
Abstract
Molecular characterization of neuraminidase (NA) gene of 25 influenza A(H3N2) virus isolates (2009-2013) archived at the Manipal Centre for Virus Research was carried out. The annual rate of amino acid substitutions in the N2 gene of influenza A(H3N2) virus isolates was 0.2-0.6%. Out of the 25 NA sequences analyzed, catalytic site mutations were observed in three isolates. Two of the mutations (D151G and E276G) were detected in functional catalytic residues, and an E227V mutation was detected in the framework residues. To the best of our knowledge, NA inhibitor resistance associated with the mutations E276G and E227V has not been reported. However, the mutation D151G, which is commonly associated with culturing of influenza A(H3N2) virus in Madin-Darby canine kidney (MDCK) cells, has been reported to result in a reduction in virus susceptibility to NA inhibitor drugs. Our study also detected mutations in antigenic residues. Some of the mutations (except D197G, K249E, A250T, S334C, and H347R/N) remained conserved in isolates of succeeding seasons. Antigenic residue mutations (D197G and S334C) have not been reported globally to date. The effect of these catalytic and antigenic mutant residues on drug and antibody binding was analyzed using three-dimensional structural analysis and biochemical assays. Antigenic variability of influenza A(H3N2) viruses is a major concern, and vaccine failures are mainly due to genetic variations in the HA gene. Our study documents that genetic changes in N2 occur at a slower rate, and this information is useful for the consideration and standardization of NA in influenza vaccines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antigenically variable pathogens cause much of the burden of infectious diseases today because these pathogens can escape from the immunity induced by either vaccination or previous infection. Influenza virus is a classic example of an antigenically variable pathogen [1]. Influenza viruses are enveloped RNA viruses with a single-stranded, negative-sense, segmented genome. The virus harbors two surface glycoproteins: hemagglutinin (HA) and neuraminidase (NA) [2]. The segmented nature of the virus facilitates exchange of entire genes between different influenza viruses during co-infection in a host, resulting in new strains with pandemic potential. These pandemic viruses later establish themselves as seasonal influenza viruses and cause annual epidemics [3]. In 1968, a new influenza A(H3N2) virus emerged and caused the Hong Kong influenza pandemic [4]. Even after four decades, this virus still remains a major cause of epidemics worldwide.
The World Health Organization (WHO) conducts meetings twice annually to review the global epidemiological situation of influenza and recommend new vaccine strains based on available data. Influenza vaccines are generally trivalent, consisting of a mixture of the influenza A and B virus strains most likely to circulate in the upcoming season (http://www.who.int/biologicals/vaccines/influenza/en/). The Centers for Disease Control and Prevention (CDC) conducts studies throughout each influenza season to determine vaccine effectiveness (http://www.cdc.gov/flu/news/flu-activity-expands.htm). Several studies have reported a reduction in vaccine effectiveness due to circulation of genetically drifted influenza A(H3N2) virus (http://www.cdc.gov/flu/news/flu-activity-expands.htm; emergency.cdc.gov/han/han00374.asp). Vaccine failures are mainly due to genetic variations in the HA gene, and therefore, current vaccines standardized primarily on the basis of HA protein content have reduced effectiveness [5]. Hence, studies on the genetic variability of the NA gene are important. In a previous study, we conducted a detailed genetic analysis of the NA gene of influenza A(H1N1)pdm09 virus isolates (2009-2012) [6]. The aim of this study was to investigate the genetic variation within the NA gene of influenza A(H3N2) virus isolates (2009-2013) archived at the Manipal Centre for Virus Research (MCVR), Manipal University. NA sequences were analyzed in comparison to reference vaccine strains to detect mutations in catalytic and antigenic-site residues. The effect of the mutations on drug and antibody binding was determined by performing structural and biochemical studies.
Materials and methods
The methodology followed was as described in our study on the genetic analysis of the NA gene of influenza A(H1N1)pdm09 isolates [6]. A brief description of the methodology is given below.
Virus isolates
The clinical samples used were received as part of the Influenza Surveillance Network Program from Southwest Indian states and were tested by real-time reverse transcription polymerase chain reaction (RT-PCR) using the WHO protocol (http://www.who.int/csr/…/WHO_Diagnostic_RecommendationsH1N1_2009). Virus from positive samples with Ct values below 30 were propagated and isolated in MDCK cells. A hemagglutination assay was performed on positive cultures, and the viruses were harvested and stored at -80 °C. A total of 25 archived isolates (passage no. 0) were selected randomly from the 2009-2013 seasons (a minimum of two isolates per year) and used in the present study.
The CDC neuraminidase inhibitor susceptibility reference virus panel used for the NAI assay was obtained from the Influenza Reagent Resource, Manassas, VA (http://www.influenzareagentresource.org/).
