide bond (Table 3). However, the detailed comprehensive analysis revealed that, for both Fmoc–β-SAA–OHs, PyBOP/DIEA is the method of choice! We found that if conditions are optimized, each amide coupling can be as high as 80–100% and that the overall coupling efficacy for the –GXXG– tetrapeptides reaches 78% and 76% (Tables 2 and 3). In general, using PyBOP/DIEA, (i) high coupling efficacy is achieved for all the types of couplings (–GX–, –XX–, and –XG–), (ii) reaction can be terminated within 3 h or less so racemization does not have to be feared (Albericio and Carpino 1997; Frérot et al. 1991). In conclusion, both for the furanoid (1) and pyranoid (2) Fmoc–β-SSA–OH prototypes, the application of PyBOP/DIEA is strongly supported. Moreover, with the PyBOP/DIEA-mediated effective coupling, it was possible to reduce the excess of the costly Fmoc–β-SSA–OHs from the previously proposed 3 to 1.5 molar equivalent (Mándity et al. 2014) making peptide synthesis even more cost-effective, nevertheless robust and efficient.
Conclusions
Solid phase peptide synthesis conditions were optimized to effectively couple Fmoc–β-SAA–OHs (X = 1 and 2), prototypes of different ring size sugar amino acids, to produce chimera peptides. We found that the key step of success is the active ester formation. Therefore, we have focused both on reactivity and active ester stability by probing a furanoid (Fmoc–RibAFU(ip)–OH, 1) and a pyranoid (Fmoc–GlcAPU(Me)–OH, 2) β-amino acid derivative using the most common reagents, namely, HOBt/EDCI/DIEA, HATU/DIEA, PyBOP/DIEA and HOBt/DIC. Using time-resolved 1H-NMR measurements for deciphering the optimum conditions for –GX–, –XX–, and –XG– amide bond formations, and the difficult couplings of the –GXXG– α/β-model peptides were successfully achieved with SPPS. We found the PyBOP/DIEA reagent pair to be the best among the probed coupling reagents, providing 80 to 100% coupling efficacy and an overall > 76% yield for both β-SAA containing tetrapeptides. Efficacy of the most hindered and thus most problematic –XX– bond formation with PyBOP/DIEA can be as high as 91 or even 99%.
The present comprehensive analysis has revealed that PyBOP may be the best choice to successful coupling H–β-SAA–OHs, providing now the possibility of making various β/β-homo- and α/β-heterooligomers for spectroscopic and pharmaceutical purposes.
Experimental section
Analytical data for all compounds (HPLC chromatograms, 1H NMR and MS spectra); 1H NMR spectra and figures of active ester formation of all coupling agents can be found in Supporting Information, in the online version.
Reagents and instrumentations
Reagents, materials and solvents were obtained from Alfa Aesar, Sigma-Aldrich, Merck, Reanal, or VWR. For moisture-sensitive reactions, the solvents were distilled with the standard procedures or dried on molecular sieves (3 Ǻ). Products were analyzed by reverse-phase HPLC on a Phenomenex Jupiter C-18 column using the water/acetonitrile mixtures of 0.1% TFA in water (A) and 0.08% TFA, and 95% acetonitrile in water (B), and UV detection completed at 220 and 280 nm. Products were identified with Bruker Esquire 3000+ tandem quadrupole mass spectrometer equipped with an electrospray ion source.
NMR measurements
1H-NMR experiments were performed at 298–300 K on Bruker Avance DRX 250 MHz spectrometer equipped with 5-mm SB dual probe with z-gradient, operating at 250.13 MHz for 1H and/or Bruker Avance III 700 MHz spectrometer operating at 700.17 MHz for 1H equipped with 5-mm z-gradient probe head. Spectra were recorded in [D7]DMF using the solvent residual peaks as the 1H internal reference: 2.75, 2.93, and 8.03 ppm. The sample concentrations ranged from 10 to 20 mM. Spectra evaluation was completed within the TopSpin 3.5 software.
