
Abstract
Molecular classification of cancer has entered clinical routine to inform diagnosis, prognosis, and treatment decisions. At the same time, new tumor entities have been identified that cannot be defined histologically. For central nervous system tumors, the current World Health Organization classification explicitly demands molecular testing, e.g., for 1p/19q-codeletion or IDH mutations, to make an integrated histomolecular diagnosis. However, a plethora of sophisticated technologies is currently needed to assess different genomic and epigenomic alterations and turnaround times are in the range of weeks, which makes standardized and widespread implementation difficult and hinders timely decision making. Here, we explored the potential of a pocket-size nanopore sequencing device for multimodal and rapid molecular diagnostics of cancer. Low-pass whole genome sequencing was used to simultaneously generate copy number (CN) and methylation profiles from native tumor DNA in the same sequencing run. Single nucleotide variants in IDH1, IDH2, TP53, H3F3A, and the TERT promoter region were identified using deep amplicon sequencing. Nanopore sequencing yielded ~0.1X genome coverage within 6 h and resulting CN and epigenetic profiles correlated well with matched microarray data. Diagnostically relevant alterations, such as 1p/19q codeletion, and focal amplifications could be recapitulated. Using ad hoc random forests, we could perform supervised pan-cancer classification to distinguish gliomas, medulloblastomas, and brain metastases of different primary sites. Single nucleotide variants in IDH1, IDH2, and H3F3A were identified using deep amplicon sequencing within minutes of sequencing. Detection of TP53 and TERT promoter mutations shows that sequencing of entire genes and GC-rich regions is feasible. Nanopore sequencing allows same-day detection of structural variants, point mutations, and methylation profiling using a single device with negligible capital cost. It outperforms hybridization-based and current sequencing technologies with respect to time to diagnosis and required laboratory equipment and expertise, aiming to make precision medicine possible for every cancer patient, even in resource-restricted settings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Histomolecular classification of brain tumors has entered clinical routine diagnostics as the current World Health Organization (WHO) classification explicitly demands histological findings to be refined by molecular testing [20]. Thus, pathologists rely on timely and accurate molecular testing to make an integrated diagnosis using both in situ methods and genetic information. However, high turnaround time of current implementations delays integrated diagnosis by weeks. In addition, targeted next-generation sequencing panels, microarray-based analysis of copy number (CN), and epigenetic alterations all provide high-quality data and aid in the diagnosis and therapeutic management of patients (i.e., stratification or identification of actionable targets or inclusion in clinical trials), but their high capital cost, demanding workflows and need for highly skilled personnel hinder their widespread use. Here, we demonstrate that real-time molecular genomics using nanopore sequencing is both fast and reliable to aid diagnosing cancer by unsupervised classification of CN and methylation profiles.
Nanopore sequencing interprets changes in ionic currents observed when single DNA molecules pass through a nanometer-size protein pore. This has led to the development of handheld size devices that allow sequencing outside of classical laboratory settings and even in the field [27]. While overall throughput currently lacks behind other deep sequencing technologies, nanopores allow read analysis in real-time and selective sequencing [19], both of which allow rapid generation of data. In addition, nanopores are able to discriminate not only the nucleotides of a strand of DNA but also single base modifications such as 5-methylation of cytosine [29, 35]. This allows concurrent analysis of sequence identity and methylation using native DNA.
Materials and methods
Experimental design
We performed a retrospective observational study for molecular characterization of diagnostically relevant genetic alterations using nanopore sequencing. Patients were recruited at the Pitié-Salpêtrière university hospital and have given informed consent for research use of tumor material, including genotyping. All tumor samples have been molecularly characterized previously using short-read exome sequencing, Sanger sequencing, SNP array, and/or genome-wide methylation microarray [14, 30].
