
Abstract
The phylogenetic diversity and abundance of ammonia-oxidizing beta-proteobacteria (beta-AOB) was analyzed along an anthropogenic pollution gradient from the coastal Pearl River Delta to the South China Sea using the ammonia monooxygenase subunit A (amoA) gene. Along the gradient from coastal to the open ocean, the phylogenetic diversity of the dominant genus changed from Nitrosomonas to Nitrosospira, indicating the niche specificity by these two genera as both salinity and anthropogenic influence were major factors involved. The diversity of bacterial amoA gene was also variable along the gradient, with the highest in the deep-sea sediments, followed by the marshes sediments and the lowest in the coastal areas. Within the Nitrosomonas-related clade, four distinct lineages were identified including a putative new one (A5-16) from the different sites over the large geographical area. In the Nitrosospira-related clade, the habitat-specific lineages to the deep-sea and coastal sediments were identified. This study also provides strong support that Nitrosomonas genus, especially Nitrosomonas oligotropha lineage (6a) could be a potential bio-indicator species for pollution or freshwater/wastewater input into coastal environments. A suite of statistical analyses used showed that water depth and temperature were major factors shaping the community structure of beta-AOB in this study area.
Similar content being viewed by others

Introduction
Nitrification has been known as an important step in the nitrogen (N) biogeochemical cycle because of the oxidation from ammonia to nitrate via nitrite. Nitrification also affects the global climate change through the emission of greenhouse gases nitrous oxide (N2O) and nitric oxide (NO) (Falkowski 1997; Casciotti et al. 2003; Shaw et al. 2006; Gruber and Galloway 2008). Nitrification could detoxify the excess ammonia in the anthropogenic affected estuarine and coastal ecosystems (Camargo and Alonso 2006). Nitrification is also responsible for the formation of the huge nitrate reservoir in the deep sea (Galloway et al. 2004; Lam et al. 2007; Klotz and Stein 2008). Ammonia-oxidizing bacteria (AOB) and the recently discovered ammonia-oxidizing archaea (AOA) (Venter et al. 2004; Schleper et al. 2005) which are affiliated to Thaumarchaeota (Brochier-Armanet et al. 2008; Spang et al. 2010) are involved in the nitrification process. AOA have been surveyed in a broad range of environments based on the culture-independent molecular methods (Francis et al. 2005; Prosser and Nicol 2008; Schleper and Nicol 2010). Though AOA were proposed as the dominant group to be responsible for the aerobic nitrification in a wide range of environments (Prosser and Nicol 2008; Schleper and Nicol 2010), the relative contributions of this group to the nitrification is still in debate and AOB remains a more robust model system for microbial ecology investigation (Kowalchuk and Stephen 2001; Horz et al. 2004; Ward 2005). AOB taxonomically falls into two monophyletic groups, beta- and gamma-proteobacteria. Beta-AOB mainly consists of two genera, Nitrosomonas and Nitrosospira in which Nitrosomonas genus could be further divided into several lineages (Purkhold et al. 2000, 2003). Three newly defined clusters affiliated to Nitrosospira genus and one belonged to Nitrosomonas has been proposed recently (Dang et al. 2010). Although gama-AOB may have a much wide distribution (Ward and O'Mullan 2002), gamma-proteobacteria such as Nitrosococcus oceani are minor members of the marine sediment ammonia-oxidizing communities (Nold et al. 2000) with lower diversity in most marine environments analyzed (O’Mullan and Ward 2005) or no positive results (Ward et al. 2000).