Peptide synthesis
For the SPPS using Fmoc–RibAFU(ip)–OH (1) 2-Cl–Trt–Cl resin, while for that with Fmoc–GlcAPU(Me)–OH (2) RAM-Tentagel® resin was used. Resins were swollen in DCM. For the RAM-Tentagel® resin, the first step was the removal of the Fmoc group with common method (2% piperidine and 2% DBU in DMF, 3 + 17 min). The successful cleavage was analyzed by the Kaiser test. Coupling of the Fmoc–Gly–OH to the 2-Cl–Trt–Cl resin was accomplished using Fmoc–Gly–OH (1.5 eqv. to the nominal capacity of the resin ~ 1.6 mmol/g) dissolved in DMF and DIEA (3.75 eqv.) was added to the solution, reaction lasted for 1 h. Coupling of the Fmoc–Gly–OH to RAM-Tentagel® resin was made using Fmoc–Gly–OH (3 eqv. to the nominal capacity of the resin ~ 0.24 mmol/g) dissolved in DMF, HOBt (3 eqv.) and DIC (3 eqv.) was added to the solution, reaction lasted for 1 h. After coupling, the resins were washed with 3× DMF, 3× DCM, 3× MeOH and 1× diethyl ether and dried in vacuo. The capacity of the resin was determined by spectrometric measurement of the amount of Fmoc chromophore (Fmoc-piperidine adduct) released upon treatment of the resin with 20% piperidine in DMF (Chan and White 2000). The Fmoc group was removed by 2% piperidine and 2% DBU in DMF. The successful cleavage was analyzed by the Kaiser test.
The model peptides were synthesized by Fmoc chemistry using HOBt/DIC, HATU/DIEA, PyBOP/DIEA, or HOBt/EDCI/DIEA reagent pairs with repeated coupling if specified (Tables 2, 3). Capacity of the resins was determined by the above described method. The Fmoc group was removed by 2% piperidine and 2% DBU in DMF, but, in the case of Fmoc–GlcAPU(Me)–OH (2) instead of 3 + 17 min, cleavage time 10 + 40 min was used.
The final cleavage from 2-Cl–Trt–Cl resin was carried out with the AcOH–MeOH–DCM 1:1:8 mixture (5–10 mL/g resin) for 3 h. Resin was washed with 3× DCM, 3× iPrOH and 1× diethyl ether. The solvent was removed in vacuo. The final cleavage from RAM-Tentagel® resin was carried out with 50% TFA, 45% DCM, 2.5% TIS, and 2.5% H2O mixture (5–10 mL/g resin) for 3 h. Resin was washed with 2× DMF, 3× DCM and 2× MeOH and solvent was removed in vacuo. The crude products were precipitated with diethyl ether.
Compound 6: HPLC: 4.5 min, ESI–MS: m/z calculated for C20H30N4O11 [M+H]+ 503.2, found: 503.1.
Compound 7: HPLC: 19.4 min, ESI–MS: m/z calculated for C46H55N5O12 [M+H]+ 870.3, found: 870.6.
Abbreviations
- H–RibAFU(ip)–OH:
-
1,2-O-isopropylidene-3-amino-3-deoxy-α-d-ribofuranuronic acid
- H–XylAFU(ip)–OH:
-
1,2-O-isopropylidene-3-amino-3-deoxy-α-d-xylofuranuronic acid
- H–GlcAPU(Me)–OH:
-
Methyl 2,3-di-O-benzyl-4-amino-4-deoxy-α-d-glucopyranoside uronic acid
- Fmoc–β-SAA–OH:
-
Fmoc protected β-Sugar Amino Acid
- H–β-SAA–OH:
-
β-Sugar Amino Acid
- –β-SAA–:
-
β-Sugar Amino Acid in peptide bond
- Fmoc–β-SAA–OBt:
-
Fmoc protected β-amino acid HOBt ester
- Fmoc–β-SAA–OAt:
-
Fmoc protected β-amino acid HATU ester
References
Albericio F, Carpino LA (1997) Coupling reagents and activation. Methods Enzymol 289:104–126
Andreini M, Taillefumier C, Chretien F, Thery V, Chapleur Y (2009) Synthesis and solution conformation of homo-β-peptides consisting of N-mannofuranosyl-3-ulosonic acids. J Org Chem 74:7651–7659
Beke T, Csizmadia IG, Perczel A (2004) On the flexibility of β-peptides. J Comput Chem 25:285–307
Cabrele C, Martinek TA, Reiser O, Berlicki L (2014) Peptides containing β-amino acid patterns: challenges and successes in medicinal chemistry. J Med Chem 57:9718–9739
Chakraborty TK, Jayaprakash S, Diwan PV, Nagaraj R, Jampani SRB, Kunwar AC (1998) Folded conformation in peptides containing furanoid sugar amino acids. J Am Chem Soc 120:12962–12963