Nanopore whole genome sequencing
DNA quality of fresh-frozen tumor tissue was determined using NanoDrop (Thermo Fisher Scientific) and samples were quantified using a QuantiFluor dsDNA assay (Promega, Madison, WI, USA). For whole genome sequencing, libraries were prepared using Rapid 1D Sequencing Kit (SQK-RAD001, SQK-RAD002, or SQK-RBK001, Oxford Nanopore Technologies, UK) following the manufacturer’s instructions. Briefly, 200 ng of tumor DNA was fragmented using a transposase and subjected to adapter ligation. Sequencing was performed using R9 or R9.4 flow cells on a MinION Mk 1B device (Oxford Nanopore) with the MinKNOW software (versions 1.0.5–1.5.12), respectively. For samples run with R9.4 sequencing chemistry, basecalling was performed using Albacore 1.1.0 (Oxford Nanopore). For R9 chemistries, online EPI2ME basecalling (Metrichor Ltd, Oxford, UK) was performed.
Template reads were exported as FASTA using nanopolish or poretools version 0.6 [18] and aligned to the hg19 human reference genome using BWA MEM 0.7.12 with the “−x ont2d” option [17]. Due to compatibility issues of data generated with R9 chemistries, only samples with R9.4 flow cells were used for copy number analysis and methylation-based classification.
Copy number analysis
For copy number analysis, the QDNAseq package version 1.8.0 [33] and R/Bioconductor, version 3.3, were used. Reads with a minimum mapping quality of 20 were sorted into 1000 kbp bins. Bins with missing reference sequence were excluded from analysis. To account for region- and technology-specific artifacts, public nanopore WGS data for the NA12878 human reference genome were processed identically and subtracted from the normalized tumor sample bin counts. Circular binary segmentation was performed as implemented in the DNAcopy package requiring an alpha value <0.05 to accept change points. Arm-level copy number calls were made by calculating the segment length weighted mean log ratio per chromosome arm.
Methylation analysis
To identify 5-methylation of cytosines, we used a recently published algorithm based on a hidden Markov model which has been trained using in vitro methylated E. coli DNA [35]. Training models for R9 sequencing chemistries were kindly provided by Jared Simpson. We modified the original implementation of nanopolish 0.6.0 to allow methylation calling from different basecalling groups. For classification, the subset of CpG sites overlapping with sites covered by Illumina 450K BeadChip microarrays was used. Beta values in the training set were dichotomized using a cut-off value of 0.6.
Structural variant detection
For detection of structural variants in amplified regions, we aligned nanopore FASTQ files from sample 3427T to the human reference genome, build GRCh37, using LAST (version 744) with settings: −Q 0. The last-train function was used with 1000 nanopore reads (~10 million bases) as input to adapt the alignment scoring parameters (−p) for error-prone nanopore reads. LAST alignment files (MAF) were converted to BAM files using the maf-convert function. BAM files were used as input for NanoSV [36] (available at https://github.com/mroosmalen/nanosv) with default settings.
Amplicon sequencing
Amplicons were designed to cover one or multiple exons of canonical transcripts of IDH1, IDH2, TP53, H3F3A, and the TERT promoter region. Oligonucleotide primers (Thermo Fisher Scientific) were then designed using Primer3 with the following non-default parameters (T min 59 °C, T opt 60 °C, T max 61 °C, and maximum mononucleotide repeat length = 3) to yield product sizes of 489–2902 bp (Table S1).
25 ng of genomic DNA was amplified using 0.02 U/µl Q5 polymerase (New England Biolabs, Ipswich, MA, USA), 200 µM dNTPs, 500 nM forward and reverse primers, and Q5 reaction buffer with high GC enhancer in a total reaction volume of 20 µl. Thermal cycling was performed as follows: 98 °C initial denaturation for 2 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 65 °C for 20 s and extension at 72 °C for 90 s, as well as a final extension at 72 °C for 2 min. Amplicons were analyzed using a Caliper LabChip GX DNA 5K assay (Perkin Elmer, Waltham, MA, USA). PCR products were purified using NucleoFast 96 PCR plates (Macherey–Nagel, Düren, Germany).
For amplicon sequencing, Ligation Sequencing Kit 1D (SQK-LSK108, Oxford) was used following the manufacturer’s protocol. Briefly, 1 µg of pooled amplicon DNA was subjected to end repair and dA-tailing. 250 ng of end-repaired DNA (equivalent to 0.2 pmol of 2 kbp fragments) was then used as input for adapter ligation. For real-time monitoring of sequencing depth, reads were streamed to the BWA aligner using npReader [6] with jHDF5 2.11.0 and coverage was calculated using BEDTools [28]. For variant calling, reads were realigned on the event level and variants called using VarScan 2.4.3 [15]. Variants were annotated using SnpEff version 4.3i [9] and ExAC release 0.3.1 germline variants [16] before filtering for coding or hotspot mutations with a minimum mutant allele frequency >0.2.