In the estuarial, coastal and deep-sea ecosystems, beta-AOB are ecologically significance based on numerous studies through culture-independent molecular detection of ammonia monooxygenase subunit A gene (amoA) which could provide reliable information about the beta-AOB community structure and their response to environmental changes (de Bie et al. 2001; Bollmann and Laanbroek 2002; Caffrey et al. 2003, 2007; Francis et al. 2003; Bernhard et al. 2005, 2007; O’Mullan and Ward 2005; Beman and Francis 2006; Freitag et al. 2006; Hunter et al. 2006; Urakawa et al. 2006a, b; Nakagawa et al. 2007; Mosier and Francis 2008; Sahan and Muyzer 2008; Santoro et al. 2008; Dang et al. 2010; Wankel et al. 2011). Nevertheless, the partial information from different ecological niches could not guarantee how the beta-AOB community structure, abundance, and distribution respond to the environmental change on a larger scale (Kowalchuk and Stephen 2001; Freitag et al. 2006; Urakawa et al. 2006a, 2006b). On the other hand, although long-standing efforts to explore the driving forces including salinity, pH, ammonium, and O2 concentrations on selecting differential ecophysiological lineages of beta-AOB, a thorough understanding of these relationships is not available (Bernhard et al. 2007; Dang et al. 2010), especially in the Pearl River Estuary which is a complex dynamic environments impacted strongly by anthropogenic activities and also the most rapidly developing activity of the world economy in the past more than two decades, and the South China Sea (SCS) (Huang et al. 2003; Dai et al. 2008; Harrison et al. 2008; Zhou et al. 2011). Recently, studies on the nitrifying bacterial in the Pearl River Estuary and the SCS provided an opportunity to comprehensively review the beta-AOB community structures along a gradient of surface sediments from the Pearl River Estuary, coastal to deep-sea sites (Cao et al. 2011e; Jin et al. 2011; Li et al. 2011) because surface sediments is a major site for nitrification due to a relatively high beta-AOB abundance and activity (Satoh et al. 2007). In addition, another study conducted in Jiaozhou Bay of North China indicated that the sediment beta-AOB community could serve as bio-tracers or bio-indicators to reflect the continental influence (Dang et al. 2010). The beta-AOB in the surface sediments from the Pearl River Estuary to the SCS could be employed as a model for this test.
The phylogenetic diversity of beta-AOB was surveyed and the abundance of both groups of ammonia oxidizers including AOA and beta-AOB was also evaluated in the northern part of the SCS with a deep rhombus-shaped basin in the eastern part employed amoA gene as a functional genetic biomarker. Comparative analyses of beta-AOB community structures were also conducted with those from the Pearl River Estuary and the Mai Po Nature Reserve marshes of Hong Kong. Along the gradient formed by the surface sediments from wetland, estuary to the coastal and deep sea, the community structure changes of beta-AOB were detected and dominated lineages were identified so as to draw a general view about the spatial distributions of beta-AOB in this area under increasing pressure of anthropogenic influences.
Materials and methods
Sampling and molecular experiments
Seven surface sedimentary samples were collected on the SCS Open Cruise in July 2008. When sediment cores were retrieved on board, air-exposed outer surfaces of the sediment were discarded and the central core was taken with sterile stainless steel knife and put into plastic bags for storage at −20°C immediately. All samples from the SCS could be divided into two groups, shallow and deep-sea ones from the coastal areas to the deep sea as shown in Fig. S1 and Table S1 in the ESM. The detailed descriptions of the sampling sites involved in this study are also available elsewhere (Cao et al. 2011a, b, c, d, e). Additional samples from this large area covering the anthropogenic pollution gradient were reported in previous studies and also included in the current analysis to expand our dataset (Cao et al. 2011a, b, c, d, e; Jin et al. 2011; Li et al. 2011).
