
Abstract
Coenzyme A (CoA) and its derivatives such as acetyl-CoA are essential metabolites for several biosynthetic reactions. In the yeast S. cerevisiae, five enzymes (encoded by essential genes CAB1-CAB5; coenzyme A biosynthesis) are required to perform CoA biosynthesis from pantothenate, cysteine, and ATP. Similar to enzymes from other eukaryotes, yeast pantothenate kinase (PanK, encoded by CAB1) turned out to be inhibited by acetyl-CoA. By genetic selection of intragenic suppressors of a temperature-sensitive cab1 mutant combined with rationale mutagenesis of the presumed acetyl-CoA binding site within PanK, we were able to identify the variant CAB1 W331R, encoding a hyperactive PanK completely insensitive to inhibition by acetyl-CoA. Using a versatile gene integration cassette containing the TPI1 promoter, we constructed strains overexpressing CAB1 W331R in combination with additional genes of CoA biosynthesis (CAB2, CAB3, HAL3, CAB4, and CAB5). In these strains, the level of CoA nucleotides was 15-fold increased, compared to a reference strain without additional CAB genes. Overexpression of wild-type CAB1 instead of CAB1 W331R turned out as substantially less effective (fourfold increase of CoA nucleotides). Supplementation of overproducing strains with additional pantothenate could further elevate the level of CoA (2.3-fold). Minor increases were observed after overexpression of FEN2 (encoding a pantothenate permease) and deletion of PCD1 (CoA-specific phosphatase). We conclude that the strategy described in this work may improve the efficiency of biotechnological applications depending on acetyl-CoA.
Key points
• A gene encoding a hyperactive yeast pantothenate kinase (PanK) was constructed.
• Overexpression of CoA biosynthetic genes elevated CoA nucleotides 15-fold.
• Supplementation with pantothenate further increased the level of CoA nucleotides.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coenzyme A (CoA) is an indispensable and ubiquitous metabolic cofactor of numerous enzymes requiring activated carboxylic acids as CoA thioesters. Especially acetyl-CoA is needed as the energy source of the citric acid cycle and as a building block for the biosynthesis of fatty acids, sterols, and several other compounds. Acetyl-CoA is also of fundamental importance for protein acetylation with special emphasis on histone acetylation as a mechanism for regulating chromatin accessibility in the course of gene activation (summarized by Galdieri et al. 2014). While higher organisms synthesize cytoplasmic acetyl-CoA by cleavage of citrate (exported from mitochondria, catalyzed by the ATP-citrate lyase ACL), Saccharomyces yeasts use acetyl-CoA synthetase for ATP-dependent activation of acetate (constitutive nucleocytosolic isoenzyme Acs2). Acetyl-CoA at the junction of anabolic and catabolic metabolism must be considered a metabolite whose concentration is indicative of the cellular nutritional and energetic status (reviewed by Pietrocola et al. 2015). For yeast and cultured mammalian cells, fluctuation of acetyl-CoA concentration and its intracellular distribution has been proposed as an indicator of “fed” or “fasted” states with a decisive influence on fundamental aspects of cellular physiology (e.g., entry into a new cell division cycle; Shi and Tu 2013; 2015). Under conditions of oxidative stress, CoA can also trigger a regulatory response by covalent modification of proteins such as peroxiredoxins at cysteine residues (CoAlation; Baković et al. 2019). In biotechnology, acetyl-CoA is an essential compound for improving several strategies of metabolic engineering which may enable microbial biosynthesis of biofuels (n-butanol and farnesene; Schadeweg and Boles 2016a, b; Meadows et al. 2016; Tippmann et al. 2016) as well as pharmaceutical compounds (hydrocortisone, polyketide antibiotics, cannabinoids, and the antimalarial drug artemisinin; Szczebara et al. 2003; Paddon et al. 2013; Luo et al. 2019). The importance of malonyl-CoA for pathways related to biosynthesis of fatty acids and polyketides has been recently reviewed (Milke and Marienhagen 2020).
Comparative genomic studies have clearly shown that the biosynthesis of CoA utilizes five universal reactions, using pantothenate, cysteine, and ATP as its substrates (reviewed by Leonardi et al. 2005b; Spry et al. 2008; Theodoulou et al. 2014). While vertebrates depend on the uptake of exogenous pantothenate (vitamin B5) as a precursor, most bacteria, yeasts, fungi, and plants are able to synthesize pantothenate de novo, using the carbon backbone of amino acids. In the yeast Saccharomyces cerevisiae, uptake of pantothenate by the high-affinity plasma membrane H+-symporter Fen2 has been described (Stolz and Sauer 1999). Although some strains of S. cerevisiae may be auxotrophic for pantothenate, others are competent for de novo biosynthesis, using polyamines such as spermine as an unusual source of β-alanine which is required to form pantothenate from pantoate (White et al. 2001).
The initial and presumably rate-limiting step of CoA biosynthesis is ATP-dependent phosphorylation of pantothenate by a pantothenate kinase (PanK). While bacterial PanK enzymes are strongly inhibited by unacylated CoA (coaA gene product from E. coli; Vallari et al. 1987), eukaryotic pantothenate kinases (which are poorly, if at all, related to bacterial enzymes) are sensitive against inhibition by acetyl-CoA (first shown for the PanK of the fungus Aspergillus nidulans: Calder et al. 1999). A temperature-sensitive mutant of S. cerevisiae initially isolated because its fatty acid synthase (FAS) was pantetheine-free later turned out as defective for pantothenate kinase (G351S missense mutation). The corresponding wild-type gene CAB1 (coenzyme A biosynthesis) exhibits substantial similarity to the enzyme of A. nidulans (43.8% identity) and is expressed at a low level. Although transcription of CAB1 was not substantially affected by the carbon source or by availability of amino acids (Olzhausen et al. 2009), its upstream region contains two sequence motifs reminiscent of the sterol-response element (SRE) which is bound by activators Upc2 and Ecm22 (Brohée et al. 2011). Thus, it should be possible to strongly elevate yeast PanK activity by expression of CAB1 using a heterologous promoter. While lower eukaryotes contain a single PanK gene copy, four PANK genes with different tissue-specific patterns of transcription could be identified in mammals, encoding enzymes with distinct cellular localization and sensitivity against CoA and acetyl-CoA (Zhang et al. 2005). Importantly, a human neurodegenerative disorder has been associated with a defect of the PANK2 gene, encoding the mitochondrial isoenzyme in mammals (Hayflick 2014).
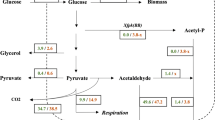
Similar to CAB1, genes encoding the remaining four enzymes of yeast CoA biosynthesis are also essential for cellular viability. 4′-Phosphopantothenate as the product of the PanK reaction is used for formation of 4′-phosphopantothenoylcysteine (PPC) which is subsequently decarboxylated to give 4′-phosphopantetheine (PP). While these biosynthetic steps are catalyzed by a bifunctional enzyme in E. coli (PPC synthetase PPCS and PPC decarboxylase PPCDC encoded by coaBC), individual enzymes exist in S. cerevisiae (encoded by CAB2 and CAB3, respectively; Olzhausen et al. 2009) and higher eukaryotes (Daugherty et al. 2002). Exome sequencing in human patients gave evidence that PPCS variants may cause cardiomyopathy (Iuso et al. 2018). In addition to Cab3, yeast heterotrimeric PPCDC also contains subunits Hal3 (= Sis2) and Vhs3 which were initially described as negative regulators of phosphatase Ppz1 involved in halotolerance (Ruiz et al. 2009; Abrie et al. 2015). Thus, Hal3 and Vhs3 are “moonlighting” proteins at least one of which is essential for yeast viability. PP then reacts with the adenylyl moiety of ATP to give dephospho-CoA (DPC), and finally DPC is phosphorylated, forming CoA. Yeast monofunctional enzymes PP adenylyltransferase (PPAT) and DPC kinase (DPCK) encoded by CAB4 and CAB5, respectively (coaD and coaE in E. coli), are required to complete CoA biosynthesis, while a bifunctional CoA synthase exists in mammals (Zhyvoloup et al. 2002). Mutations in the human COASY gene are associated with neurodegeneration (Dusi et al. 2014). Besides its enzymatic PPCDC activity, Cab3 physically interacts with Cab2, Hal3, Vhs3, Cab4, and Cab5 (but not with Cab1/PanK) and thus functions as a scaffold of the yeast CoA synthesizing protein complex (CoA-SPC) with a molecular weight of about 330 kDa (Olzhausen et al. 2013). Assembly of such a CoA biosynthetic complex has been recently also shown for mammalian cell lines (Bakovic et al. 2021). There is evidence that Drosophila and mice may use PP as an extracellular precursor which is used to complete biosynthesis of CoA in the absence of PANK, PPCS, and PPCDC (Srinivasan et al. 2015; reviewed by Sibon and Strauss 2016). Figure 1 summarizes the biosynthesis of CoA in S. cerevisiae and shows metabolic pathways requiring acetyl-CoA together with some applications in biotechnology.