Chan WC, White PD (2000) Fmoc solid phase peptide synthesis—a practical approach. Oxford University Press, Oxford
Chandrasekhar S, Reddy SM, Jagadeesh B, Prabhakar A, Ramana Rao MHV, Jagannadh B (2004) Formation of a stable 14-helix in short oligomers of furanoid cis-β-sugar-amino acid. J Am Chem Soc 126:13586–13587
Coste J, Le-Nguyen D, Castro B (1990) PyBOP®: a new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett 31:205–208
Csordás B, Nagy A, Harmat V, Zsoldos-Mády V, Leveles I, Pintér I, Farkas V, Perczel A (2016) Origin of problems related to Staudinger reduction in carbopeptoid syntheses. Amino Acids 48:2619–2633
Feher-Voelger A, Borges-González J, Carillo R, Morales EQ, González-Platas J, Martín T (2014) Synthesis and conformational analysis of cyclic homooligomersfrom pyranoid e-sugar amino acids. Chem Eur J 20:4007–4022
Frérot E, Coste J, Pantaloni A, Dufour MN, Jouin P (1991) PyBOP® and PyBroP: two reagents for the difficult coupling of the α,α-dialkyl amino acid. Aib. Tetrahedron 47(2):259–270
Giri AG, Jogdand GF, Rajamohanan PR, Pandey SK, Ramana CV (2012) Synthesis and structural characterization of homochiral homo-oligomers of cis-γ-methoxy-substituted cis- and trans Furanoid-β-Amino acids. Eur J Org Chem 13:2656–2663
Giuliano MW, Maynard SJ, Almeida AM, Reidenbach AG, Guo L, Ulrich EC, Guzei IA, Gellman SH (2013) Evaluation of a cyclopentane-based γ-amino acid for the ability to promote α/γ peptide secondary structure. J Org Chem 78:12351–12361
Goldschmidt Gőz V, Pintér I, Harmat V, Perczel A (2018) Approaches to pyranuronic β-sugar amino acid building blocks of peptidosaccharide foldamer. Eur J Org Chem 3:355–361
Gorrea E, Pohl G, Nolis P, Celis S, Burusco KK, Branchadell V, Perczel A, Ortuno RM (2012) Secondary structure of short β-peptides as the chiral expression of monomeric building units: a rational and predictive model. J Org Chem 77(21):9795–9806
Gruner SAW, Truffault V, Voll G, Locardi E, Stöckle M, Kessler H (2002) Design, synthesis, and NMR structure of linear and cyclic oligomers containing novel furanoid sugar amino acids. Chem Eur J 8:4365–4376
Guichard G, Huc I (2011) Synthetic foldamers. Chem Commun 47:5933–5941
Herradón B, Seebach D (1989) Mono- and dialkylation of derivatives of (1R,2S)-2-hydroxycyclopentanecarboxylic acid and -cyclohexanecarboxylic acid via bicyclic dioxanones: selective generation of three contiguous stereogenic centers on a cyclohexane ring. Helv Chim Acta 72:690–714
Horne WS, Gellman SH (2008) Foldamers with heterogeneous backbones. Acc Chem Res 41:1399–1408
Kessler H, Diefenbach B, Finsinger D, Geyer A, Gurrath M, Goodman SL, Hölzemann G, Haubner R, Jonczyk A, Müller G, Graf von Roedern E, Wermuth J (1995) Design of superactive and selective integrin receptor antagonists containing the RGD sequence. Lett Pept Sci 2:155–160
Kiss L, Fülöp F (2014) Synthesis of carbocyclic and heterocyclic β-aminocarboxylic acids. Chem Rev 114:1116–1169
Kiss L, Mándity IM, Fülöp F (2017) Highly functionalized cyclic β-amino acid moieties as promising scaffolds in peptide research and drug design. Amino Acids 49:1441–1455
Long DD, Hungerford NL, Smith MD, Brittain DEA, Marquess DG, Claridge TDW, Fleet GWJ (1999) From sequencamers to foldamers? Tetrameric furanose carbopeptoids from cis- and trans 5-aminomethyl-tetrahydrofuran-2-carboxylates. Tetrahedron Lett 40:2195–2198
Mándity IM, Fülöp F (2015) An overview of peptide and peptoid foldamers in medicinal chemistry. Expert Opin Drug Discov 10(11):1163–1177
Mándity IM, Olasz B, Ötvös SB, Fülöp F (2014) Continuous-flow solid-phase peptide synthesis: a revolutionary reduction of the amino acid excess. Chem Sus Chem 7(11):3172–3176