RT-PCR for amplification of the full-length NA gene of influenza A(H3N2) viruses
Primers that amplify the NA gene of influenza A(H3N2) viruses were selected from published literature [7]. A QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) was used for extraction of RNA from the positive culture supernatants. cDNA synthesis and PCR amplification were performed using a OneStep RT-PCR Kit (QIAGEN, Hilden, Germany). Amplification products were analyzed in a 0.8% agarose gel and were further purified using a PCR Purification Kit (QIAGEN, Hilden, Germany).
DNA sequencing
Purified PCR products were cloned using an InsTAcloneTM PCR Cloning Kit (Thermo Scientific, Vilnius, Lithuania). Plasmid DNA was isolated using a QIAprep Spin Miniprep Kit (QIAGEN, Hilden, Germany) and sequenced using a BigDye Terminator (version 3.1) Cycle Sequencing Kit (Applied Biosystems, Foster, CA, USA). A 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) was used for analyzing purified sequences. In order to eliminate sequencing errors, each isolate was sequenced in triplicate using both M13 forward and M13 reverse sequencing primers.
Sequence analysis
For sequence analysis, NA sequences of WHO influenza A(H3N2) vaccine strains (2003-2015) from the Influenza Research Database (http://www.fludb.org/) were used as reference strains. The NA nucleotide sequences from this study have been deposited in the GenBank database (Table 1). The MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) program was used for multiple sequence alignment, and a phylogenetic tree was constructed by the neighbor-joining method with bootstrap analysis (n = 1000), using Molecular Evolutionary Genetic Analysis (MEGA 5.2) software.
Structural modeling
Swiss-Model (http://swissmodel.expasy.org/), an automated protein structure homology modeling program [8] was used for building homology models. Protein Data Bank (PDB) files from modeling results were visualized using PyMOL software (http://www.pymol.org). Inter-chain contacts between the antibody/drug and the NA protein were analyzed using contact software available in the CCP4i suite [9].
Neuraminidase assay (NA) and neuraminidase inhibition (NAI) assay
The NA activity of all of the mutant isolates was determined, followed by an NAI assay in the presence of a drug for catalytic residue mutant isolates or an NAI antibody assay for antigenic residue mutant isolates. The NA and NAI assays were both performed in duplicate.
Oseltamivir carboxylate (RO0640802-002) was supplied by F. Hoffmann-LA Roche Ltd, Basel, Switzerland. The isolates with mutated residues were passaged until the HA titre reached 16 or greater. To determine the NA activity, a twofold dilution of the virus was incubated with substrate 20-(4-methylumbelliferyl)-a-D-N-acetylneuraminic acid (MUNANA; Sigma, MO, USA), and results were recorded using a fluorescent reader (FLx800, Biotek, Winooski, VT, USA). The standard virus dose for the NAI assay was calculated according to the virus dilution at which NA activity was equivalent to the fluorescence of 10 μM 4-methyllumbelliferon sodium salt (Sigma, MO, US).
In the NAI assay, the concentration of drug required to inhibit the NA activity at the standard virus dose by 50% (IC50) was determined using oseltamivir (1000, 100, 10, 1, 0.1, and 0.01 nM). Results were analyzed according to the standard operating procedure (SOP) provided by the Health Protection Agency (HPA), London (http://www.isirv.org/site/index.php/special-interest-groups/antiviral-group-home). The mean and standard deviation were calculated for duplicate tests, and the results were analysed using the Welch t-test.
Neuraminidase inhibition antibody assay
An NAI antibody assay was performed using pooled convalescent serum samples from patients who tested positive for influenza A(H3N2) virus. The standard concentration of virus was mixed with a twofold dilution of serum, and the experiment was performed according to the SOP provided by HPA, London (http://www.isirv.org/site/index.php/special-interest-groups/antiviral-group-home ).
An enzyme-linked lectin assay to measure NAI antibody titres was performed according to the protocol standardised by Couzens et al. [10] Twofold dilutions of pooled serum sample (heat treated) were added to a fetuin-coated plate. An equal volume of the standardised mutant virus isolate was then added and incubated at 37 °C for 16-18 h. After incubation, peanut agglutinin conjugated to horseradish peroxidase (Sigma, MO, USA) was added, and the plates were incubated at room temperature for 2 h. o-phenylenediamine dihydrochloride was then added as a substrate to all wells, and the plate was incubated in the dark for 10 min. The reaction was stopped by adding 1 N H2SO4, and the plates were read at 490 nm. The highest dilution that resulted in at least 50% inhibition of the maximum signal was determined.