Microarray methylation profiling
Samples for Illumina Infinium BeadChip 450K profiling were prepared as described before [14]. Briefly, 500 ng of DNA was subjected to bisulfite conversion. Hybridization and imaging were performed by IntegraGen (Evry, France). Raw IDAT files were preprocessed using the GenomeStudio software (Illumina, San Diego, CA, USA). Processed methylation data from previously characterized samples [14] were retrieved via ArrayExpress (accession E-MTAB-3903). Beta values were used for all the subsequent analysis steps.
Statistics
All data analysis was done using R/Bioconductor version 3.3 [13]. Hierarchical clustering was used for arranging probes in the depicted classification training set. Random forest classification as implemented in the R/randomForest package, version 4.6–12, was run with default parameters. Sequence concordance was calculated using the Genome Analysis Toolkit’s Genotype Concordance tool, version 3.7 [21].
Data and material availability
Raw sequencing data are available via the European Genome–phenome Archive (accession EGAS00001002213). Microarray-based methylome data are available at ArrayExpress (E-MTAB-5797). TCGA data were retrieved from the UCSC Cancer Browser [11] or the TCGA FireBrowse website (http://www.firebrowse.org). Pipelines, scripts, and supplementary data to reproduce all results presented in this work are available at https://gitlab.com/pesk/glioma.nano-seq.
Results
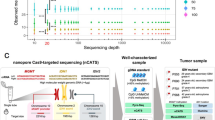
To meet the needs of the WHO 2016 classification of CNS tumors, we designed 1-day workflows for CN, methylation, and point mutation profiling using nanopore sequencing (Fig. 1a). We first subjected tumor DNA from molecularly well-characterized brain tumors [14, 30] to low-pass whole genome sequencing (WGS) using a commercially available, handheld size nanopore sequencing device. With the aim of widespread implementation in routine diagnostics in mind, we used a transposon-based library preparation kit, which reduces sample preparation time to less than hour. In a cohort of 28 patients (Table 1), low-pass WGS for 6 h performed yielded a mean mapped read depth from <0.01X to 0.24X (Table S1), depending on the sequencing chemistry and input DNA fragment size. Nanopores decipher DNA sequence of single molecules as they present to the pore, generating long reads of variable length, whose distribution is determined by DNA extraction and fragmentation method. We observed typical mean read lengths around 2 kb (Fig. 1b). As library preparation does not involve PCR amplification, no GC bias is introduced and the GC content distribution of the reads resembles closely that of the human reference genome (Fig. 1c).

Copy number profiling using nanopore low-pass whole genome sequencing. a Same-day workflows to simultaneously characterize copy number variation (CNV) and methylation profiles or single nucleotide variants, respectively. Tumor DNA is subjected to quality control (QC), and then, 250 ng input material is used for library preparation for either whole genome sequencing (WGS) or PCR-based deep amplicon sequencing. b Representative read length distribution of mapped reads. Note log scale on X axis. c Representative distribution of GC content of reads in comparison with the hg19 human reference genome. A randomly drawn subsample of the entire reference genome split into 1000 bp fragments is shown. d Copy number profile showing log2 transformed, normalized read counts per 1000 kbp window (grey) with running mean (red) and segmentation results (blue). e Comparison of nanopore WGS with matched SNP arrays. Heatmaps indicate copy number calls (losses and deletions in blue, and gains and amplifications in red) across the genome
Copy number profiling
We then used WGS data to generate CN profiles. Reads were counted in 1000 kb windows, normalized and subjected to circular binary segmentation (Fig. 1c). No correction of GC bias or mappability is necessary for nanopore reads; however, the long reads cause alignment artifacts with current reference genomes in regions with repetitive sequence such as centromeres. Still, the resulting CN profiles closely resembled matched SNP array-based profiles (Fig. 1d). Importantly, codeletion of chromosome 1p/19q as a diagnostic criterion for oligodendrogliomas implemented in the 2016 WHO classification of CNS tumors was detected in three out of four affected samples (Fig. S1). The remaining sample did not yield sufficient read depth (<0.01) due to low input DNA quality (Table S1). High-level focal amplifications of EGFR, PDGFRA, and CDK4 were detected in affected glioblastoma samples (Table 1). In contrast, focal deletions, such as CDKN2A, were frequently missed by segmentation. Beyond diagnostic needs, we could reconstruct the double minute nature of an EGFR amplification (case 3427T), identify the exact genomic breakpoint using algorithmic structural variant discovery [36], and confirm the latter by Sanger sequencing (Fig. S2).