Biosynthesis of CoA in S. cerevisiae, metabolic pathways requiring acetyl-CoA and applications of acetyl-CoA in molecular biotechnology. Acetyl-CoA as a substrate of the glyoxylate cycle essential for utilization of C2-substrates (e.g., ethanol) is not shown. Abbreviations of cellular compartments: ER, endoplasmic reticulum; MIT, mitochondria; NUC, nucleus; PM, plasma membrane. Enzymes of CoA biosynthesis: PanS, pantothenate synthase; PanK, pantothenate kinase; PPCS, 4′-phosphopantothenoylcysteine synthetase; PPCDC, 4′-phosphopantothenoylcysteine decarboxylase; PPAT, 4′-phosphopantetheine adenylyltransferase; DPCK, dephospho-CoA kinase
Since all yeast CAB genes are expressed at a low level, gene overexpression may be considered a means for engineering of the CoA biosynthetic pathway. However, it appears reasonable to assume that inhibition of PanK activity by the final product of the pathway prevents an increased metabolic flux towards CoA, as previously shown in E. coli (Song and Jackowski 1992). We thus devised a strategy to overcome the presumed PanK inhibition by CoA/acetyl-CoA and were indeed able to identify a hyperactive and constitutive enzyme variant (W331R). Stable overexpression of this variant in combination with additional genes of CoA biosynthesis and physiological modifications of growth media finally allowed us to construct yeast strains in which the intracellular concentration of CoA nucleotides was increased almost 40-fold above the wild-type level.
Materials and methods
Yeast strains and media
All strains of S. cerevisiae used in this work were derived from regulatory wild-type strains JS91.15–23 (Olzhausen et al. 2009) and BY4741 (Brachmann et al. 1998) by successive transformation with gene cassettes encoding TPI1-CAB fusions (see below). Complete genotypes of major strains are shown in Table 1 (an extended list of all strains and genotypes is available as Supplementary Table S1).
Synthetic complete (SC) media were prepared by using 0.17% yeast nitrogen base as a source of vitamins (final concentration of pantothenate: 0.84 μM). Concentration of ammonium sulfate, amino acids, and bases has been described (Olzhausen et al. 2009). Transformants were selected with SCD-Leu medium (2% glucose, without leucine). For improved supply of a pantothenate precursor, β-alanine was added to a final concentration of 200 mg/l (2.25 mM). Concentration of pantothenate was elevated 50-fold (20 mg/l; 42 μM) or 100-fold (40 mg/l; 84 μM) for certain cultivations.
Plasmid constructions
Standard cloning vectors pRS415, pRS416 (Sikorski and Hieter 1989), p415-MET25, p416-MET25, and p426-MET25HA (Mumberg et al. 1994) were used for plasmid constructions employed in this work (Genetic markers are given in Supplementary Table S2).
Allelic variants of CAB1 obtained by selection of conditional mutant JS91.14–24 (cab1ts G351S) at 37 °C were amplified by PCR using reading frame primers CAB1-Bam-Start + CAB1-Hind-Stop and chromosomal DNA prepared from suppressor strains obtained. Amplification products were cleaved with BamHI + HindIII and inserted into expression plasmid p416-MET25 to give various pLS12-S plasmids. As a wild-type reference, pTM8 was constructed accordingly.
Multi-copy expression plasmids pSBS5 (CAB1) and pJO73 (murine PanK3 gene) have been described (Olzhausen et al. 2009). Variants of CAB1 for functional analysis were obtained by site-directed mutagenesis (see below) and inserted into p415-MET25 and p426-MET25HA.
The construction of plasmid pLEUTEX3 used for integration of CoA biosynthetic genes activated by the TPI1 promoter is described in detail in the Supplementary Information. Coding regions of CAB1, CAB1 W331R, CAB2, CAB3, CAB4, CAB5, HAL3aa260-495 (= HAL3PD; PPCDC domain), and FEN2 were amplified with gene-specific primers and individually inserted into the multi-cloning site of pLEUTEX3 to give TPI1-CAB fusions. For site-specific recombination with selected chromosomal loci, flanking sequences of about 400 bp were amplified and ligated to both sides of pLEUTEX plasmids. Recombination of gene expression cassettes with the desired genomic position was verified by analytical PCR, using chromosomal DNA of transformants and specific verification primers outside the flanking regions. After each transformation, the selection marker loxP-LEU2-loxP was removed by activation of the GAL1-dependent cre recombinase on plasmid pSH62 (cultivation in SCGal-His; Güldener et al. 1996).
Site-directed mutagenesis
To introduce missense mutations into the CAB1 coding region at selected sites, the QuikChange Site-Directed Mutagenesis Kit from Agilent was used. Plasmid pJO20 was derived from pUC19 by insertion of the CAB1 coding region as a BamHI + HindIII fragment and subsequently used as a template for mutagenesis. Sequences of oligonucleotides used for mutagenesis are shown in Supplementary Table S3. After confirmation of the desired mutation by DNA sequencing, CAB1 reading frame cassettes were transferred to various expression plasmids.
Plasmid shuffling
Missense variants of CAB1 and human COASY were functionally studied in S. cerevisiae using the plasmid shuffling strategy (Sikorski and Boeke 1991). Entire genes CAB1, CAB4, and CAB5, respectively, were inserted into ARS CEN URA3 vector pRS416, and the resulting rescue plasmids pJO57, pGE7, and pGE9 were individually transformed into wild-type strain JS91.15–23. Chromosomal copies of these CAB genes were subsequently deleted, using gene disruption cassettes released from plasmids pSBS7 (cab1Δ::HIS3), pSB2 (cab4Δ::HIS3), and pSB5 (cab5Δ::HIS3), respectively (Olzhausen et al. 2009). The resulting strain JS19.1 (cab1Δ::HIS3 [CAB1]) was then transformed with ARS CEN LEU2 plasmid pKH45 (CAB1, positive control) and related plasmids containing various missense mutations at selected positions. Similarly, the complementation of null mutations cab4Δ and cab5Δ by pGE8 (CAB4), pGE10 (CAB5), and pLS20 (hCOASY) was tested by transformation of LSY20 (cab4Δ::HIS3 [CAB4]) and LSY21 (cab5Δ::HIS3 [CAB5]), respectively. Transformants were incubated on synthetic medium containing 5-fluoroorotic acid (FOA) to counter-select against URA3 rescue plasmids.
Enzyme assay
Assay of pantothenate kinase has been described (Olzhausen et al. 2009). In brief, the assay depends on ATP-dependent conversion of D-[1-14C] pantothenate (55 mCi/mMol; Biotrend, Cologne, Germany) to phosphopantothenate and its binding to DEAE cellulose ion exchange filter disks which were subsequently analyzed by scintillation counting (6 assays for each PanK variant). Since strain JS91.14–24 used as a recipient for transformation also contains a conditional PanK, assays were performed at 37 °C.
To investigate the influence of CoA and its thioesters on the activity of PanK from yeast (Cab1) and mouse (PanK3 isoenzyme) in the absence of cellular metabolites, crude extracts from transformants of strain JS91.14–24 were partially purified by ion exchange chromatography. Prior to protein binding, DEAE sepharose CL-6B (Sigma-Aldrich) was equilibrated with binding buffer (100 mM Tris/HCl + 2.5 mM MgCl2, pH 7.4). Crude extracts (5 mg/ml protein) were incubated with DEAE sepharose at 4 °C, washed 5 times with the 2.5-fold volume of binding buffer, and finally treated with elution buffer containing 600 mM NaCl. Salt was removed by microdialysis against binding buffer. Inhibitory concentrations of CoA and its thioesters on PanK activity were calculated by using the GraphPad Prism software (San Diego, USA).
Measurements of metabolites
Yeast strains were harvested at a density of 2 × 107 cells/ml, collected by centrifugation and used for preparation of crude extracts by intensive agitation with glass beads for 6 min. Removal of proteins by precipitation with perchloric acid (final concentration: 400 mM), neutralization with K2CO3, and subsequent quantification of metabolites CoA and acetyl-CoA by enzyme-coupled reactions has been performed as described (Bergmeyer 1974). In brief, CoA and acetyl-CoA were assayed together by using the citrate synthase reaction which catalyzes the formation of citrate from acetyl-CoA and oxaloacetate. Oxaloacetate is provided by the NAD-dependent malate dehydrogenase reaction which also serves as the photometric indicator reaction (increase of absorbance by NADH at 340 nm). All CoA is converted to acetyl-CoA by phosphotransacetylase with acetylphosphate as a substrate. To distinguish between CoA and acetyl-CoA, N-ethylmaleimide was used to block the free SH group of CoA. Validity of the assay was verified by analysis of CoA + acetyl-CoA samples of known concentration and by adding defined concentrations of coenzymes to yeast crude extracts. Amounts of CoA + acetyl-CoA were calculated with respect to protein concentrations determined prior to protein removal from crude extracts. Routinely, each strain was cultivated three times, and cell extracts derived were assayed in triplicate. Details on the assay conditions are provided as Supplementary Method.