Bacterial amoA gene sequences were amplified using the primer set (amoA-1F: 5′-GGGGTTTCTACTGGTGGT-3′ and amoA-2R: 5′-CCCCTCKGSAAAGCCTTCTTC-3′) (Rotthauwe et al. 1997) through polymerase chain reaction (PCR) amplification of the total genomic DNA extracted from sediment samples as described earlier (Cao et al. 2011a, b, c, d, e; Li et al. 2011). In addition to the bacterial amoA gene sequences retrieved from the seven samples in the present study, more amoA gene sequences from a number of other studies over the same large area were also downloaded from the GenBank and used in the analyses (Cao et al. 2011b, e; Jin et al. 2011; Li et al. 2011). The abundance (gene copy numbers) of archaeal and bacterial amoA genes were determined with specific primers (Arch-amoAF: 5′-STAATGGTCTGGCTTAGACG-3′ and Arch-amoAR: 5′-GCGGCCATCCATCTGTATGT-3′ for AOA; amoA-1F and amoA-2R for AOB) (Rotthauwe et al. 1997; Francis et al. 2005) by quantitative PCR (Q-PCR) in 25-μl reaction volume including 1 μl template DNA, 0.2 mg/ml BSA, 200 nM of each primer, and 12.5 μl of SYBR Premix (Takara, Hong Kong). The protocol used was described previously by Chen et al. (2008). All reactions were run in triplicate. A serial dilution of plasmids containing clone 704BA-6 for AOA and clone 704BB-6 for AOB were separately used as standards in Q-PCR assays (Cao et al. 2011b, d).
Phylogenetic and statistical analyses in the surface sediments from SCS
The bacterial amoA gene sequences from this study were firstly evaluated by the rarefaction analysis, and richness estimators (Chao1 and Shannon) and diversity index (Simpson) were simultaneously calculated using DOTUR (Schloss and Handelsman 2006) prior to other analyses. To keep the congruence between different studies, the distance cut-off was set at 5% for operational taxonomic units (OTUs) definition in DOTUR (Schloss and Handelsman 2006).
Bacterial amoA gene sequences recovered from the present study and the relevant ones retrieved from GenBank including those from the Mai Po Nature Reserve marshes and the Pearl River Estuary were transferred into MEGA 5.0 (Tamura et al. 2011) and merged for editing and further analyses (Cao et al. 2011e; Jin et al. 2011; Li et al. 2011). All sequences were aligned by Clustal X 1.81 (Higgins et al. 1997) and the phylogenetic analysis was conducted in MEGA 5.0 (Tamura et al. 2011). Phylogenetic tree was constructed based on the neighbor-joining criterion and credible values for every node were evaluated with bootstrap method via 1,000 replicates in MEGA 5.0 (Tamura et al. 2011).
Principal coordinates analyses (PCoA), Jackknife Environment Clusters analyses were conducted in an online software UniFrac (http://bmf2.colorado.edu/unifrac/index.psp) (Lozupone et al. 2006) which employs the genetic distances to evaluate the community similarity based on the gene sequences data. The bacterial amoA gene sequence communities from the SCS surface sediments and other former studies were classified on the basis of the environmental variables. The environment clusters tree was projected in MEGA 5.0 (Tamura et al. 2011). Canonical correspondence analysis (CCA) was executed in CANOCO 4.5 to determine the correlations between bacterial amoA gene sequence community compositions from northern SCS and physicochemical parameters (ter Braak and Smilauer 2002). In addition, Pearson moment correlations were also evaluated with Microsoft Excel to relate the physicochemical factors with the diversity and richness indices of bacterial amoA genes.
Nucleotide sequences accession numbers
The bacterial amoA gene sequences retrieved in this study from the seven surface sediment samples were deposited in the GenBank with the accession numbers JN575094-JN575260.
Results
Environmental conditions in the study areas
Eutrophication is a widely known environmental issue in the Pearl River Delta and the adjacent coastal areas of the northern SCS because of rapid economic growth driven by manufacturing for the world and also urbanization, resulting in a high load of dissolved inorganic nitrogen (Huang et al. 2003; Dai et al. 2008; Harrison et al. 2008; Zhou et al. 2011). Mai Po Nature Reserve marshes in Hong Kong receive a large quantity of wastewater from the adjacent areas with highly contaminated sediments showing high contents of inorganic and organic nitrogen and heavy metals (Cheung et al. 2003; Liang and Wong 2003; Laboratory of Environmental Toxicology 2009) and an apparent decline of infauna diversity over the last 7 years (Shen et al., 2010). In this study, four types of ecosystem sediments were identified based on the environmental conditions, namely marshes, estuarine, coastal and deep-sea sediments. The salinity was on an average of 27.8 ± 7.1‰ in the estuarine coastal plume, 32.9 ± 1.3‰ on the continental shelf, and 33.5 ± 0.8‰ in the open ocean (He et al. 2009). However, salinity in the Mai Po Nature Reserve was highly variable seasonally and lower (∼17‰) than the other areas mentioned above (Cao et al. 2011e; Li et al. 2011). Generally, the salinity increases from the marshes to the estuarine and the open sea. In contrast, organic contaminants and heavy metals, e.g., mercury (Hg) in the sediments displayed a decreasing trend from the Pearl River Delta estuary to the open sea (Shi et al. 2010). High nitrification rates were also observed in the Pearl River Estuary and the adjacent areas indicating the importance of nitrifying microbes (Dai et al. 2008).