Results
Molecular evolution of the NA gene
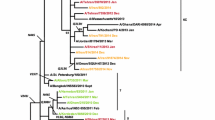
The nucleotide sequence of the NA gene was determined for 25 influenza A(H3N2) virus isolates, including five isolates from 2009, eight isolates from 2010, six isolates from 2011, two isolates from 2012, and three isolates from 2013. The annual rate of amino acid substitution in isolates from the 2009-2010, 2010-2011, 2011-2012, and 2012-2013 seasons was 0.5%, 0.4%, 0.2%, and 0.6%, respectively. Phylogenetic analysis of NA protein sequences of influenza A(H3N2) virus isolates showed that isolates from the 2009 season formed two subclades consisting of two and three isolates each (Fig. 1). All of the isolates from the 2010 season clustered in a single subclade except isolate A/Calicut/MCVR6154/2010(H3N2). Out of the five isolates from the 2011 season, four isolates clustered in a phylogenetic subclade, and isolate A/Malappuram/MCVR7610/2011(H3N2) clustered with vaccine strain A/Victoria/361/2011(H3N2) in a separate subclade. The two isolates from the 2012 season formed a subclade in the phylogenetic tree. Out of the four isolates from the 2013 season, three isolates clustered together, and isolate A/Calicut/MCVRAA2095/2013(H3N2) formed a subclade with vaccine strains A/Texas/50/2012(H3N2) and A/Switzerland/9715293/2013(H3N2). The amino acid sequence similarity between NA sequences from the 2009, 2010 and 2011 seasons and A/Perth/16/2009(H3N2), vaccine strain for 2010-2011 and 2011-2012 seasons, was 99.06%, 98.6%, and 98.2%, respectively. The amino acid sequence similarity between sequences from 2012, 2013 and A/Victoria/361/2011(H3N2), the vaccine strain for 2012-2013 and 2013-2014, was 98.82% and 98.6%, respectively. The NA sequences of all 25 influenza A(H3N2) virus isolates were analyzed in comparison to the reference vaccine strain A/Perth/16/2009(H3N2) and the results are summarized in Table 2. The non-synonymous mutations S367N+K369T+I464L were found to appear towards the end of the 2009 season and were later conserved in all of the isolates of the succeeding seasons. The two isolates from the 2009 season lacking S367N+K369T+I464L substitutions possessed I26T+Y40C substitutions. The L81P substitution was found in all 2011-2013 isolates. All isolates from the 2010 season possessed the R400K substitution. The N402D substitution was observed in all isolates from 2011-2013 and one isolate from the 2010 season. The amino acid substitutions D93G+Y155F+D251V+S315G were detected in three isolates, and only the D93G substitution was detected in one isolate from the 2013 season. The Y155F substitution was also detected in some of the 2012 isolates.

Phylogenetic analysis of NA gene sequences of influenza A(H3N2) virus isolates. The phylogenetic tree was generated using the neighbour-joining method (bootstrap values = 1000) with a distance scale of 0.005. Reference vaccine strains from the 2003-2015 influenza seasons are indicated by blue colored square boxes
Variations in NA catalytic site residues
The catalytic site of the NA protein consists of eight functional residues (Arg118, Asp151, Arg152, Arg224, Glu276, Arg292, Arg371 and Tyr406) and 11 framework residues (Glu119, Arg156, Trp178, Ser179, Asp/Asn 198, Ile222, Glu227, His274, Glu277, Asn 294, and Glu425), which are conserved among all influenza A and B isolates [11]. Out of the 25 NA sequences analyzed, catalytic site mutations were observed in three isolates (Table 3). Two of the mutant residues (D151G and E276G) are functional residues, whereas the mutant residue E227V is one of the framework residues. NA inhibitor drug resistance associated with the mutation D151G has been documented [12]. However, the mutations E276G and E227V have not been reported to be associated with reduction in virus susceptibility to NA inhibitor drugs.
Structural and biochemical analysis of the NA-D151G mutant isolate
Amino acid residue D151 has been suggested to be involved in the protonation of the sialic acid substrate during the binding of the NA [13]. Crystal structure studies of an NA-D151G mutant in complex with oseltamivir carboxylate have documented that the mutation results in lack of interaction with the drug due to the loss of a hydrogen bond (H-bond) [14]. D151 makes an H-bond (2.8Å and 3.7Å) with the drug (oseltamivir), and the mutation D151G results in the loss of the H-bond, destabilizing the drug binding. In our study, isolate A/Thrissur/MCVR4244/2010(H3N2) was found to contain the D151G mutation. For analysis, the three-dimensional (3D) model of the NA-D151G mutant isolate (template PDB: 4GZT) was superimposed onto the 3D homology model of vaccine strain A/Perth/16/2009(H3N2) built by Swiss-Model in the automated mode using the template PDB: 4GZP. The results of the structural analysis were in agreement with the published crystallography results [14], and the mutant residue was lacking the side chain that forms the contact (H-bond) with oseltamivir (Fig. 2a). The NA activity of the isolate A/Thrissur/MCVR4244/2010(H3N2) determined in the presence of substrate 20-(4-methylumbelliferyl)-a-D-N-acetylneuraminic acid (MUNANA; Sigma, MO, US) was unaffected by the mutation D151G, and the isolate was also sensitive to oseltamivir (Table 4 and Fig. 3b).