Methylation profiling
A major advantage of nanopore sequencing is the ability to detect base modifications, especially 5-methylation of cytosines, in native DNA without need for bisulfite conversion. Epigenomic changes are functionally important in cancer, but also aid in delineating cancer entities. For example, IDH mutations cause a global hypermethylation of CpG islands [25], a phenotype of utmost prognostic importance in neuro-oncology. We thus aimed to detect the G-CIMP phenotype from nanopore reads.
First, we compared methylation events in CpG sites identified by nanopore sequencing to matched methylome microarrays. Good correlation was observed between single read methylation status of a given CpG site and its corresponding beta value in microarray data (Fig. 2a). Next, we applied random forest (RF) classification to predict IDH mutation.

Methylome profiling by nanopore sequencing of native tumor DNA. a Comparison of methylation calls from nanopore sequencing with matched Illumina 450K microarray-based data. Beta value distributions for CpG sites that were identified as unmethylated (red) or methylated (blue), respectively, by nanopore WGS are shown. b “Random taiga” simulation of classification error as a function of the number of randomly sampled CpG sites. Each dot represents the class-specific error rate of an ad hoc generated random forest using a random subset of N CpG sites (indicated on X axis) from the TCGA lower grade glioma Illumina 450K cohort as training set. Lines indicate the mean of five independent simulations. c Methylation profiles from nanopore sequencing discriminate IDH-mutant and wild-type tumors. Bar plots indicate vote distribution from ad hoc random forest classification. The TCGA low-grade glioma cohort was used as a training set. Illumina 450K-based beta values were dichotomized using >0.6 as threshold
RF classification is a commonly used machine-learning algorithm based on randomly generated (weak) decision trees [3]. Majority votes then integrate decisions from the entire forest to provide robust classification. The challenge with low-pass WGS data is that it is not known beforehand which CpG sites will be sequenced and the classifier can be built upon. Therefore, we generated random forests ad hoc. With increasing numbers of probed CpG sites, we expect the classifier’s error rate to decrease. To test the feasibility of this approach, we simulated multiple random forests for a given number of CpG sites using the low-grade glioma cohort [5] from The Cancer Genome Atlas (TCGA) and determined misclassification rate for this “random taiga” (Fig. 2b). The simulations show that the mean class error rate to predict IDH and 1p/19q status does not improve for more than approximately 500 CpG sites. This amount of data is reliably sampled within 6 h of nanopore sequencing. Thus, information with respect to a cancer’s entity is redundantly encoded in the methylome and this fact can be exploited for classification from sparse, randomly sampled CpG sites.
Using the same training set, we then predicted IDH status in our samples from nanopore-based methylation calls. Due to the low read depth (usually N = 1), methylation calls from nanopore WGS were binary. To enable classification using microarray-based training data, beta values were dichotomized as described in previous applications of RF in methylation data [5, 7]. All samples were correctly classified (Fig. 2c).
Supervised pan-cancer classification
Next, following the idea of a machine-learning-based molecular classification of tumors to fully recognize molecular entities and rule out interobserver variability [32], we sought to investigate whether nanopore CN and methylation profiles can be used to classify tumor samples on a pan-cancer level. As a training set for all analyses, we used public microarray-based methylation data from primary brain tumors (adult and pediatric glioblastomas, lower grade gliomas, and medulloblastomas) and tumors that frequently metastasize to the brain (melanoma, breast, lung, bladder, prostate, colon, and clear cell renal carcinoma) [1, 2, 4, 5, 12, 23, 24, 37,38,39,40]. Where CN data were available, too, SNP array-based CN profiles were aggregated to chromosome arm level and added to the training set (Fig. 3a). The resulting classifiers for any set of CpG sites in our cohort usually yielded an overall out-of-bag classification error rate ≪5%.