Miscellaneous procedures
For amplification of CAB genes by PCR, proofreading-competent Pwo DNA polymerase was used. Western blot analysis of epitope-tagged PanK variants was performed with peroxidase-conjugated anti-HA antibody (Roche Diagnostics) and luminol-containing detection system. CAB1 inserts of plasmids obtained by selection for second-site suppressor mutations of the cab1G351S allele or site-directed mutagenesis were completely sequenced to confirm the presence of the desired mutations and the absence of unwanted alterations (performed by LGC Genomics, Berlin, Germany).
Results
Intragenic suppressor mutations of a temperature-sensitive cab1 mutant allele
In a previous work, we could show that the mutant allele cab1 G351S encodes a temperature-sensitive variant of S. cerevisiae pantothenate kinase (Olzhausen et al. 2009). Interestingly, overexpression of a MET25-cab1 G351S promoter fusion using a multi-copy plasmid led to slow but substantial growth of transformants (not shown). Because of this residual activity, we hypothesized that selection for growth of strain JS91.14–24 (chromosomal cab1ts G351S mutation) at the nonpermissive temperature might allow identification of intragenic second-site mutations which reconstitute PanK enzyme activity. Thirty revertants which could grow at 37 °C on rich medium (YEPD) as well as on synthetic medium (SCD) similar to a wild-type strain were selected for further analysis. Chromosomal DNA was prepared from these strains and used for amplification of the CAB1 coding region. After fusing these CAB1 alleles with the MET25 promoter of a single-copy vector, resulting plasmids were introduced into the cab1ts strain. Twenty-seven plasmids could complement this mutation, indicating that they contain an intragenic suppressor mutation which again enables biosynthesis of a functional PanK. Comparative sequence analysis of the cloned CAB1 variants showed that they represent four distinct intragenic second-site mutations (A22G, F103V, D114E, and W331L). As shown in Fig. 2, transformants of the cab1ts G351S mutant strain containing additional CAB1 variants A22G G351S, F103V G351S, D114E G351S, and W331L G351S exhibit a substantial increase of PanK activity (with W331L G351S being most effective) and were able to grow at 37 °C similar to the wild-type. We conclude that variants A22G, F103V, D114, and W331L are critical for PanK activity of the yeast enzyme.

Phenotypic analysis of intragenic cab1 G351S revertants. Single-copy plasmid p416-MET25 (empty vector, negative control) and expression plasmids containing CAB1 wild-type (pTM8) and allele variants G351S W331L (pLS12-S8), G351S D114E (pLS12-S9), G351S F103V (pLS12-S10), and G351S A22G (pLS12-Y6) were transformed into strain JS91.14–24 (ura3 cab1ts G351S). Serial dilutions of transformants were spotted on selective synthetic medium (SCD) and incubated at 30 °C and 37 °C, respectively. Pantothenate kinase (PanK) activity in protein extracts prepared from transformants was assayed at 37 °C. For each assay, 75 μg of total protein was used. PanK activities are given in cpm 1-14C-phosphopantothenate formed per μg protein. Standard deviations are shown in parentheses
Inhibition of S. cerevisiae PanK by acetyl-CoA
It has been reported that the enzyme activity of eukaryotic pantothenate kinases from fungal and mammalian organisms is mainly regulated by acetyl-CoA and less effective by CoA (Calder et al. 1999; Zhang et al. 2005). We thus investigated whether CoA and its thioesters influence the activity of the CAB1 encoded PanK from S. cerevisiae. Strain JS91.14–24 was transformed with a multi-copy expression plasmid containing a MET25-CAB1 fusion and subsequently used to prepare partially purified PanK from a protein extract. PanK assays were performed at 37 °C in the presence of varying concentrations of CoA and its thioesters acetyl-CoA, malonyl-CoA, and palmitoyl-CoA. As is apparent from Fig. 3b, acetyl-CoA clearly inhibits PanK activity (IC50 = 36 μM; inhibitory concentration leading to 50% reduction of PanK activity). In contrast, some inhibition was also observed with CoA, malonyl-CoA, and palmitoyl-CoA, but significantly higher concentrations were needed (IC50 = 283 μM, 812 μM, and 891 μM, respectively; Fig. 3a, c, d).

Influence of CoA and its acyl derivatives on pantothenate kinases. Multi-copy expression plasmids pSBS5 (MET25-CAB1; wild-type; a–d), pJO73 (MET25-PANK3; murine wild-type; e), and pEB27 (MET25-CAB1 W331R; hyperactive yeast allele; f) were transformed into strain JS91.14–24 (cab1ts). To remove contaminating soluble yeast metabolites, crude extracts of transformants were partially purified by PanK binding to DEAE sepharose, elution, and subsequent dialysis. Varying concentrations of CoA (a), acetyl-CoA (b, e, f), malonyl-CoA (c), and palmitoyl-CoA (d) as indicated were added to PanK assay mixtures. Relative PanK activities (%) refer to enzyme activities in the absence of CoA inhibitors (100%). Standard deviations for at least four independent assays per inhibitor concentration are shown
Previously, inhibitory concentrations for acetyl-CoA of murine isoenzymes PanK1β (IC50 = 10 μM) and PanK3 (IC50 = 1 μM; Zhang et al. 2005) have been reported. For a direct comparison with the yeast enzyme, we thus studied acetyl-CoA-dependent activity of PanK3 from Mus musculus (IC50 = 1 μM; Fig. 3e) and could demonstrate that the single PanK from S. cerevisiae is clearly less sensitive against inhibition by acetyl-CoA than mammalian PanK3.
Site-directed mutagenesis of the PanK acetyl-CoA binding region
Inhibition of PanK activity by acetyl-CoA must be considered a key mechanism regulating the CoA biosynthetic pathway, thus preventing overproduction of CoA. We thus reasoned that overexpression of CAB genes may not be sufficient to increase the cellular CoA level. However, this problem may be overcome by construction of a PanK variant which is no longer affected by acetyl-CoA.
To obtain such a PanK variant, we took advantage of the crystal structure obtained for the human PanK3 isoenzyme in the presence of acetyl-CoA (Hong et al. 2007). Structural modelling indicated that binding sites of pantothenate as a substrate and acetyl-CoA as a competitive feedback inhibitor partially overlap (cf. Supplementary Fig. S1). While several amino acid side chains are presumably required for interaction with substrate and inhibitor, some residues may be specifically involved in binding of acetyl-CoA. Thus, it should be possible to generate PanK variants of the wild-type which are not affected for binding of pantothenate but do no longer interact with acetyl-CoA, uncoupling binding of substrate and inhibitor. Following alignment of human PanK3 and yeast Cab1 sequences, we selected residues for site-directed mutagenesis presumably important for binding of pantothenate, ATP, and/or acetyl-CoA (N155, S158, R173, Y326, F330, W331, A233 and I234). It should be emphasized that W331 forming hydrophobic interactions with the methyl group of acetyl-CoA has been independently identified by selecting suppressors of the cab1ts mutant allele G351S (see above). CAB1 variant W331L was constructed because it corresponds to the second-site suppressor of the G351S allele but now in the sequence context of an intact enzyme. We also constructed a W331R variant which should abolish binding of acetyl-CoA even more effectively. Arginine instead of tryptophan was selected because the PanK from Staphylococcus aureus (coaA gene product) which is moderately related to the yeast enzyme (18% identity, 29% similarity; cf. Supplementary Fig. S2) but insensitive to CoA and acetyl-CoA (Leonardi et al. 2005a) contains this amino acid (R244) at the position corresponding to yeast W331 (W340 in human PanK3).
CAB1 reading frame cassettes containing the desired mutations were inserted into a multi-copy expression plasmid, activated by the MET25 promoter. Protein extracts from transformants of strain JS91.14–24 (ura3 cab1ts) were used to assay PanK activities. Using the plasmid shuffling strategy (Sikorski and Boeke 1991), CAB1 variants were also tested for functional complementation of a cab1 null mutation. We thus transformed a wild-type strain with a single-copy URA3 plasmid containing the authentic CAB1 gene, deleted the chromosomal copy of CAB1, and subsequently introduced single-copy LEU2 plasmids encoding CAB1 variants. Use of a synthetic medium containing FOA (5-fluoroorotic acid) allows for counter-selection of plasmids with URA3 as a selection marker so that growth entirely depends on the functionality of CAB1 variants present on LEU2 plasmids.
CAB1 variants S158V, R173A, and I234E failed to complement a cab1 null mutation (no growth on FOA-containing synthetic medium; Supplementary Fig. S3) and encoded PanK enzymes had completely or almost completely lost their activity (Fig. 4a). All gene variants tested could be stably expressed in strain JS91.14–24 (Fig. 4b). Variants N155V and A233E were able to complement the cab1 null allele and showed residual PanK activity (31% and 22% of the wild-type level, respectively). Loss of aromatic residues affecting interaction with the acetyl moiety of acetyl-CoA (double variant Y326A F330A and W331L) led to slight or substantial increase of PanK activity. Importantly, Cab1 W331R turned out as a hyperactive PanK variant the activity of which was more than fourfold increased compared with the wild-type enzyme. We finally investigated whether the activity of this PanK variant responds to acetyl-CoA. In contrast to the wild-type enzyme, Cab1 W331R was completely insensitive to increased concentrations of acetyl-CoA (compare Fig. 3b and f). We conclude that the hyperactive and completely deregulated PanK variant W331R may be a suitable tool for engineering the CoA biosynthetic pathway.