Structural models of influenza A(H3N2) virus isolate NA protein-oseltamivir complexes. The 3D model of the NA-D151G mutant isolate (template PDB: 4GZT), NA-E276G (template PDB: 4GZP), and NA-E227V (template PDB: 4GZP) were superimposed onto the 3D model of vaccine strain A/Perth/16/2009(H3N2) (template PDB: 4GZP) Superimposed structural models of mutant N2 virus isolates [a: D151G (magenta); b: E276G (yellow); c: E227V (cyan)] and wild-type vaccine strain (green) are shown as sticks bound to oseltamivir (green). Dotted lines indicate hydrogen bonds (Å)

Neuraminidase and neuraminidase inhibition assay of NA proteins with mutant catalytic residues. Analysis curves of NA and NAI assays by the point-to-point method. The x-axis represents virus dilution (a) or the concentration of oseltamivir (b). The y-axis represents relative fluorescence units (RFU), and duplicate curves are represented in straight and dotted lines. Horizontal lines indicate RFU cutoff (a) or IC50 cutoff values (duplicate; b)
Structural and biochemical analysis of the NA-E276G mutant isolate
Residues E276 and R224 of the NA protein re-orient to form a salt bridge that is essential to accommodate the bulky hydrophobic pentyl ether side chain of oseltamivir [15]. In our study, the E276G mutation was detected in isolate A/Calicut/MCVR7104/2011(H3N2). Although no structural model for E276G was available, a study on recombinant viruses expressing NA protein with substitutions at position 276 has been published [16]. A 3D homology model of the mutant with the E276G substitution was built using the Swiss-Model automated modeling tool (template PDB: 4GZP). Analysis of superimposed 3D models of the NA-E276G mutant isolate and the vaccine strain A/Perth/16/2009(H3N2) built by Swiss-Model in the automated mode using the template PDB: 4GZP revealed that the mutation of E276 to G276 resulted in the loss of a salt-bridge with residue R224 that is essential for accommodating oseltamivir (Fig. 2b). Biochemical analysis demonstrated that the NA activity and sensitivity of the isolate to oseltamivir were unaffected by this mutation (Table 4 and Fig. 3b).
Structural and biochemical analysis of the NA-E227V mutant isolate
Residue E227 plays an important role in stabilizing the NA active site by interacting with other residues as well with the sialic acid substrate [16]. To the best of our knowledge, this mutation had not been detected earlier, and therefore crystallography studies on an NA-E227V mutant protein have not been done. However, studies on recombinant virus expressing an NA protein with a substitution at position 227 have been reported [16]. The 3D NA protein model of isolate A/Trivandrum/MCVR2977/2010(H3N2) was built using the Swiss-Model automated modeling tool (template PDB: 4GZP). The 3D model of NA-E227V mutant was superimposed onto the 3D model of vaccine strain A/Perth/16/2009(H3N2), which was built using the template PDB: 4GZP. Structural analysis revealed that residue E227 interacts with oseltamivir (4.9 Å) and residue E277 (5 Å) through a weak H-bond. Mutant residue V227 lost contact with oseltamivir as well as residue E277, and the distance between V227 to E277 increased to 6.8 Å (Fig. 2c). Biochemical assays showed that the NA activity of the isolate was unaffected, but the IC50 was increased by approximately fourfold in comparison to the standard wild-type strain A/Washington/01/2007(H3N2) (Table 4 and Fig. 3b).
Variations in NA gene antigenic site residues
The crystal structure of the NA protein of influenza virus A/Memphis/31/98(H3N2) in complex with the Fab region of monoclonal antibody Mem5 (PDB: 2AEP) has been determined at 2.1 Å resolution [17]. For analysis of NA antigenic site residues, experimentally determined B-cell epitopes from previously reported studies as well as from the Immune Epitope Database and Analysis Resource (IEDB) were used (Table 5).
Analysis of NA sequences in comparison with vaccine strain A/Perth/16/2009(H3N2) showed that antigenic residue mutations were present in all isolates (Table 6). However, some of the antigenic changes, except mutations D197G, K249E, A250T, S334C, and H347R/N, were found in all of the strains isolated in a particular year, and also, some changes were conserved in isolates of succeeding seasons. The NA sequence of vaccine strain A/Perth/16/2009 (H3N2) was also compared with the NA sequence of isolate A/Memphis/31/98(H3N2), as some of the epitopes listed in Table 5 were determined by studies of the crystal structure of the 1998 strain in complex with antibody. Sequence analysis showed mutations D147N, H150R, H197D, E199K, K221E, R249K, F370S and Q431E in the vaccine strain.
Structural analysis of NA gene antigenic residue mutations
For analysis, the 3D structure of vaccine strain A/Perth/16/2009(H3N2) was superimposed onto the crystal structure of the 1998 strain and visualized using PyMOL. Inter-chain contact analysis using the CCP4i suite showed that antibody heavy chain residues 31, 33, 52, 53, 56, 96, 97, 98, 99, 100 and light chain residues 31, 32, 49, 50, 52, 53, 92, 94 interact with the NA protein. Structural analysis showed that the mutations H150R, H197D, E199K, K221E and R249K in 2009 vaccine strain altered the interactions between the NA protein and antibody. The mutation H150R resulted in the loss of an H-bond (3.3 Å) with heavy chain residue Asp31 of the Mem5 antibody. The H197D mutation resulted in a loss of interaction with the heavy-chain residue Thr56 and also increased the distance of H-bonding with residue Arg52 (4.8 Å and 4.4 Å). Similarly, the mutations E199K, K221E and R249K altered interactions with antibody residues Tyr33 (7.1 Å), Thr99 (5.3 Å) and Ser52 (6.9 Å), respectively (Fig. 4a, b).