Sediments of the SCS surveyed in this study could be divided into two groups, shallow and deep-sea ones because temperature and water depth are evidently clustered into two groups. Besides, other environmental factors also showed variable trend (Table S1 in the Electronic supplemental materials (ESM)) and previous studies (Cao et al. 2011b, c), for instance the decrease of ammonium concentrations from the Pearl River Delta areas to the SCS (Dai et al. 2008; Cao et al. 2011b, c; Jin et al. 2011) (Table S1 in the ESM). On the other hand, within the seven sites from this study, site CF5 exhibited extremely high concentration of inorganic nitrogen, especially NH +4 , followed by site 201 with the second highest (Table S1 in the ESM).
Molecular diversity of bacterial amoA gene sequences
Higher diversities of bacterial amoA gene sequences were observed in deep-sea sites 08CF7 and 525 while lower ones in coastal sites with shallow overlying water, sites 201, 510, and 708 based on the indices for samples from the SCS (Table 1) (Cao et al. 2011b, c). Within each clone library, the number of OTU recovered was variable from 2 to 13 based on rarefaction analysis (Fig. 1) (Cao et al. 2011b, c). A total of 351 sequences from the SCS (including others from the previous studies) (Cao et al. 2011b) were combined for analyses, and 26 OTUs were identified based on 5% cutoff for nucleotide sequences. For the Pearl River Estuary, the diversity of bacterial amoA genes was difficult to be evaluated and compared with others because only one clone library containing all samples was constructed with 26 sequences but with obvious diversity information (Jin et al. 2011). In the Mai Po Nature Reserve marsh sediments, the diversity indices were slightly higher than in the coastal areas, but lower than in the deep-sea sediments of the SCS (Cao et al. 2011e; Li et al. 2011).

Rarefaction curves of bacterial amoA gene sequences retrieved from the northern South China Sea with 5% distance cut-off
Phylogenetic diversity of the beta-AOB amoA gene
Two larger clades separately grouped with known sequences of beta-AOB in the Nitrosomonas and Nitrosospira genera based on the phylogenetic analysis of amoA gene sequences (Fig. 2). Most of the sequences from the Mai Po Nature Reserve marshes were in the Nitrosomonas-like clade. In contrast, a few sequences from the Pearl River Estuary, coastal and deep-sea of the Northern SCS (all in cluster 6a) were also grouped into this clade. As in the former studies, three lineages, Nitrosomonas sp. Nm143 (cluster 143), Nitrosomonas oligotropha (cluster 6a), and Nitrosomonas marina (cluster 6b) were delineated in the current phylogeny (Purkhold et al. 2003; Dang et al. 2010). Another new cluster, tentatively named cluster A5-16 from Jiaozhou Bay is also present in this clade (Dang et al. 2010; Jin et al. 2011). Two sequences from the Pearl River Estuary sediments were grouped into this cluster, which appeared at the basal position and clearly separated from the Nitrosomonas europaea, Nitrosomonas communis, and Nitrosomonas cryotolerans lineages (Dang et al. 2010).