Functional analysis of CAB1 variants. a Activity of PanK variants constructed by site-directed mutagenesis. The coding region of wild-type CAB1 was mutagenized at the positions indicated and gene variants were inserted into a multi-copy expression vector containing the MET25 promoter together with the HA epitope. The resulting plasmids pSBS5 (CAB1 wild-type), pEB5 (CAB1 Y326A F330A), pEB6 (cab1 I234E), pEB8 (CAB1 W331L), pEB22 (CAB1 N155V), pEB23 (cab1 S158V), pEB25 (cab1 R173A), pEB26 (CAB1 A233E), and pEB27 (CAB1 W331R) were transformed into strain JS91.14–24 (cab1ts). Transformants were cultivated at 30 °C until the mid-log growth phase. Pantothenate kinase (PanK) activity in protein extracts prepared from transformants was assayed at 37 °C. For each assay, 75 μg of total protein was used. PanK activities are given in cpm 1-14C-phosphopantothenate formed per μg protein. Standard deviations are indicated by error bars. b Stable expression of PanK variants was investigated by Western blot analysis of protein extracts prepared from transformants using anti-HA antibodies
Functional analysis of human COASY in S. cerevisiae
To facilitate pathway engineering of yeast CoA biosynthesis, we supposed that the mammalian gene COASY encoding the bifunctional CoA Synthase could be possibly used instead of individual genes CAB4 and CAB5. Assaying COASY for functional complementation of null mutations cab4 and cab5 should provide evidence whether such a strategy is helpful. For this analysis, we again used the strategy of plasmid shuffling (as described above for functional studies of CAB1 variants). Wild-type strains with single-copy URA3 rescue plasmids containing CAB4 or CAB5 were used to introduce null mutant alleles cab4Δ::HIS3 and cab5Δ::HIS3, respectively. To assay for functional complementation, a cDNA encoding the human COASY gene (564 aa) driven by the yeast MET25 promoter was inserted into a single-copy LEU2 plasmid. Corresponding LEU2 plasmids containing authentic yeast genes CAB4 and CAB5, respectively, were constructed and used as a positive control. As shown in Fig. 5a, hCOASY could partially complement a cab4Δ mutation (although clearly less effective than CAB4), while only residual growth was observed with hCOASY in the cab5Δ mutant strain. Human CoA synthase could be efficiently synthesized in S. cerevisiae (Fig. 5b), indicating that this result is not a consequence of failure to express the heterologous gene. Since full-length hCoasy is associated with mitochondria (Zhyvoloup et al. 2003), we repeated this experiment with a variant lacking aa 1–29 being responsible for this targeting in mammalian cell lines. However, the truncated hCOASY variant encoding aa 30–564 was completely unable to complement null mutations cab4Δ and cab5Δ, respectively (not shown). We thus conclude that the bifunctional COASY gene cannot be used to simplify engineering of the CoA biosynthetic pathway in yeast. A similar strategy can be considered with bacterial coaBC, encoding a bifunctional enzyme corresponding to Cab2 + Cab3. Although coaBC could complement null mutations cab2 and cab3, transformants showed slower growth, compared with authentic genes CAB2 and CAB3, respectively (not shown).

Functional analysis of human CoA Synthase gene (COASY) in S. cerevisiae. a For plasmid shuffling, strains LSY20 (cab4Δ + rescue plasmid pGE7 [ARS CEN URA3 CAB4]; upper part) and LSY21 (cab5Δ + rescue plasmid pGE9 [ARS CEN URA3 CAB5]; lower part) were transformed with the ARS CEN LEU2 plasmid pLS20 containing the human COASY gene activated by the MET25 promoter. Plasmids pGE8 (CAB4) and pGE10 (CAB5) served as positive controls, empty vector YCp111 as a negative control. FOA: 5-Fluoroorotic acid. b Stable biosynthesis of full-length hCoasy and truncated variant in transformants of S. cerevisiae. Expression plasmids pLS14 and pLS15 encoding HA-tagged length variants hCoasy1-564 and hCoasy30-564, respectively, were transformed into strain JS91.15–23. For comparison of expression efficiencies, S. cerevisiae Cab3 of similar size has been also analyzed (expression plasmid pJO3). Protein extracts were analyzed by immuno-blotting using anti-HA antibodies
Construction of strains stably overexpressing CoA biosynthetic genes
To elevate the cellular level of CoA and/or acetyl-CoA, we wished to overproduce all enzymes being important for this pathway. This should be achieved by stable integration of additional gene copies, activated by a strong control region. Our previous characterization of CAB genes showed that these genes are expressed at a moderate or low level and that expression is not substantially influenced by the carbon source or availability of amino acids (Olzhausen et al. 2009). Consequently, use of an effective control region to elevate CAB gene transcription in combination with the allele CAB1 W331R may allow increasing the biosynthesis of CoA and/or acetyl-CoA which now should no longer inhibit the initial PanK reaction of the pathway. We thus selected the strong TPI1 promoter of the glycolytic enzyme triosephosphate isomerase for high-level expression of genes CAB1 (wild-type and W331R variant), CAB2, CAB3, HAL3, CAB4, CAB5, and FEN2. Although PPCDC is a heterotrimeric enzyme in yeast, co-overexpression of CAB3 and HAL3 should be sufficient for a substantially elevated enzyme activity (Ruiz et al. 2009). Since Hal3 is a moonlighting enzyme with an additional metabolic function, only aa 260–495 of Hal3 which are relevant for its PPCDC function (Hal3PD; Abrie et al. 2012) were overproduced. To study the possible influence of increased supply of pantothenate, we decided to also overexpress FEN2, encoding the pantothenate transporter of the plasma membrane (Stolz and Sauer 1999).
For stable chromosomal integration of up to seven additional genes, we constructed plasmid pLEUTEX3, containing LEU2 as a selection marker flanked by two loxP sites. Thus, LEU2 can be removed later by induction of the cre recombinase (marker rescue). The plasmid also contains TPI1 promoter and CYC1 terminator, separated by the HA epitope and a versatile cloning site for insertion of genes of CoA biosynthesis. Finally, flanking sequences from a suitable chromosomal site must be added to enable integration of the expression cassette at a position of choice by homologous recombination. Details of the construction procedure are provided as supplementary description, and functional elements of this cassette are shown in Supplementary Fig. S4. Because of the mobile LEU2 selection marker, repeated integration events of TPI1-CAB gene fusions are possible.
Using basic plasmid pLEUTEX3, a number of expression cassettes with individual coding regions of CoA biosynthetic genes activated by TPI1 were constructed. For subsequent site-specific integration, flanking sequences from genes with an extended upstream region were selected. Thus, positions of integration should not interfere with expression of nearby genes, preventing a possibly reduced fitness of transformants with additional CAB genes. Two upstream fragments of about 400 bp from selected genomic sites were amplified by PCR and ligated to the expression cassettes on both sides (selected positions: “upstream” regions of genes ADH3, CUP9, ENO1, GAT1, HIP1, MRH1, und ZPS1). Release of the complete linear cassette including its flanking regions by cleavage with restriction enzymes and subsequent transformation of a leu2 mutant should allow its stable integration by homologous recombination at the desired gene locus (Rothstein 1991). Plasmids derived from pLEUTEX3 which were subsequently used to integrate additional copies of CoA biosynthetic genes are listed in Table 2.
Increased production of acetyl-CoA by overexpression of CAB genes
After having completed the construction of 8 pLEUTEX plasmids (7 different genes and CAB1 wild-type and W331R variant, respectively), linear expression constructs were integrated one after another into leu2 strains JS91.15–23 and BY4741, representing different strain backgrounds. Our previous work on CoA biosynthesis was performed with strain JS91.15–23 (Olzhausen et al. 2009), while BY4741 has been routinely used for functional gene studies (Brachmann et al. 1998). To investigate whether such differences possibly influence our intended engineering of CoA biosynthesis, both strains were treated in parallel. The order of integration events (CAB2 – CAB3 – CAB4 – CAB5 – CAB1 [wild-type or W331R variant] – HAL3PD – FEN2) is apparent from the complete list of constructed strains (cf. Table 1 of Supplementary Information). Successful integration of each expression construct at the correct genomic positions was verified by analytic PCR with gene-specific primers (not shown). Transformants also contained episomal plasmid pSH62, encoding a GAL1-cre fusion which allowed galactose-inducible synthesis of cre recombinase for removal of loxP-flanked selection marker LEU2 after each step (Güldener et al. 1996).
Strains MGY22 (derived from JS91.15–23) and MGY23 (derived from BY4741) were obtained by multiple rounds of gene integration and contain additional genes CAB1 W331R, CAB2, CAB3, HAL3PD, CAB4, and CAB5. Both strains (together with their isogenic reference strains without additional CAB genes) were cultivated in YPD-rich medium until mid-log phase and subsequently used for preparation of protein-free extracts. To quantify CoA biosynthesis in these metabolite extracts, an enzymatic assay simultaneously recording CoA + acetyl-CoA was used (Bergmeyer 1974). To distinguish between both nucleotides, NEM (N-ethylmaleimide) was added to certain assays, blocking the free SH group of CoA. As a result, only acetyl-CoA is able to react in the assay system.