Pan-cancer classification using copy number and methylation profiles. a Training set composed of TCGA samples from nine cancer entities using arm-level averaged copy number (CN) information (CN loss blue, CN gain red) and dichotomized methylation data. For illustration purposes, only 200 random CpG sites were sampled, clustered, and plotted. b–d Classification of samples subjected to WGS using R9.4 flow cells using ad hoc random forests (500 trees per sample). Bar plots show vote distributions based on copy number only (b), methylation (c), or both modalities (d). e, f Methylation-based pan-cancer classification of medulloblastoma (e) and a brain metastasis of a lung adenocarcinoma (f). BRCA breast cancer, BLCA bladder urothelial carcinoma, COAD colon adenocarcinoma, KIRC kidney renal cell carcinoma, LUNG lung squamous cell and adenocarcinoma, SKCM skin cutaneous melanoma, PRAD prostate adenocarcinoma, MB medulloblastoma, K27 diffuse midline glioma H3 K27M mutant, G34 pediatric glioblastoma, H3 G34R mutant
We first subjected seven glioma samples with CN and methylation profiles generated by nanopore sequencing to ad hoc RF classification. When we compared classification using CN alone (Fig. 3b), methylation only (Fig. 3c) or both modalities together (Fig. 3d), using the joint approach improved overall accuracy: all (7/7) samples were correctly classified.
Then, we subjected two medulloblastoma (MB) cases to classification (here, only methylation training data were available). Both samples were identified as MB and also the genetic subtype according to the WHO classification was predicted correctly as WNT-activated (case MB683) or non-SSH-activated/non WNT-activated (i.e., group 4, case 8372T) (Fig. 3e). Next, we attempted classification of brain metastasis and could predict the pulmonary origin in one case (Fig. 3f). We also selected a metastasis of a breast adenocarcinoma in the posterior fossa for study which immunohistochemically showed expression of GFAP and S100, so it was misleading for the diagnosis of carcinoma. Pan-cancer classification based on nanopore WGS correctly identified this sample as breast cancer (Table 1, Fig. S1).
Several cases were not classifiable (requiring a > 5 percentage points’ difference of the majority vote to the next best vote) or misclassified (Table 1). These cases had often lower DNA quality with respect to fragment size (Table S1). One GBM sample that was not classifiable had low tumor purity when estimated from matched transcriptomic profiles using the ESTIMATE algorithm [41] (Fig. S3a). This also resulted in false-negative calling of copy number CN alterations using fixed thresholds, even though they were present at visual inspection (Fig. S3b).
Amplicon sequencing
Finally, we explored deep amplicon nanopore sequencing for identification of single nucleotide variants. We designed an amplicon panel covering hotspot exons in IDH1, IDH2, and H3F3A, all coding exons of TP53 and, additionally, the TERT promoter (pTERT) region. Due to the long reads delivered by nanopore sequencing, this could be achieved with only nine PCR reactions (Table S2). Mutations in these genes (with exception of pTERT) inform molecular diagnosis of glioma and medulloblastoma, and are demanded for diagnosis in the 2016 WHO classification of CNS tumors [20]. Sufficient read depth is a critical parameter for variant calling with defined sensitivity and specificity. We thus implemented a real-time analysis pipeline that allowed monitoring of read depth and to stop sequencing when sufficient information to make a diagnosis has been collected (Fig. 4a). In samples run as single samples with real-time monitoring, a sequencing depth of 1000X in all target regions could repeatedly be achieved within 2–20 min of sequencing. Mean overall coverage >1000X could be achieved in single runs, but was lower in runs using barcoding PCR for multiplexing (Fig. 4b).