Phylogenetic tree split into Nitrosomonas- (a) and Nitrosospira-related (b) ones constructed based on bacterial amoA gene sequences from the various surface sediments in the Mai Po Nature Reserve marshes sediments, the Pearl River Estuary and the northern South China Sea sediments with most similar sequences from GenBank through the neighbor-joining (NJ) method. Numbers at the nodes represent the significant bootstrap values (>50) via 1,000 replicates test. Branch lengths correspond to sequence differences as indicated by the scale bar (2% nucleotide substitution)
In the Nitrosospira-like clade (Fig. 2b), except for one sequence from site 407 which was affiliated to the cultured Nitrosospira sp. ka3 (Aakra et al. 1999), others were not grouped within any cultured beta-AOB amoA gene sequences cluster, similar to those retrieved before from various estuarine, coastal, and deep-sea sediments (Bernhard et al. 2007; Hayashi et al. 2007; Kim et al. 2008; Dang et al. 2010). Three tentatively named clusters 13, 14 and 15 were evident in this study based on the nomenclature by Avrahami and Conrad (2003) and Dang et al. (2010). Clusters 13 was constituted by amoA gene sequences from coastal marine environments as proposed in the previous study, e.g., the sequences from sites 201, 510, 704, 706, 707, 708, and 709 (Bernhard et al. 2007; Kim et al. 2008; Dang et al. 2010). Cluster 14 was mainly comprised of environmental amoA gene sequences from deep-sea marine sediments (Hayashi et al. 2007; Dang et al. 2010). On the other hand, cluster 15 with another synonym as “Nitrosospira-like cluster B” was composed by the amoA gene sequences mainly from estuarine and coastal sediments (Francis et al. 2003; Kim et al. 2008; Dang et al. 2010).
Community analyses of the beta-AOB assemblages
A few groups were identified from the UniFrac environment clustering analysis based on phylogenetic diversity of bacterial amoA gene sequences (Fig. 3a). Samples from the Mai Po Nature Reserve wetland and the Pearl River Estuary were distinguished separately. In the wetland assemblage, sequences from sample D-May separated from others, consistent with our previous analysis (Cao et al. 2011e). The coastal sites in the SCS were divided into two sub-classes: one comprised by the sequences from sites 704 and 425 and another constituted by sites 510, 706, 707, 708, and 709. The deep-sea sites in the SCS formed one distinct class. Similar classification was revealed in the UniFrac PCoA analysis (Fig. 3b). The first principal coordinate (P1), which explained 60.63% of the total community variability, unambiguously distinguished the amoA assemblages of wetland from those of the Pearl River Estuary and the SCS.

PCoA (a) and hierarchical clustering analysis (UPGMA algorithm with 100 replicates Jackknife supporting test) (b) for the bacterial amoA gene sequences from the Mai Po Nature Reserve marshes sediments, the Pearl River Estuary and the northern South China Sea sediments on the basis of the online software UniFrac
An all-environment UniFrac significance test indicated a significant difference (P = 0.002) among the all amoA assemblages in the current study. Pairwise UniFrac significance test indicated that the amoA assemblage of site 407 was marginally significantly different from 702 (P = 0.02), 510 from 707 (P = 0.03), 510 from 707 to 709 (P = 0.03), and A-NOV from D-NOV (P = 0.04). Those of sites CF5 with 525 (P = 0.09) and 706 with 707 (P = 0. 6), were of different community composition.
Environmental factors to shape the distribution of the beta-AOB
Pearson moment analyses provided clues to the environmental parameters that shape the spatial distribution of beta-AOB in the northern SCS. Both depth and temperature showed weaker relationships with the diversity of bacterial amoA genes (Table S2 in the ESM). The concentration of nitrite was correlated with the diversity of bacterial amoA genes. Correlations of the bacterial amoA assemblages with environmental parameters analyzed by CCA indicated that depth and temperature via the CCA axis 1 separated all the samples into two groups, namely shallow and deep-sea sediments groups, consistent with the UniFrac community classification. In addition, the concentration of inorganic nitrogen could contribute to the distribution of the deep-sea sediments, 08CF7, 525 and CF5, while the concentration of nitrite separated 201, 704, and 707 (Fig. 4).