Importantly, strong overexpression of 6 CoA biosynthetic genes (CAB1 W331R CAB2 CAB3 HAL3PD CAB4 CAB5) resulted in a 15-fold increase of CoA nucleotides in the JS strain background when transformants were grown in YPD-rich medium (0.9 μMol/g protein in the reference strain vs. 13.5 μMol/g protein in strain MGY22 with additional CAB genes; Fig. 6). Almost identical results were obtained with transformants in the BY background (17-fold increase compared with the reference strain). The level of CoA nucleotides was slightly reduced after NEM had been added to extracts from the reference strain (regular CAB gene dosage), indicating that most CoA exists as acetyl-CoA which is insensitive against NEM. This finding appears plausible, considering the inhibition of PanK by acetyl-CoA and much weaker by CoA (see above, Fig. 3a, b). In the overproducing strain MGY22, acetyl-CoA constitutes 78% of total CoA. Interestingly, cultivation of strain MGY22 in synthetic SCD medium led to a substantially lower level of CoA nucleotides as found after growth in YPD (2.6-fold increase vs. 15-fold increase; Fig. 6). In contrast, CoA biosynthesis in the SCD-grown reference strain is similar to the same strain grown in YPD. This finding provides evidence for a limitation of precursor molecules when the overproducing strain is grown in standard SCD, preventing a level of CoA biosynthesis as observed in rich medium.

Influence of CAB gene dosage variation on biosynthesis of CoA nucleotides. CoA and acetyl-CoA were simultaneously measured by enzymatic analysis in protein-free cell extracts of reference strains JS91.15–23/BY4741 and transformants MGY22/MGY23. Additional TPI1-dependent genes introduced into strains are indicated by + . Transformants were cultivated in rich medium (YPD) or synthetic complete medium (SCD), both containing 2% glucose as a carbon source. NEM (N-ethylmaleimide) was added to certain reactions to distinguish between CoA and acetyl-CoA. For comparison, transformants of two different strain backgrounds (JS and BY) were investigated. Standard deviations are indicated by error bars
As an estimation of CAB gene overexpression by TPI1-dependent transcription, we comparatively assayed PanK activity in strains JS91.15–23 (regular gene dosage, PanK sensitive to acetyl-CoA) and MGY22 (contains the deregulated hyperactive PanK encoded by TPI1-CAB1 W331R and additional TPI1-CAB genes). It turned out that the activity of pantothenate kinase in protein extracts of MGY22 was about 13-fold increased relative to the reference (JS91.15–23: 72 cpm/μg protein, MGY22: 950 cpm/μg protein).
Influence of genetic and physiological variations on production of acetyl-CoA
After having shown that overexpression of CoA biosynthetic genes led to a strong increase of acetyl-CoA in protein-free cell extracts, we wished to study the relative contribution of the genes involved in more detail. Since engineering of the CoA biosynthetic pathway led to similar results in both strain backgrounds tested, subsequent studies focused on strain JS91.15–23 and its derivatives. As described above, variant CAB1 W331R present in strain MGY22 encodes a PanK which is completely insensitive against inhibition by acetyl-CoA. We thus investigated whether overexpression of wild-type CAB1 is able to similarly increase CoA biosynthesis as observed with the deregulated variant. As is shown in Fig. 7, TPI1-dependent expression of wild-type CAB1 in combination with 5 additional genes of CoA biosynthesis (strain MGY24) was also able to increase the production of CoA + acetyl-CoA (4.1-fold, compared with the reference strain). However, this increase remained substantially below the level obtained in the presence of CAB1 W331R (15-fold increase), proving that release of PanK from inhibition by acetyl-CoA is an essential prerequisite for efficient engineering of CoA biosynthesis. We next studied whether gene variant CAB1 W331R may be solely responsible for elevation of CoA nucleotides. Indeed, strain TUY1 containing only the TPI1-CAB1 W331R expression cassette and regular dosage of the remaining CoA biosynthetic genes showed a clearly increased level of CoA + acetyl-CoA (4.8-fold increase) which nevertheless was lower than the yield observed with strain MGY22. We also investigated the influence of PPCDC subunits on CoA biosynthesis, comparing two strains with and without a TPI1-HAL3PD expression cassette. In strain MGY18 devoid of this cassette, CoA + acetyl-CoA were 13-fold elevated with respect to the reference (Fig. 7), arguing for only a minor contribution of additional Hal3PD to the CoA biosynthetic pathway.

Various combinations of additional CAB genes and influence on biosynthesis of CoA nucleotides. CoA and acetyl-CoA were simultaneously measured by enzymatic analysis in protein-free cell extracts of reference strain JS91.15–23 and transformants MGY22/MGY24/TUY1/MGY18. Additional TPI1-dependent genes introduced into strains are indicated by + . All transformants were cultivated in rich medium (YPD). Standard deviations are indicated by error bars
Our finding that production of CoA nucleotides in MGY22 after cultivation in standard synthetic medium (SCD) was substantially below the level observed with YPD-rich medium strongly indicates limited supply with a critical nutrient. We thus supplemented standard SCD with precursor pantothenate (20 mg/l; 50-fold increase relative to the contribution of yeast nitrogen base) which can be taken up by plasma membrane transporter Fen2 (Sauer and Stolz 1999). In a similar experiment, β-alanine was also used for supplementation (200 mg/l; possibly imported by general amino acid permease Gap1 or a related permease; Van Zeebroeck et al. 2014). While imported pantothenate can be directly used by PanK, β-alanine must react with pantoate (catalyzed by pantothenate synthase, PAN6) to finally give pantothenate. The availability of β-alanine may be critical for pantothenate biosynthesis because yeast, in contrast to E. coli, does not contain an aspartate decarboxylase gene and generates β-alanine inefficiently from polyamines (White et al. 2001).
Interestingly, even in the unmodified reference strain, supplementation of SCD medium with β-alanine or pantothenate could elevate the amount of CoA + acetyl-CoA (2.3 and 4.8-fold; Fig. 8). Growth of strain MGY22 in SCD supplemented with β-alanine stimulated CoA biosynthesis about fourfold, while additional pantothenate increased the amount of CoA nucleotides almost tenfold (28.4 μMol/g protein; Fig. 8), exceeding even the productivity observed after cultivation in YPD-rich medium (13.5 μMol/g protein; Fig. 6). Although the concentration of pantothenate in rich medium is unknown, it may be still limiting. Indeed, supplementation of YPD with additional 20 mg/l pantothenate resulted in a further increase of CoA + acetyl-CoA (30.7 μMol/g protein; Fig. 8). Together, these results confirm that the availability of pantothenate is a critical factor for the metabolic flux towards CoA.

Influence of media supplementation on biosynthesis of CoA nucleotides. CoA and acetyl-CoA were simultaneously measured by enzymatic analysis in protein-free cell extracts of reference strain JS91.15–23 and transformant MGY22. Additional TPI1-dependent genes introduced into MGY22 are indicated by + . Transformants were cultivated in synthetic complete medium (SCD; 0.4 mg/l Ca-pantothenate; 0.84 μM) or rich medium (YPD), both containing 2% glucose as a carbon source. Media were supplemented to final concentrations of 200 mg/l β-alanine (βA; 2.25 mM) and 20 mg/l Ca-pantothenate (Pan, 50-fold compared with standard-SCD, 42 μM), respectively. Standard deviations are indicated by error bars
Assuming that CoA biosynthesis can be further improved by an additional copy of the pantothenate permease gene, we finally decided to introduce a TPI1-FEN2 expression cassette into MGY22. The resulting strain TUY4 was then cultivated in media enriched with pantothenate (50- and 100-fold, respectively). As is shown in Fig. 9, increase of the FEN2 gene dosage could indeed further elevate the concentration of CoA nucleotides when pantothenate was present in non-limiting amounts. This increase was more apparent when transformants MGY22 and TUY4 were cultivated with 100-fold pantothenate (40 mg/l; + 27%) compared with 50-fold pantothenate (20 mg/l; + 16%). Supplementation with even higher concentrations of pantothenate could not substantially elevate the cellular yield of CoA nucleotides (not shown).

Influence of FEN2 overexpression and deletion of PCD1 on biosynthesis of CoA nucleotides. CoA and acetyl-CoA were simultaneously measured by enzymatic analysis in protein-free cell extracts of transformants MGY22, TUY4 (+ FEN2), LRY1 (Δpcd1), and LRY2 (+ FEN2 Δpcd1). Additional TPI1-dependent genes are indicated by + . Transformants were cultivated in synthetic complete medium (SCD), containing 2% glucose as a carbon source. Media were supplemented to final concentrations of 20 mg/l or 40 mg/l Ca-pantothenate (Pan, 50- or 100-fold compared with standard-SCD, 42 μM or 84 μM). Deletion of the genomic PCD1 gene encoding a CoA phosphatase is shown by Δ. Standard deviations are indicated by error bars
The cellular level of CoA may not only be affected by its biosynthesis but also by degradation of the nucleotide structure. The nudix hydrolase encoded by PCD1 encodes a pyrophosphatase which is effective against oxidized derivatives of CoA (CoA disulfide), but the purified enzyme also showed a substantial specificity for CoA and acetyl-CoA (Cartwright et al. 2000). Thus, we finally introduced a pcd1Δ null mutant allele into strains MGY22 and TUY4 (to give LRY1 and LRY2, respectively) and comparably investigated whether the concentration of CoA nucleotides changed. Indeed, with both strains, we observed a further increase of CoA + acetyl-CoA (by 11–15%; Fig. 9), indicating that some hydrolysis even of intact CoA occurs when PCD1 is functional.