NA protein of influenza A(H3N2) virus in complex with 1998 antibody Mem5. (a) The NA protein of the A/Memphis/31/98 strain (green) in complex with the Mem5 antibody (magenta; PDB: 2AEP) is shown as a cartoon. Residues of the NA protein and antibody that interact to form a complex are shown as sticks. (b) NA protein of vaccine strain A/Perth/16/2009(H3N2) (template PDB: 4GZP) with mutated residues shown in sticks (yellow). (c) Mutant isolate A/Calicut /MCVR5938/2010(H3N2) with D197G mutation (blue). (d) Mutant isolate A/Calicut/MCVRAA2090/2013(H3N2) K249E and A250T mutations (grey)
Three-dimensional structures of mutant NA proteins were superimposed onto the 3D structure of vaccine strain A/Perth/16/2009 (H3N2) in order to determine whether the antigenic residue mutations in isolates from the 2009-2013 seasons further affected the binding of the Mem5 antibody. Out of the five antigenic residue mutants detected in our study, two residues (S334 and H347) do not interact with the antibody against A/Memphis/31/98(H3N2), and thus the effect of these mutations on antibody binding could not be determined by structural analysis. Structural analysis of the NA-D197G mutant showed that the mutation resulted in a complete loss of interaction with antibody residue Arg52 (Fig. 4c). However, structural analysis of the NA protein of isolate A/Calicut/MCVRAA2090/2013(H3N2) showed that the mutations K249E and A250T may not affect antibody binding (Fig. 4d).
An NA assay performed in presence of the substrate MUNANA demonstrated that antigenic residue mutations did not affect the NA activity of three isolates. The results of an NAI antibody assay that was performed to test the effect of these mutations on antibody binding were negative. since no reduction in NA activity was observed in the presence of pooled serum containing antibodies against influenza A(H3N2) virus. The results of an NA assay in the presence of the large substrate fetuin were unaffected in three isolates, similar to the NA assay in the presence of MUNANA. The NA activity was very low for isolate A/Calicut/MCVRAA2090/2013(H3N2) with both of the substrates, and repeated passaging did not increase the NA activity, and thus the NAI assay could not be performed. An NAI assay with fetuin as substrate showed reduction in NA activity in the presence of pooled serum containing antibodies against influenza A(H3N2) virus, indicating that the antigenic mutations H347R/N and D197G might not affect antibody binding.
Discussion
The neuraminidase protein of influenza viruses is the second most abundant major surface glycoprotein and contributes to virus replication by its sialidase activity, which facilitates spread of infection among host cells. NA is not only a major target for influenza antivirals but also has gained significant importance as an antigen that needs to be standardized in influenza vaccines [18]. Continuous monitoring of variations in the NA gene is essential to detect mutations that might contribute to drug resistance or affect antibody binding.
In this study, detailed genetic analysis of the NA gene of 25 archived influenza A(H3N2) virus isolates (2009-2013) was carried out. The annual rate of amino acid substitutions in the N2 gene of influenza A(H3N2) virus isolates was 0.2-0.6%, which was similar to the substitution rates (0.3-0.7%) of the N1 gene of influenza A(H1N1)pdm09 virus isolates [6] during the same seasons. However, studies have reported that amino acid substitutions in H3 genes are higher than in H1 genes [19]. The mutations S367N+K369T, and I464L occured in almost 88% and 84% of influenza A(H3N2) virus isolates, respectively. Mutations at 367SKI369 have been reported to create a potential glycosylation site that might affect antigenic and other properties (flusurver.bii.a-star.edu.sg/). The mutations I26T and Y40C were detected in isolates from the 2009 season that lacked the S67N+K369T+I464L mutations. Byarugaba et al., reported that 2009 influenza A(H3N2) virus isolates differed from the 2008 isolates by the mutations I26T and Y40C [20]. The significance of these mutations is unknown, since the mutations were not conserved in isolates of succeeding seasons. Previous reports suggest that mutations in the HA gene along with the NA substitutions L81P and D93G, which occurred around 2011, might have given rise to influenza A(H3N2) virus epidemics in 2011 [21]. In our study, the L81P mutation was detected in 2011 isolates, whereas the D93G mutation was detected in 2013 isolates. In the Indian subcontinent during the 2011 and 2013 season, influenza A(H3N2) viruses were the dominating circulating strains of influenza viruses, and thus the mutations L81P and D93G might have contributed to antigenic variation and predominance. The mutations D93G+Y155F+D251 were detected along with S315G mutation in our study. However, previous study showed that the D93G+Y155F+D251V combination started to arise in May 2013, and these mutations did not alter susceptibility of NA to inhibitor drugs [22]. The mutation S315G detected in our study has been reported in influenza A(H3N2) viruses circulating in northern India during 2013 season [23]. Detailed analysis of NA gene sequences from India might provide better insights if such substitutions were more common in Indian isolates.