CCA ordination plots for the environmental factors and the ammonia-oxidizing bacterial lineages representing by amoA gene sequences analyzing from the CANOCO software
Bacterial and archaeal quantification analyses of the amoA gene
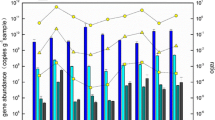
Beta-AOB amoA gene ranged from 4.24 × 104 to 1.99 × 106 copies per gram of sediment (wet weight), and the highest one was only more than two orders than the lowest one (Fig. 5). The AOA amoA gene abundance ranged from 1.6 × 105 to 2.45 × 108 copies/g of sediment (wet weight), the highest and the lowest differed by 3-fold (Fig. 5). The ratios of AOA/beta-AOB amoA gene copy numbers ranged from 2.69 to 2.65 × 103. The PCR amplification efficiencies were 91–95% (average, 93%) for archaeal amoA and 88–92% (average, 90%) for beta-proteobacterial amoA. Correlation coefficient (R 2) for both assays on average were 0.98 ± 0.01 (standard deviation). Generally, the archaeal amoA gene abundance was higher than bacteria at each site. The Q-PCR results showed a heterogeneous distribution of the sediment archaeal and bacterial amoA gene abundance and the ratio of them among the sampling sites of the SCS, in that the sites CF5 and 425 showed higher gene abundance than the others. The ratio was higher at site 425.

Relative abundance of archaeal and beta-proteobacterial amoA gene. The error bar represents the standard deviation of triplicate Q-PCR reactions
Discussion
Beta-AOB phylogenetic diversity along the pollution gradient
In the surface sediments from the wetland in the Pearl River Delta to the coastal and deep sea in the SCS, the phylogenetic diversity of beta-AOB delineated by amoA gene sequences was spatially distributed from the dominance by Nitrosomonas- to Nitrosospira-related clades supported by the UniFrac analyses. Most sequences from the SCS, both in coastal and deep-sea sediments, were affiliated to Nitrosospira. All amoA gene sequences from two previous studies at the Mai Po Nature Reserve wetland were also exclusively clustered into the Nitrosomonas-related clade (Cao et al. 2011e; Li et al. 2011). In each clade, a few specific beta-proteobacterial amoA gene lineages were identified and each lineage potentially occupied a distinct habitat through adaptation. And a few lineages identified before were delineated again with much more similar sequences from the SCS this time.
The lineage designated as cluster A5-16, because of one specific OTU A5-a-16 identified here, was clustered with another two clones, B51 and B58 (shared about 90% identity with clone A5-a-16) retrieved from the Pearl River Estuary (Dang et al. 2010; Jin et al. 2011). And CCA analysis in a previous study suggested that beta-AOB in this lineage might participate in the in situ nitrification in sediments (Dang et al. 2010). The amoA gene sequences from this lineage shared large genetic distance with others (0.21 to 0.37, percent nucleotide divergent, data not shown) indicating the novelty of this lineage and they were grouped with a few cultured AOB stains, e.g., N. europaea and N. communis which had been detected in anthropogenic influenced areas with heavy metals and NH +4 –N before (Fig. 2a) (Stein et al. 2007 and references therein). These three sequences diverged from the close relatives with a few non-synonymous substitutes, indicating the possible specific adaptation of beta-AOB for these habitats and these two sequences from the Pearl River Estuary have only four non-synonymous substitutes from the OTU A5-a-16, resulting from the large geographical distance (Fig. S2 in the ESM). As the OTU A5-a-16 occurred at the location with high discharge from the adjacent river and the wastewater treatment plants (WWTPs) (Dai et al. 2008), the Pearl River Estuary experienced strong anthropogenic impact. In contrast, this lineage was not retrieved from the highly polluted Mai Po Nature Reserve wetland (Cao et al. 2011e; Li et al. 2011). This could be resulted from the distinctiveness between wetland with vegetations and estuarine sediments or the lower abundance of this lineage in the environments (Dang et al. 2010; Jin et al. 2011). Up to date, this lineage has only been detected in the estuarine environments and may represent a novel lineage to be further investigated, especially in pure culture study.