Discussion
Engineering of a pathway not only requires overexpression of the biosynthetic genes involved but has also taken into account that activity of enzymes may be affected by feedback inhibition by the end-product of the pathway. Similar to what has been shown for E. coli (Rock et al. 2003), this work demonstrates that biochemical regulation of PanK is a key mechanism controlling the metabolic flux towards CoA and its major thioester derivative acetyl-CoA. Taking advantage of the crystal structure of E. coli CoaA in the presence of the feedback inhibitor CoA, Rock et al. (2003) constructed and characterized a CoaA variant (R106A) which exhibited 54% activity of the wild-type enzyme but was completely insensitive against inhibition by CoA. As a result, the intracellular level of CoA increased about fourfold. For a mammalian cell line transfected with a mPANK1β expression construct, a CoA level 13 times higher than with control cells was reported (Rock et al. 2000).
In this work, we show that the S. cerevisiae PanK (Cab1) is inhibited by acetyl-CoA with an IC50 concentration similar to what has been reported for the enzyme from A. nidulans (36 μM for ScPanK and 32 μM for AnPanK; Calder et al. 1999). We used a dual strategy to identify variants of Cab1 which are no longer inhibited by acetyl-CoA: (I) Genetic selection for intragenic revertants of the conditional mutant cab1 G351S with restored growth at 37 °C led to identification of the double mutation G351S W331L. PanK activity of the encoded enzyme reached 53% of the wild-type level, explaining why the growth defect of the single mutation could be suppressed. We hypothesized that a mutation improving activity of a defective enzyme may also positively affect the activity of the wild-type enzyme. (II) W331 is a strictly conserved residue of eukaryotic pantothenate kinases involved in binding of acetyl-CoA (Hong et al. 2007). Release from competitive inhibition of PanK may be achieved by replacing amino acids which specifically influence binding of acetyl-CoA but leave substrate binding sites unaffected. We thus replaced amino acids involved in inhibitor binding by residues presumed to be non-functional and assayed activity and regulation of the resulting yeast PanK variants. Indeed, Cab1 variants with the double mutation Y326A F330A or the single mutation W331L exhibited elevated PanK activities, confirming that it is indeed possible to uncouple binding of inhibitor and substrates. Importantly, variant W331R increased the activity of PanK by a factor of four and rendered the enzyme completely insensitive against inhibition by acetyl-CoA. The observed hyperactivity indicates that PanK variant W331R not only prevents binding of the inhibitor acetyl-CoA but may also facilitate substrate access. It should be mentioned that Hong et al. (2007) obtained variants of human PanK3 some of which also showed elevated enzyme activity (e.g., 1.85-fold increase of PanK3 R78C).
Although the CoA biosynthetic pathway is universal, structural organization of the enzymes involved may differ. This is apparent for the bifunctional mammalian enzyme COASY, corresponding to individual gene products Cab4 and Cab5 in S. cerevisiae. However, it turned out that COASY could only partially complement a cab4 null mutation and was unable to replace Cab5. Consequently, for the intended engineering of CoA biosynthesis, only authentic genes from S. cerevisiae were used. Although the acetyl-CoA-sensitive PanK encoded by CAB1 is clearly of major regulatory importance, we nevertheless decided to introduce additional copies of at least 6 genes (CAB1 W331R, CAB2, CAB3, HAL3PD, CAB4, and CAB5) overexpression of which should guarantee substantially elevated activities of 5 enzymes, thus preventing possible bottlenecks in the course of CoA biosynthesis. This was achieved by fusing CAB coding regions with the strong control region of the glycolytic gene TPI1 and subsequent stable integration of fusion genes at selected genomic positions, using a marker rescue strategy which allows repeated use of the selection marker LEU2.
Successive transformation of two S. cerevisiae strains representing different genetic backgrounds with 6 expression cassettes, cultivation of the resulting transformants in rich medium, and quantification of CoA + acetyl-CoA in protein-free extracts revealed that these CoA nucleotides were indeed strongly overproduced in both strains at a similar level (MGY22: 15-fold; MGY23: 17-fold). As supposed, CAB1 variant W331R encoding a hyperactive PanK insensitive against inhibition by acetyl-CoA substantially contributed to this increase. This was shown by comparative analysis of a strain identical to MGY22 but overexpressing the CAB1 wild-type gene (MGY24; fourfold increase). Although the CAB1 W331R allele is of major importance for an elevated level of CoA nucleotides, the remaining CAB genes must be also overexpressed to achieve maximal overproduction (as shown by strain TUY1 with merely an extra copy of CAB1 W331R; 4.8-fold overproduction).
Comparative cultivation of the CoA overproducer MGY22 in different media revealed that growth in synthetic medium SCD was unable to ensure an increased level of CoA nucleotides as observed after growth in rich medium YPD (2.6-fold increase vs. 15-fold). This deficiency could be overcome by supplementation of SCD with β-alanine or, even more effective, with pantothenate as the immediate precursor of the pathway, leading to a level of CoA nucleotides even higher as observed after growth in YPD.
In the yeast Schizosaccharomyces pombe, overexpression of the liz1+ gene homologous to the high-affinity H+-pantothenate symporter FEN2 of S. cerevisiae could elevate the transport capacity of pantothenate 430-fold above the wild-type level (Stolz et al. 2004). In addition to 6 extra copies of CoA biosynthetic genes, we thus also introduced a TPI1-FEN2 fusion into strain MGY22. Cultivating the resulting strain TUY4 in media heavily supplemented with pantothenate (50- and 100-fold), CoA nucleotides indeed showed a further increase (by 16% and 27%, respectively).
The intracellular concentration of CoA nucleotides may not only be influenced by their biosynthesis but also by degradation. Presumably, hydrolysis by a phosphatase (Pcd1 in yeast; Cartwright et al. 2000) is the initial step for complete degradation of CoA, finally releasing ADP, pantothenate, and cysteamine (Naquet et al. 2020). Introduction of a pcd1Δ null allele into strains MGY22 and TUY4 again elevated the level of CoA nucleotides, confirming degradation as an important regulatory step which contributes to the CoA steady-state level. When strain LRY2 (overexpression of 7 additional genes with a pcd1Δ null mutation) was cultivated in pantothenate-supplemented SCD, we were able to detect the highest level of CoA nucleotides found in the course of this work (42.6 μMol/g protein) which means an almost 39-fold increase, compared with the unmodified reference strain grown in standard SCD.
Because of this substantial overproduction described, our strains may be helpful tools to improve the metabolic flux towards biosynthetic pathways depending on acetyl-CoA. Phenotypic characterization of CoA-overproducing strains revealed that they are fully viable and are able to utilize carbon sources glucose, ethanol, or acetate with the same efficiency as the wild-type. No morphological differences concerning cell size and budding pattern could be observed by microscopic inspection.
Previously, other strategies for improvement of biosynthetic pathways dependent on acetyl-CoA were used. Hong et al. (2019) overexpressed CAB1 (regulatory wild-type) + CAB3 + ATF1 (encoding an alcohol acetyl transferase) to increase the yield of ethyl acetate and other esters in S. cerevisiae. For improved biosynthesis of the biofuel n-butanol by S. cerevisiae, Schadeweg and Boles (2016a) decided to overproduce pantothenate kinase from E. coli (which is inhibited by CoA, but not by acetyl-CoA; MET25-coaA fusion) or the pantothenate permease (MET25-FEN2 fusion). Indeed, both strategies were able to improve n-butanol production. To elevate cellular supply with β-alanine, Schadeweg and Boles (2016b) also overexpressed the amine oxidase gene FMS1 which is rate-limiting for its biosynthesis from spermine (White et al. 2001), giving a further increase of n-butanol yield. In other work, introduction of bacterial genes encoding subunits of a cytosolic pyruvate dehydrogenase (without mitochondrial localization sequences) into a strain devoid of alcohol dehydrogenase genes (adhΔ) allowed a direct conversion of glycolytic pyruvate to acetyl-CoA, leading to an increased yield of n-butanol (Lian et al. 2014). The level of n-butanol could be also elevated after heterologous expression of the ATP-dependent citrate lyase gene ACL1 from the oleaginous yeast Yarrowia lipolytica, enabling to cleave citrate into oxaloacetate and acetyl-CoA. Chen et al. (2013) were able to improve conversion of acetaldehyde into acetyl-CoA by co-expression of ALD6 (encoding NADP-dependent aldehyde dehydrogenase) and a bacterial acetyl-CoA synthetase gene (acsSEL641P of Salmonella enterica; resistant against inhibition by acetylation). While Hong et al. (2019) and Schadeweg and Boles (2016a, b) partially intervened into the CoA biosynthetic pathway, these latter strategies increased the level of acetyl-CoA by shifting the ratio among CoA nucleotides without affecting total CoA concentration. Since the genetic manipulations described in this work affect all steps of CoA biosynthesis, the resulting substantial elevation especially of the acetyl-CoA concentration could be advantageous for existing and future strategies of metabolic engineering in S. cerevisiae.