Real-time amplicon sequencing of single nucleotide variants. a Representative coverage plot of target regions in IDH1, IDH2, H3F3A, TP53, and TERT promoter region over time. The time needed to achieve 1000X depth in all amplicons is indicated. Note log scale on Y axis. b Mean read depth over all amplicons in samples processed individually or as barcoded multiplex libraries. Of note, FFPE samples were sequenced as part of a multiplex library. c Comparison of selected variant calls from nanopore sequencing (filtered for coding or hotspot mutations with minimum allele frequency >0.2) with reference calls from Sanger or Illumina sequencing
In all samples, coding mutations were reliably detected as compared to routine diagnostics based on Sanger sequencing, immunohistochemistry or a next-generation sequencing (NGS) panel (Fig. 4c). Nanopore sequencing reads have historically shown high error rates, especially in homopolymer contexts. We, therefore, compared nanopore consensus sequences to matched short-read whole exome data in five cases. Overall concordance was 97.8–98.6% before functional filtering. Even though at low number (<5 per sample) after filtering for coding mutations, false-positive variants were present. Most of these mutations occurred in multiple samples, indicating a context-specific error (Table S3). Improved base calling algorithms are thus needed to reduce the time to manually review mutations for false positives.
Technical aspects
Nanopore sequencing is highly scalable due to low capital cost of the device (use of multiple sequencers) and reuse of flow cells. To exclude carry-over and cross-contamination in sequential sequencing runs and for scalability, we evaluated barcoding and multiplexing for both WGS and amplicon workflows (Table S1, Fig. 4b). For WGS, up to four samples were combined without major protocol changes and permitting convenient overnight runs (e.g., one sample for 6 h and two samples for 12 h). Barcoding of amplicon libraries and multiplexing 12 samples greatly reduces per-assay price at the cost of additional PCR and quality control steps. Finally, we explored use of DNA derived from formalin-fixed paraffin-embedded tissue (FFPE). PCR amplicons were generated from two FFPE samples with identical input amount and protocol. As expected from the usually highly fragmented DNA, PCR yields were lower, especially for large amplicons (>1 kbp). This could only partly be compensated by extending sequencing time. For nanopore WGS, transposase-based library preparation is not compatible with fragment size distribution of FFPE-derived DNA samples. We thus performed a different ligation protocol to test WGS in one FFPE sample. While read yield was acceptable (Table S1), the resulting copy number profile was noisy and hard to interpret (Fig. S1). In summary, nanopore sequencing is compatible with FFPE samples, but clearly not recommended due to inferior performance.
Discussion
Histomolecular classification promises to significantly improve diagnosis, prognosis, and treatment decision making of cancer patients by aiding in clearly delineating distinct (molecular) entities and identifying targetable genomic alterations for personalized treatment. It is, therefore, crucial to ensure widespread implementation of appropriate technology in clinical routine for patient benefit. We explored the potential of nanopore sequencing to comprehensively characterize genetic alterations.
CN alterations could be detected in brain tumor samples using ultra low-pass WGS. While overall resolution is lower than current SNP arrays or NGS approaches, arm-level alterations and high-level focal alterations are reliably recapitulated. Most importantly, detection of 1p/19q-codeletion fulfills diagnostic needs for the current WHO 2016 classification of CNS tumors. While WGS using rapid, transposase-based library preparation works very well with high molecular weight DNA, some of the clinical routine fresh-frozen tumor DNA samples were highly fragmented and yielded insufficient results. Quality of input DNA thus seems to be pivotal. For use of FFPE material, changes to the protocol and further optimization are needed.
Methylation data can directly be obtained from the same WGS data set which makes time-consuming bisulfite conversion and specialized methylation assays (sequencing or hybridization-based) expendable. Very recently, it has been shown in the context of meningioma that classification of tumors using methylome data alone is sufficient or superior to make a correct diagnosis [32]. With low genome coverage, we obtained sparse random sampling of CpG sites. We show that this information is sufficient to subtype gliomas into IDH-mutant vs. wild-type samples and that cancer entities from different tissue origins can be distinguished in a few hours. This may aid in the differential diagnosis of primary brain tumors vs. brain metastases and greatly facilitate staging and the search for unknown primary tumors [22]. However, as diagnosis is inferred from relatively sparse data, it precludes inter-patient comparison and reuse of data with currently obtainable coverage in the (relatively short) time frame of 6 h of sequencing.