Montalbetti CAGN, Falque V (2005) Amide bond formation and peptide coupling. Tetrahedron 61:10827–10852
Nagy A, Csordás B, Zsoldos-Mády V, Pintér I, Farkas V, Perczel A (2017) C-3 epimers of sugar amino acids as foldameric building blocks: improved synthesis, useful derivatives, coupling strategies. Amino Acids 49:223–240
Pandey SK, Jogdand GF, Oliveira JCA, Mata RA, Rajamohanan PR, Ramana CV (2011) Synthesis and structural characterization of homochiral homo-oligomers of parent cis- and trans-Furanoid-βamino acids. Chem Eur J 17:12946–12954
Pilsl LKA, Reiser O (2011) α/β-Peptide foldamers: state of the art. Amino Acids 41:709–718
Pohl G, Gorrea E, Branchadell V, Ortuńo RM, Perczel A, Tarczay Gy (2013) Foldamers of β-peptides: conformational preference of peptides formed by rigid building blocks the first MI-IR spectra of a triamide nanosystem. Amino Acids 45(4):957–973
Risseeuw MDP, Overhand M, Fleet GWJ, Simone MI (2013) A compendium of cyclic sugar amino acids and their carbocyclic and heterocyclic nitrogen analogues. Amino Acids 45:613–689
Sharma GVM, Nagendar P, Ramakrishna KVS, Chandramouli N, Choudhary M, Kunwar AC (2008) Three-residue turns in α/β-peptides and their application in the design of tertiary structures. Chem Asian J 3:969–983
Sharma GVM, Reddy PS, Chatterjee D, Kunwar AC (2011) Synthesis and structural studies of homooligomers of geminally disubstituted β2,2-amino acids with carbohydrate side chain. J Org Chem 76:1562–1571
Simone MI, Soengas R, Newton CR, Watkin DJ, Fleet GWJ (2005) Branched tetrahydrofuran α,α-disubstituted-δ-sugar amino acid scaffolds from branched sugar lactones: a new family of foldamers? Tetrahedron Lett 46:5761–5765
Suhara Y, Yamaguchi Y, Collins B, Schnaar RL, Yanagishita M, Hildreth JEK, Shimada I, Ichikawa Y (2002) Oligomers of glycamino acid. Bioorg Med Chem 10:1999–2013
Torres E, Gorrea E, Da Silva E, Nolis P, Branchadell V, Ortuño RM (2009) Prevalence of eight-membered hydrogen-bonded rings in some bis(cyclobutane) beta-dipeptides including residues with trans stereochemistry. Org Lett 11(11):2301–2304
Torres E, Gorrea E, Burusco KK, Da Silva E, Nolis P, Rúa F, Boussert S, Díez-Pérez I, Dannenberg S, Izquierdo S, Giralt E, Jaime C, Branchadell V, Ortuño RM (2010) Folding and self-assembling with beta-oligomers based on (1R,2S)-2-aminocyclobutane-1-carboxylic acid. Org Biomol Chem 8(3):564–575
Valeur E, Bradley M (2009) Amide bond formation: beyond the myth of coupling reagents. Chem Soc Rev 38:606–631
von Roedern EG, Kessler H (1994) A sugar amino acids as a novel peptidomimetic. Angew Chem Int Ed 33:687–689
Acknowledgements
Open access funding provided by Eötvös Loránd University (ELTE). The authors wish to thank Dóra K. Menyhárd and Ernő Keszei for consulting, Anita Kapros for the MS measurements, and András Láng and Dániel Horváth for helping 700 MHz NMR measurements. These research projects were supported by the European Union and the State of Hungary and co-financed by the European Regional Development Fund (VEKOP-2.3.2-16-2017-00014). This paper was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (V. Farkas) and MedInProt Grant Facilitating Access to Instruments from the Hungarian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: P. Meffre.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nagy, A., Goldschmidt Gőz, V., Pintér, I. et al. α/β-Chimera peptide synthesis with cyclic β-sugar amino acids: the efficient coupling protocol. Amino Acids 51, 669–678 (2019). https://doi.org/10.1007/s00726-019-02702-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-019-02702-9