Data availability
Original data are available upon request. Additional information is provided in the Supplementary Material.
Code availability
Not applicable.
Change history
13 February 2022
Open Access funding enabled and organized by Projekt DEAL has been added as funding.
29 January 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00253-022-11803-7
References
Abrie JA, González A, Strauss E, Ariño J (2012) Functional mapping of the disparate activities of the yeast moonlighting protein Hal3. Biochem J 442:357–368. https://doi.org/10.1042/BJ20111466
Abrie JA, Molero C, Ariño J, Strauss E (2015) Complex stability and dynamic subunit interchange modulates the disparate activities of the yeast moonlighting proteins Hal3 and Vhs3. Sci Rep 5:15774. https://doi.org/10.1038/srep15774
Baković J, López Martínez D, Nikolaou S, Yu BYK, Tossounian MA, Tsuchiya Y, Thrasivoulou C, Filonenko V, Gout I (2021) Regulation of the CoA biosynthetic complex assembly in mammalian cells. Int J Mol Sci 22:1131. https://doi.org/10.3390/ijms 22031131
Baković J, Yu BYK, Silva D, Chew SP, Kim S, Ahn SH, Palmer L, Aloum L, Stanzani G, Malanchuk O, Duchen MR, Singer M, Filonenko V, Lee TH, Skehel M, Gout I (2019) A key metabolic integrator, coenzyme A, modulates the activity of peroxiredoxin 5 via covalent modification. Mol Cell Biochem 461:91–102. https://doi.org/10.1007/s11010-019-03593-w
Bergmeyer HU (1974) Methods of enzymatic analysis. Vol. 4, 2nd edition, Academic Press, New York
Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115–132. https://doi.org/10.1002/(SICI)1097-0061(19980130)14:2%3c115::AID-YEA204%3e3.0.CO;2-2
Brohée S, Janky R, Abdel-Sater F, Vanderstocken G, André B, van Helden J (2011) Unraveling networks of co-regulated genes on the sole basis of genome sequences. Nucleic Acids Res 39:6340–6358. https://doi.org/10.1093/nar/gkr264
Calder RB, Williams RS, Ramaswamy G, Rock CO, Campbell E, Unkles SE, Kinghorn JR, Jackowski S (1999) Cloning and characterization of a eukaryotic pantothenate kinase gene (panK) from Aspergillus nidulans. J Biol Chem 274:2014–2020. https://doi.org/10.1074/jbc.274.4.2014
Cartwright JL, Gasmi L, Spiller DG, McLennan AG (2000) The Saccharomyces cerevisiae PCD1 gene encodes a peroxisomal nudix hydrolase active toward coenzyme A and its derivatives. J Biol Chem 275:32925–32930. https://doi.org/10.1074/jbc.M005015200
Chen Y, Daviet L, Schalk M, Siewers V, Nielsen J (2013) Establishing a platform cell factory through engineering of yeast acetyl-CoA metabolism. Metab Eng 15:48–54. https://doi.org/10.1016/j.ymben.2012.11.002
Daugherty M, Polanuyer B, Farrell M, Scholle M, Lykidis A, de Crécy-Lagard V, Osterman A (2002) Complete reconstitution of the human coenzyme A biosynthetic pathway via comparative genomics. J Biol Chem 277:21431–21439. https://doi.org/10.1074/jbc.M201708200
Dusi S, Valletta L, Haack TB, Tsuchiya Y, Venco P, Pasqualato S, Goffrini P, Tigano M, Demchenko N, Wieland T, Schwarzmayr T, Strom TM, Invernizzi F, Garavaglia B, Gregory A, Sanford L, Hamada J, Bettencourt C, Houlden H, Chiapparini L, Zorzi G, Kurian MA, Nardocci N, Prokisch H, Hayflick S, Gout I, Tiranti V (2014) Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet 94:11–22. https://doi.org/10.1016/j.ajhg.2013.11.008
Galdieri L, Zhang T, Rogerson D, Lleshi R, Vancura A (2014) Protein acetylation and acetyl coenzyme a metabolism in budding yeast. Eukaryot Cell 13:1472–1483. https://doi.org/10.1128/EC.00189-14
Güldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24:2519–2524. https://doi.org/10.1093/nar/24.13.2519
Hayflick SJ (2014) Defective pantothenate metabolism and neurodegeneration. Biochem Soc Trans 42:1063–1068. https://doi.org/10.1042/BST20140098
Hong BS, Senisterra G, Rabeh WM, Vedadi M, Leonardi R, Zhang YM, Rock CO, Jackowski S, Park HW (2007) Crystal structures of human pantothenate kinases. Insights into allosteric regulation and mutations linked to a neurodegeneration disorder. J Biol Chem 282:27984–27993. https://doi.org/10.1074/jbc.M701915200
Hong KQ, Fu XM, Dong SS, Xiao DG, Dong J (2019) Modulating acetate ester and higher alcohol production in Saccharomyces cerevisiae through the cofactor engineering. J Ind Microbiol Biotechnol 46:1003–1011. https://doi.org/10.1007/s10295-019-02176-4
Luso A, Wiersma M, Schüller HJ, Pode-Shakked B, Marek-Yagel D, Grigat M, Schwarzmayr T, Berutti R, Alhaddad B, Kanon B, Grzeschik NA, Okun JG, Perles Z, Salem Y, Barel O, Vardi A, Rubinshtein M, Tirosh T, Dubnov-Raz G, Messias AC, Terrile C, Barshack I, Volkov A, Avivi C, Eyal E, Mastantuono E, Kumbar M, Abudi S, Braunisch M, Strom TM, Meitinger T, Hoffmann GF, Prokisch H, Haack TB, Brundel BJJM, Haas D, Sibon OCM, Anikster Y (2018) Mutations in PPCS, encoding Phosphopantothenoylcysteine Synthetase, cause autosomal-recessive dilated cardiomyopathy. Am J Hum Genet 102:1018–1030. https://doi.org/10.1016/j.ajhg.2018.03.022
Leonardi R, Chohnan S, Zhang YM, Virga KG, Lee RE, Rock CO, Jackowski S (2005a) A pantothenate kinase from Staphylococcus aureus refractory to feedback regulation by coenzyme A. J Biol Chem 280:3314–3322. https://doi.org/10.1074/jbc.M411608200
Leonardi R, Zhang YM, Rock CO, Jackowski S (2005b) Coenzyme A: back in action. Prog Lipid Res 44:125–153. https://doi.org/10.1016/j.plipres.2005.04.001
Lian J, Si T, Nair NU, Zhao H (2014) Design and construction of acetyl-CoA overproducing Saccharomyces cerevisiae strains. Metab Eng 24:139–149. https://doi.org/10.1016/j.ymben.2014.05.010
Luo X, Reiter MA, d’Espaux L, Wong J, Denby CM, Lechner A, Zhang Y, Grzybowski AT, Harth S, Lin W, Lee H, Yu C, Shin J, Deng K, Benites VT, Wang G, Baidoo EEK, Chen Y, Dev I, Petzold CJ, Keasling JD (2019) Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 567:123–126. https://doi.org/10.1038/s41586-019-0978-9
Meadows AL, Hawkins KM, Tsegaye Y, Antipov E, Kim Y, Raetz L, Dahl RH, Tai A, Mahatdejkul-Meadows T, Xu L, Zhao L, Dasika MS, Murarka A, Lenihan J, Eng D, Leng JS, Liu CL, Wenger JW, Jiang H, Chao L, Westfall P, Lai J, Ganesan S, Jackson P, Mans R, Platt D, Reeves CD, Saija PR, Wichmann G, Holmes VF, Benjamin K, Hill PW, Gardner TS, Tsong AE (2016) Rewriting yeast central carbon metabolism for industrial isoprenoid production. Nature 537:694–697. https://doi.org/10.1038/nature19769
Milke L, Marienhagen J (2020) Engineering intracellular malonyl-CoA availability in microbial hosts and its impact on polyketide and fatty acid synthesis. Appl Microbiol Biotechnol 104:6057–6065. https://doi.org/10.1007/s00253-020-10643-7
Mumberg D, Müller R, Funk M (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res 22:5767–5768. https://doi.org/10.1093/nar/22.25.5767
Naquet P, Kerr EW, Vickers SD, Leonardi R (2020) Regulation of coenzyme A levels by degradation: the ‘Ins and Outs’. Prog Lipid Res 78:101028. 10.1016/ j.plipres.2020.101028
Olzhausen J, Moritz T, Neetz T, Schüller HJ (2013) Molecular characterization of the heteromeric coenzyme A-synthesizing protein complex (CoA-SPC) in the yeast Saccharomyces cerevisiae. FEMS Yeast Res 13:565–573. https://doi.org/10.1111/1567-1364.12058
Olzhausen J, Schübbe S, Schüller HJ (2009) Genetic analysis of coenzyme A biosynthesis in the yeast Saccharomyces cerevisiae: identification of a conditional mutation in the pantothenate kinase gene CAB1. Curr Genet 55:163–173. https://doi.org/10.1007/s00294-009-0234-1
Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, Polichuk DR, Teoh KH, Reed DW, Treynor T, Lenihan J, Fleck M, Bajad S, Dang G, Dengrove D, Diola D, Dorin G, Ellens KW, Fickes S, Galazzo J, Gaucher SP, Geistlinger T, Henry R, Hepp M, Horning T, Iqbal T, Jiang H, Kizer L, Lieu B, Melis D, Moss N, Regentin R, Secrest S, Tsuruta H, Vazquez R, Westblade LF, Xu L, Yu M, Zhang Y, Zhao L, Lievense J, Covello PS, Keasling JD, Reiling KK, Renninger NS, Newman JD (2013) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496:528–532. https://doi.org/10.1038/nature12051
Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 21:805–821. https://doi.org/10.1016/j.cmet.2015.05.014
Rock CO, Calder RB, Karim MA, Jackowski S (2000) Pantothenate kinase regulation of the intracellular concentration of coenzyme A. J Biol Chem 275:1377–1383. https://doi.org/10.1074/jbc.275.2.1377
Rock CO, Park HW, Jackowski S (2003) Role of feedback regulation of pantothenate kinase (CoaA) in control of coenzyme A levels in Escherichia coli. J Bacteriol 185:3410–3415. https://doi.org/10.1128/JB.185.11.3410-3415.2003
Rothstein R (1991) Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol 194:281–301. https://doi.org/10.1016/0076-6879(91)94022-5
Ruiz A, González A, Muñoz I, Serrano R, Abrie JA, Strauss E, Ariño J (2009) Moonlighting proteins Hal3 and Vhs3 form a heteromeric PPCDC with Ykl088w in yeast CoA biosynthesis. Nat Chem Biol 5:920–928. https://doi.org/10.1038/nchembio.243
Schadeweg V, Boles E (2016a) n-Butanol production in Saccharomyces cerevisiae is limited by the availability of coenzyme A and cytosolic acetyl-CoA. Biotechnol Biofuels 9:44. https://doi.org/10.1186/s13068-016-0456-7
Schadeweg V, Boles E (2016b) Increasing n-butanol production with Saccharomyces cerevisiae by optimizing acetyl-CoA synthesis, NADH levels and trans-2-enoyl-CoA reductase expression. Biotechnol Biofuels 9:257. https://doi.org/10.1186/s13068-016-0673-0
Shi L, Tu BP (2013) Acetyl-CoA induces transcription of the key G1 cyclin CLN3 to promote entry into the cell division cycle in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 110:7318–7323. https://doi.org/10.1073/pnas.1302490110
Shi L, Tu BP (2015) Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol 33:125–131. https://doi.org/10.1016/j.ceb.2015.02.003
Sibon OCM, Strauss E (2016) Coenzyme A: to make it or uptake it? Nat Rev Mol Cell Biol 17:605–606. https://doi.org/10.1038/nrm.2016.110
Sikorski RS, Boeke JD (1991) In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol 194:302–318. https://doi.org/10.1016/0076-6879(91)94023-6
Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27
Song WJ, Jackowski S (1992) Cloning, sequencing, and expression of the pantothenate kinase (coaA) gene of Escherichia coli. J Bacteriol 174:6411–6417. https://doi.org/10.1128/jb.174.20.6411-6417.1992
Spry C, Kirk K, Saliba KJ (2008) Coenzyme A biosynthesis: an antimicrobial drug target. FEMS Microbiol Rev 32:56–106. https://doi.org/10.1111/j.1574-6976.2007.00093.x
Srinivasan B, Baratashvili M, van der Zwaag M, Kanon B, Colombelli C, Lambrechts RA, Schaap O, Nollen EA, Podgoršek A, Kosec G, Petković H, Hayflick S, Tiranti V, Reijngoud DJ, Grzeschik NA, Sibon OC (2015) Extracellular 4’-phosphopantetheine is a source for intracellular coenzyme A synthesis. Nat Chem Biol 11:784–792. https://doi.org/10.1038/nchembio.1906
Stolz J, Sauer N (1999) The fenpropimorph resistance gene FEN2 from Saccharomyces cerevisiae encodes a plasma membrane H+-pantothenate symporter. J Biol Chem 274:18747–18752. https://doi.org/10.1074/jbc.274.26.18747
Stolz J, Caspari T, Carr AM, Sauer N (2004) Cell division defects of Schizosaccharomyces pombe liz1- mutants are caused by defects in pantothenate uptake. Eukaryot Cell 3:406–412. https://doi.org/10.1128/EC.3.2.406-412.2004
Szczebara FM, Chandelier C, Villeret C, Masurel A, Bourot S, Duport C, Blanchard S, Groisillier A, Testet E, Costaglioli P, Cauet G, Degryse E, Balbuena D, Winter J, Achstetter T, Spagnoli R, Pompon D, Dumas B (2003) Total biosynthesis of hydrocortisone from a simple carbon source in yeast. Nat Biotechnol 21:143–149. https://doi.org/10.1038/nbt775
Theodoulou FL, Sibon OC, Jackowski S, Gout I (2014) Coenzyme A and its derivatives: renaissance of a textbook classic. Biochem Soc Trans 42:1025–1032. https://doi.org/10.1042/BST20140176
Tippmann S, Scalcinati G, Siewers V, Nielsen J (2016) Production of farnesene and santalene by Saccharomyces cerevisiae using fed-batch cultivations with RQ-controlled feed. Biotechnol Bioeng 113:72–81. https://doi.org/10.1002/bit.25683
Vallari DS, Jackowski S, Rock CO (1987) Regulation of pantothenate kinase by coenzyme A and its thioesters. J Biol Chem 262:2468–2471
Van Zeebroeck G, Rubio-Texeira M, Schothorst J, Thevelein JM (2014) Specific analogues uncouple transport, signalling, oligo-ubiquitination and endocytosis in the yeast Gap1 amino acid transceptor. Mol Microbiol 93:213–233. https://doi.org/10.1111/mmi.12654
White WH, Gunyuzlu PL, Toyn JH (2001) Saccharomyces cerevisiae is capable of de novo pantothenic acid biosynthesis involving a novel pathway of β-alanine production from spermine. J Biol Chem 276:10794–10800. https://doi.org/10.1074/jbc.M009804200
Zhang YM, Rock CO, Jackowski S (2005) Feedback regulation of murine pantothenate kinase 3 by coenzyme A and coenzyme A thioesters. J Biol Chem 280:32594–32601. https://doi.org/10.1074/jbc.M506275200
Zhyvoloup A, Nemazanyy I, Babich A, Panasyuk G, Pobigailo N, Vudmaska M, Naidenov V, Kukharenko O, Palchevskii S, Savinska L, Ovcharenko G, Verdier F, Valovka T, Fenton T, Rebholz H, Wang ML, Shepherd P, Matsuka G, Filonenko V, Gout IT (2002) Molecular cloning of CoA Synthase: The missing link in CoA biosynthesis. J Biol Chem 277:22107–22110. https://doi.org/10.1074/jbc.C200195200
Zhyvoloup A, Nemazanyy I, Panasyuk G, Valovka T, Fenton T, Rebholz H, Wang ML, Foxon R, Lyzogubov V, Usenko V, Kyyamova R, Gorbenko O, Matsuka G, Filonenko V, Gout IT (2003) Subcellular localization and regulation of coenzyme A synthase. J Biol Chem 278:50316–50321. https://doi.org/10.1074/jbc.M307763200
Acknowledgements
We thank Eva-Maria Bornholdt and Lea Rittmeier for plasmid constructions and Gudrun Ebel and Karola Hahn for technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Deutsche Forschungsgemeinschaft (DFG; SCHU856/9–1).
Author information
Authors and Affiliations
Contributions
HJS conceived the study, designed the experiments, supervised the project, and wrote the manuscript. JO constructed CAB1 variants and analyzed PanK function. MG constructed pLEUTEX plasmids and yeast strains. LS analyzed cab1ts revertants and performed PanK assays and functional studies of hCOASY in yeast. TU constructed yeast strains and analyzed CoA + acetyl-CoA in yeast extracts.
Corresponding author
Ethics declarations
Ethics approval
Not applicable (no studies with human participants or animals were performed in this study).
Consent for publication
All authors have read and approved the final manuscript.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised. ESM1 is now corrected.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Olzhausen, J., Grigat, M., Seifert, L. et al. Increased biosynthesis of acetyl-CoA in the yeast Saccharomyces cerevisiae by overexpression of a deregulated pantothenate kinase gene and engineering of the coenzyme A biosynthetic pathway. Appl Microbiol Biotechnol 105, 7321–7337 (2021). https://doi.org/10.1007/s00253-021-11523-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-021-11523-4