Finally, we used PCR-based amplicon generation followed by nanopore sequencing to identify point mutations. Using a small, but diagnostically relevant gene panel (covering target regions with a total of 12 kb), high read depth could be routinely obtained in less than 30 min of sequencing when using real-time depth monitoring. However, context-specific base calling errors introduce platform-specific errors and false variant calls that need to be carefully reviewed.
Comparison to existing technologies
Targeted next-generation sequencing panels tailored to detect mutations in brain tumors or, more generally, cancer-related genes have been employed routinely with a turnaround time of several days [8, 31]. Methylation-based classification of brain tumors by microarray allows differentiation of a wealth of different entities within 2 weeks [12, 32]. Intraoperative subtyping of gliomas is possible using allele specific PCR for key alterations (IDH1, pTERT) but remains restricted to hotspot point mutations [34]. Similarly, CN changes and mutations have been detected in cell-free DNA from CSF to allow less invasive diagnostics [10, 26]. A major drawback of all approaches is the high investment cost, need for laboratory space or expertise.
For nanopore sequencing, besides the portable sequencing device and a laptop computer, only a spectrometer for DNA quantification and a thermocycler for library preparation and amplicon generation by PCR are needed. This allows implementation of a complete molecular pathology laboratory even in resource-restricted settings or mobile environments. Per sample cost is ~$200 for WGS and ~$120 for amplicon sequencing without multiplexing. However, being a technology still under development, frequent updates in chemistry and software currently challenge routine use and need to be addressed to allow standardized diagnostics across laboratories. In addition, hybridization microarrays and targeted short-read sequencing both work relatively well with fragmented DNA from FFPE samples, while this currently poses a technical challenge for nanopore sequencing.
Our study has several limitations. First, as this is a proof-of-principle study, sample number is small and precludes accurate quantification of sensitivity or specificity to detect structural alterations and point mutations. Second, a prospective and multi-centric evaluation of the approach presented here is needed to rule out sample selection bias and demonstrate robustness across laboratories. Third, we reused flow cells to reduce per-assay cost, but washing also decreased the number of active pores and thus performance in subsequent runs.
In conclusion, same-day diagnosis of CN alterations, epigenetic modifications, and single nucleotide variants using nanopore sequencing is feasible with minimal capital cost and without need for sophisticated laboratory equipment. For CNS tumors, molecular features demanded for diagnosis by current guidelines can be obtained, which, together with histological data and grading, enable accelerated integrated diagnosis and improve patient care.
References
Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD et al (2015) The molecular taxonomy of primary prostate cancer. Cell 163:1011–1025. doi:10.1016/j.cell.2015.10.025
Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB et al (2015) Genomic classification of cutaneous melanoma. Cell 161:1681–1696. doi:10.1016/j.cell.2015.05.044
Breiman L (2001) Random forests. Mach Learn 45:5–32. doi:10.1023/A:1010933404324
Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. doi:10.1016/j.cell.2013.09.034
Brat DJ, Verhaak RGW, Aldape KD, Yung WKA, Salama SR, Cancer Genome Atlas Research Network et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372:2481–2498. doi:10.1056/NEJMoa1402121
Cao MD, Ganesamoorthy D, Cooper MA, Coin LJM (2016) Realtime analysis and visualization of MinION sequencing data with npReader. Bioinformatics 32:764–766. doi:10.1093/bioinformatics/btv658
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563. doi:10.1016/j.cell.2015.12.028
Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A et al (2015) Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17:251–264. doi:10.1016/j.jmoldx.2014.12.006
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L et al (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. doi:10.4161/fly.19695
De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martínez-Ricarte F, Torrejon D et al (2015) Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun 6:8839. doi:10.1038/ncomms9839
Goldman M, Craft B, Swatloski T, Ellrott K, Cline M, Diekhans M et al (2013) The UCSC cancer genomics browser: update 2013. Nucl Acids Res 41:D949–D954. doi:10.1093/nar/gks1008
Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R et al (2013) Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol 125:913–916. doi:10.1007/s00401-013-1126-5
Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS et al (2015) Orchestrating high-throughput genomic analysis with bioconductor. Nat Methods 12:115–121. doi:10.1038/nmeth.3252