Our study detected three mutations (D151G, E227V, and E276G) in the conserved catalytic site of influenza A(H3N2) virus isolates. The mutation D151G in the NA gene is commonly associated with passaging of influenza A(H3N2) viruses in the MDCK cell lines [24]. The D151G mutation has been reported to increase the IC50 value for zanamivir [25], reduce enzyme activity, and also induce receptor-binding ability in the NA gene [14]. To the best of our knowledge, the mutations E227V and E276G have not been reported in NA sequences to date. In our study, the NA activity and oseltamivir IC50 value of isolate A/Thrissur/MCVR4244/2010(H3N2) with the D151G mutation was comparable with the reference wild-type strains. A study by Lee et al. reported that the cell-culture-induced mutation D151G was a result of adaptation of the virus, and repeated passaging gradually increased the proportion of mutant isolates [12]. Isolates used in our study had not been passaged, and sequencing results showed a background of wild-type amino acid (D151) along with the G151 mutation, suggesting the presence of a mixed population of wild-type and mutant virus. The presence of wild-type virus might have masked the NA activity. However, further passaging of the isolates for selection of mutants and re-testing with the drug zanamivir may be useful. Adhiambo et al. reported that increased levels of resistance to neuraminidase inhibitors were detected when the D151G mutation was present in combination with the H274Y mutation; however, in our study, none of the isolates were found to contain the H274Y mutation [26].
Of the three catalytic residue mutant isolates, two isolates (NA-D151G and NA-E276G) were sensitive to oseltamivir, whereas the isolate A/Trivandrum/MCVR2977/2010(H3N2), with NA-E227V mutation, had a slightly higher (approximately fourfold) IC50 value. Residues 222-230 are universally conserved among all influenza A and B viruses, since E227 forms a part of a conserved peptide, mutation at this position might affect enzyme activity and replication [27]. A study of recombinant viruses expressing NA-E227D showed that the growth of recombinant virus was affected, and thus the NA activity and NA inhibitor sensitivity could not be assessed. However, structural analysis revealed that E227 possibly interacts with the C4 amino group of oseltamivir and is therefore under selection pressure of NA inhibitors [28]. The isolate with the E227V mutation detected in our study showed an approximately fourfold higher IC50 value in comparison to the reference wild-type strain. Interestingly, our study on influenza A(H1N1)pdm09 viruses also detected a mutation at position 228 (N1 numbering). The influenza A(H1N1)pdm09 isolate had an approximately twofold higher IC50 value in comparison to the reference wild-type strain A/California/12/2012(H1N1)pdm09 [6]. Substitution of E227 with less-conserved residues such as G227/V227 has been reported to result in reversion to the wild-type residue E227 [29]. The isolate A/Trivandrum/MCVR2977/2010(H3N2), which contains an E227V mutation, was re-sequenced after passaging twice in MDCK cells. Repeated passaging resulted in reversion to the wild-type residue (E227; data not shown), suggesting that the mutation E227V is not conserved and is thus not a major concern during natural circulation. However, in a study by Ho et al., substitutions with the more-conserved residue D227 were found to reduce sensitivity to neuraminidase inhibitors [29]. Monitoring of residue E227 is important for tracking the emergence of resistance among naturally circulating viruses.
We report mutations in nine antigenic residues of the influenza A(H3N2) virus NA gene. Some of the mutations (except D197G, K249E, A250T, S334C, and H347R/N) were conserved in all isolates or were detected in all isolates of a particular season. Two of the antigenic residue mutations (D197G and S334C) have not been reported globally to date (flusurver.bii.a-star.edu.sg/). Interestingly, the isolate A/Calicut/MCVRAA2090/2013(H3N2) had the maximum number of non-synonymous mutations (14) and antigenic residue mutations (6). Crystallography studies of the N2 protein of currently circulating influenza A(H3N2) virus isolates in complex with antibody have not been done so far. Thus, the sequence and 3D model (NA protein) of vaccine strain A/Perth/16/2009(H3N2) were analyzed in comparison to the 3D model of A/Memphis/31/98(H3N2) in complex with the Fab region of monoclonal antibody Mem5. Results of structural analysis of the vaccine strain superimposed onto the crystal structure of the 1998 strain complexed with antibody Mem5 were in agreement with a previous study [30]. However, due to the unavailability of antibody crystal structures for currently circulating influenza A(H3N2) viruses, the actual effect of antigenic residue mutations (D197G, K249E, A250T, S334C, and H347R/N) on antibody binding could not be analyzed structurally. Structural analysis was performed by superimposing mutant NA proteins onto that of the vaccine strain to determine whether the antigenic residue mutations in isolates of the 2009-2013 seasons further affected the binding of Mem5 antibody. The effects of these mutations on antibody binding were analyzed by NAI antibody assay using a pooled serum containing antibodies against influenza A(H3N2) virus and MUNANA as substrate. No reduction in NA activity was observed despite high titers of HA antibody in the pooled serum (1 in 512), indicating that the antibody-binding residues are not near the catalytic site [31]. An NAI antibody assay with a larger substrate such as fetuin may serve as a useful tool for analyzing the effect of these mutations [31]. The NAI antibody assay was repeated using the large substrate fetuin and the results indicated that the antigenic mutations H347R/N and D197G might not affect antibody binding. The NA activity of isolate A/Calicut/MCVRAA2090/2013(H3N2) was very low, and the NAI assay therefore could not be performed. A future detailed study on the NA protein structure of isolate A/Calicut/ MCVRAA2090/2013(H3N2) might provide insights into the reason for the decreased NA activity and the effect of the presence of 14 non-synonymous mutations.
The increasing number of genetically drifted influenza A(H3N2) viruses emerging each year imposes challenges on drug and vaccine design. Our study suggests that genetic variations in the N2 gene occur at a lower rate and are comparable with the variations in the N1 gene, and thus, NA may serve as a significant tool for development of newer drugs and vaccines. However, it remains to be seen whether the use of drugs targeting NA or NA-based vaccines will result in further selection pressure on influenza viruses.
References
Smith DJ, Lapedes AS, de Jong JC et al (2004) Mapping the antigenic and genetic evolution of influenza virus. Science 305:371–376. doi:10.1126/science.1097211
Knipe DM, Howley PM (2013) Fields virology, 6th edn. Wolters Kluwer/Lippincott Williams & Wilkins Health, Philadelphia
Westgeest KB, Russell CA, Lin X et al (2014) Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J Virol 88:2844–2857. doi:10.1128/JVI.02163-13
Kilbourne ED (2006) Influenza pandemics of the 20th century. Emerg Infect Dis 12:9
Jagadesh A, Salam AAA, Mudgal PP, Arunkumar G (2016) Influenza virus neuraminidase (NA): a target for antivirals and vaccines. Arch Virol. doi:10.1007/s00705-016-2907-7
Jagadesh A, Salam AAA, Zadeh VR, Arunkumar G (2016) Genetic analysis of neuraminidase gene of influenza A(H1N1)pdm09 virus circulating in Southwest India from 2009 to 2012. J Med Virol. doi:10.1002/jmv.24625
Tang JW, Ngai KLK, Lam WY, Chan PKS (2008) Seasonality of influenza A(H3N2) virus: a Hong Kong perspective (1997–2006). PLoS One 3:e2768. doi:10.1371/journal.pone.0002768
Kiefer F, Arnold K, Künzli M et al (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res 37:D387–D392. doi:10.1093/nar/gkn750
Winn MD, Ballard CC, Cowtan KD et al (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi:10.1107/S0907444910045749
Couzens L, Gao J, Westgeest K et al (2014) An optimized enzyme-linked lectin assay to measure influenza A virus neuraminidase inhibition antibody titers in human sera. J Virol Methods 210:7–14. doi:10.1016/j.jviromet.2014.09.003
Xu X, Zhu X, Dwek RA et al (2008) Structural characterization of the 1918 influenza Virus H1N1 neuraminidase. J Virol 82:10493–10501. doi:10.1128/JVI.00959-08
Lee HK, Tang JW-T, Kong DH-L et al (2013) Comparison of mutation patterns in full-genome A/H3N2 influenza sequences obtained directly from clinical samples and the same samples after a single MDCK passage. PLoS One 8:e79252
Varghese JN, McKimm-Breschkin JL, Caldwell JB et al (1992) The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins Struct Funct Bioinforma 14:327–332
Zhu X, McBride R, Nycholat CM et al (2012) Influenza virus neuraminidases with reduced enzymatic activity that avidly bind sialic acid receptors. J Virol 86:13371–13383. doi:10.1128/JVI.01426-12
Smith BJ, McKimm-Breshkin JL, McDonald M et al (2002) Structural studies of the resistance of influenza virus neuramindase to inhibitors. J Med Chem 45:2207–2212. doi:10.1021/jm010528u
Yen H-L, Hoffmann E, Taylor G et al (2006) Importance of neuraminidase active-site residues to the neuraminidase inhibitor resistance of influenza viruses. J Virol 80:8787–8795. doi:10.1128/JVI.00477-06
Venkatramani L, Bochkareva E, Lee JT et al (2006) An epidemiologically significant epitope of a 1998 human influenza virus neuraminidase forms a highly hydrated interface in the NA-antibody complex. J Mol Biol 356:651–663. doi:10.1016/j.jmb.2005.11.061
Eichelberger MC, Wan H (2014) Influenza neuraminidase as a vaccine antigen. In: Oldstone MBA, Compans RW (eds) Influenza Pathog. Control, vol. II. Springer International Publishing, Cham, pp 275–299
Klein EY, Serohijos AWR, Choi J-M et al (2014) Influenza A H1N1 pandemic strain evolution: divergence and the potential for antigenic drift variants. PLoS One. doi:10.1371/journal.pone.0093632
Byarugaba DK, Ducatez MF, Erima B et al (2011) Molecular Epidemiology of influenza A/H3N2 viruses circulating in uganda. PLoS One. doi:10.1371/journal.pone.0027803
Zhong J, Liang L, Huang P et al (2013) Genetic mutations in influenza H3N2 viruses from a 2012 epidemic in Southern China. Virol J 10:345. doi:10.1186/1743-422X-10-345