Kamoun A, Idbaih A, Dehais C, Elarouci N, Carpentier C, Letouzé E et al (2016) Integrated multi-omics analysis of oligodendroglial tumours identifies three subgroups of 1p/19q co-deleted gliomas. Nat Commun 7:11263. doi:10.1038/ncomms11263
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L et al (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. doi:10.1101/gr.129684.111
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. doi:10.1038/nature19057
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. doi:10.1093/bioinformatics/btp324
Loman NJ, Quinlan AR (2014) Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30:3399–3401. doi:10.1093/bioinformatics/btu555
Loose M, Malla S, Stout M (2016) Real-time selective sequencing using nanopore technology. Nat Methods. doi:10.1038/nmeth.3930
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. doi:10.1007/s00401-016-1545-1
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi:10.1101/gr.107524.110
Moran S, Martínez-Cardús A, Sayols S, Musulén E, Balañá C, Estival-Gonzalez A et al (2016) Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 17:1386–1395. doi:10.1016/S1470-2045(16)30297-2
Network TCGA (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337. doi:10.1038/nature11252
Network TCGAR (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489:519–525. doi:10.1038/nature11404
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. doi:10.1016/j.ccr.2010.03.017
Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S et al (2016) Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J Clin Oncol. doi:10.1200/JCO.2016.66.6487
Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L et al (2016) Real-time, portable genome sequencing for Ebola surveillance. Nature 530:228–232. doi:10.1038/nature16996
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi:10.1093/bioinformatics/btq033
Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M et al (2017) Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods. doi:10.1038/nmeth.4189
Rosenberg S, Verreault M, Schmitt C, Guegan J, Guehennec J, Levasseur C et al (2017) Multi-omics analysis of primary glioblastoma cell lines shows recapitulation of pivotal molecular features of parental tumors. Neuro-Oncol 19:219–228. doi:10.1093/neuonc/now160
Sahm F, Schrimpf D, Jones DTW, Meyer J, Kratz A, Reuss D et al (2016) Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 131:903–910. doi:10.1007/s00401-015-1519-8
Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S et al (2017) DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. doi:10.1016/S1470-2045(17)30155-9
Scheinin I, Sie D, Bengtsson H, van de Wiel MA, Olshen AB, van Thuijl HF et al (2014) DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res 24:2022–2032. doi:10.1101/gr.175141.114
Shankar GM, Francis JM, Rinne ML et al (2015) Rapid intraoperative molecular characterization of glioma. JAMA Oncol. doi:10.1001/jamaoncol.2015.0917
Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W (2017) Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods. doi:10.1038/nmeth.4184
Stancu MC, Roosmalen MJ van, Renkens I, Nieboer M, Middelkamp S, Ligt J de, et al. (2017) Mapping and phasing of structural variation in patient genomes using nanopore sequencing. bioRxiv 129379. doi:10.1101/129379
Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. doi:10.1016/j.ccr.2012.08.024
The Cancer Genome Atlas Research Network (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499:43–49. doi:10.1038/nature12222
The Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543–550. doi:10.1038/nature13385
The Cancer Genome Atlas Research Network (2014) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507:315–322. doi:10.1038/nature12965
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W et al (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4:2612. doi:10.1038/ncomms3612
Acknowledgements
The authors would like to acknowledge Jared T. Simpson for providing methylation training models, Inès Detrait and Amithys Rahimian-Aghda for biobanking and sample management, Ludovic Prevost for excellent IT systems administration, and Mark van Roosmalen for assistance with running NanoSV. We are indebted to the Nanopore Human Genome Sequencing consortium for early release of data at https://github.com/nanopore-wgs-consortium/NA12878. The results published here are in part based upon data generated by The Cancer Genome Atlas (TCGA) project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov/.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This work has been supported by Deutsche Forschungsgemeinschaft (EU 162/1-1 to PE), the program “Investissements d’avenir” (ANR-10-IAIHU-06 to AI), Institut Universitaire de Cancérologie (to AI), Ligue Nationale Contre le Cancer (to AI), Institut Carnot (to KL), and Fondation ARC pour la recherche sur le cancer (n°PJA 20151203562 to FB).
Conflict of interest
The authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Euskirchen, P., Bielle, F., Labreche, K. et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol 134, 691–703 (2017). https://doi.org/10.1007/s00401-017-1743-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1743-5