Lee HK, Tang JW-T, Loh TP et al (2015) Molecular surveillance of antiviral drug resistance of influenza A/H3N2 virus in Singapore, 2009–2013. PLoS One. doi:10.1371/journal.pone.0117822
Jain A, Dangi T, Jain B et al (2015) Genetic changes in influenza A(H3N2) viruses circulating during 2011 to 2013 in northern India (Lucknow). J Med Virol 87:1268–1275. doi:10.1002/jmv.24096
Zaraket H, Kondo H, Hibino A et al (2016) Full genome characterization of human influenza A/H3N2 isolates from Asian Countries reveals a rare amantadine resistance-conferring mutation and novel PB1-F2 polymorphisms. Front Microbiol. doi:10.3389/fmicb.2016.00262
Mishin VP, Sleeman K, Levine M et al (2014) The effect of the MDCK cell selected neuraminidase D151G mutation on the drug susceptibility assessment of influenza A(H3N2) viruses. Antiviral Res 101:93–96. doi:10.1016/j.antiviral.2013.11.001
Combinatorial Effect of Two Framework Mutations (E119V and I222L) in the neuraminidase active site of H3N2 Influenza Virus on Resistance to Oseltamivir. https://www.researchgate.net/publication/50595812_Combinatorial_Effect_of_Two_Framework_Mutations_E119V_and_I222L_in_the_Neuraminidase_Active_Site_of_H3N2_Influenza_Virus_on_Resistance_to_Oseltamivir. Accessed 8 Feb 2017
Gravel C, Li C, Wang J et al (2010) Qualitative and quantitative analyses of virtually all subtypes of influenza A and B viral neuraminidases using antibodies targeting the universally conserved sequences. Vaccine 28:5774–5784. doi:10.1016/j.vaccine.2010.06.075
Yen H-L, Hoffmann E, Taylor G et al (2006) Importance of neuraminidase active-site residues to the neuraminidase inhibitor resistance of influenza viruses. J Virol 80:8787–8795. doi:10.1128/JVI.00477-06
Ho H, Hurt A, Mosse J, Barr I (2007) Neuraminidase inhibitor drug susceptibility differs between influenza N1 and N2 neuraminidase following mutagenesis of two conserved residues. Antiviral Res 76:263–266. doi:10.1016/j.antiviral.2007.08.002
Shil P, Chavan SS, Cherian SS (2011) Antigenic variability in Neuraminidase protein of Influenza A/H3N2 vaccine strains (1968–2009). Bioinformation 7:76–81
Marcelin G, Sandbulte MR, Webby RJ (2012) Contribution of antibody production against neuraminidase to the protection afforded by influenza vaccines. Rev Med Virol 22:267–279. doi:10.1002/rmv.1713
Colman PM, Varghese JN, Laver WG (1983) Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 303:41–44. doi:10.1038/303041a0
Acknowledgements
We thank the Indian Council of Medical Research (ICMR) for funding the research work (File No. 5/8/7/15/2010/ECD-1). We thank Dr TMA Endowment Chair in Translational Virology, Manipal University, for their support. We thank the Department of Science and Technology, Science and Engineering Research Board, India, for supporting the computational facilities through Grant SB/FT/LS-273/2012. We thank the Influenza Reagent Resource (IRR), Manassas, VA, for supplying the CDC Neuraminidase Inhibitor Susceptibility Reference Virus Panel (version 2.0); F. Hoffmann-LA Roche Ltd, Basel, Switzerland, for supplying oseltamivir carboxylate; and the Antiviral Group of the International Society for Influenza and Other Respiratory Viruses for providing the downloadable standard operating procedure and the result template for the NA and NAI assay. We thank the Department of Pharmacology, MCOPS, Manipal University, Manipal,for providing access to the fluorescence reader.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical clearance
The study was initiated after approval was received from the Institutional Ethics Committee (IEC), Manipal University, Manipal (Certificate IEC243/2013).
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Jagadesh, A., Salam, A.A.A., Zadeh, V.R. et al. Molecular characterization of neuraminidase genes of influenza A(H3N2) viruses circulating in Southwest India from 2009 to 2013. Arch Virol 162, 1887–1902 (2017). https://doi.org/10.1007/s00705-017-3306-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3306-4