The N. oligotropha lineage (Cluster 6a designated before) (Purkhold et al. 2003) was proposed as the major member in freshwater environments (Stehr et al. 1995; Koops and Pommerening-Röser 2001; Kowalchuk and Stephen 2001), wastewaters and activated sludges, biofilm reactors, and biofilters (Purkhold et al. 2003; Limpiyakorn et al. 2005; Lydmark et al. 2007) as the largest lineage in the Nitrosomonas-related clade (Fig. 2a). Most of the sequences from the Mai Po Nature Reserve wetland belonged to this lineage indicating the contributions from the adjacent terrestrial runoff, freshwater river or non-treated waste discharges as shown in other studies (Nicolaisen and Ramsing 2002; Caffrey et al. 2003) and directly discharges from WWTPs in Tokyo Bay (Urakawa et al., 2006b) and in other geographical locations (de Bie et al. 2001; Cébron et al. 2003, 2004; Nakamura et al. 2006). The environmental factors which could affect the distribution of these lineages have been investigated before, for instance, the affiliation with biofilms or benthic infaunal burrow walls as well as support for the survival or preservation of these lineages within the highly dynamic estuarine ecosystems with tidal or seasonal salinity fluctuation (Magalhães et al. 2005; Satoh et al. 2007). Salinity usually has inhibitory effects on estuarine sediment nitrification (Rysgaard et al. 1999; Bernhard et al. 2007), probably due to the low salt tolerance of the nitrifiers, particularly the N. oligotropha lineage (Stehr et al. 1995). As discussed above, this lineage was detected in the estuarine or coastal environments with low salinity (Stehr et al. 1995; de Bie et al. 2001; Ottosen et al. 2001; Bollmann and Laanbroek 2002; Cébron et al. 2003, 2004; Francis et al. 2003; Bernhard et al. 2005; Magalhães et al. 2005; Nakamura et al. 2006; Satoh et al. 2007; Dang et al. 2010). These findings provided stronger evidence to support that freshwater flush or wastewater discharge should be the main factor shaping the freshwater beta-AOB community in brackish and marine ecosystems. The dominant distribution of these beta-AOB originated from freshwater in the coastal environments could provide an effective means to track the original sources with the microbial composition analysis (Cébron et al. 2003, 2004; Urakawa et al. 2006a, 2006b). As the N. oligotropha lineage is usually associated with WWTP effluents or rivers, environmental N. oligotropha clones might be potentially served as bio-indicators of pollution in coastal environments. In the present study, some sequences from the coastal sediments, sites 201 and 510, were also grouped into this lineage (Fig. 2a). Collectively, this study provides additional new evidence on the use of this lineage as the bio-indicators for pollution monitoring and tracking in coastal environments from the Pearl River Delta and the SCS.
The three newly defined amoA gene sequence lineages of the Nitrosospira clade (Dang et al. 2010) contained only environmental sequences from estuarine, coastal, and deep-sea sediments, and these sequences and clusters may represent novel beta-AOB species and lineages (Avrahami and Conrad 2003; Bernhard et al. 2007; Hayashi et al. 2007; Kim et al. 2008; Dang et al. 2010). Clusters 13 was constituted by amoA gene sequences from coastal marine environments as proposed in the previous study, e.g., the sequences from sites 201, 510, 704, 706, 707, 708, and 709 (Bernhard et al. 2007; Kim et al. 2008; Dang et al. 2010). Cluster 14 is mainly comprised of environmental amoA gene sequences from deep-sea marine environments (Hayashi et al. 2007; Meyer and Kuever 2008; Dang et al. 2010). As proposed in the former study, sequences from this lineage were related to high salinity niche (Dang et al. 2010). However, in contrast with other lineages shown in the phylogenetic tree, salinity could not be the sole factor, and other factors, including depth, pressure, oxygen and temperature could contribute collectively to the distributions of this lineage in deep-sea and separated them from the others. Cluster 15 with another synonym as “Nitrosospira-like cluster B” in another study was composed of the amoA gene sequences mainly from estuarine and coastal sediment environments, especially estuaries (Francis et al. 2003; Kim et al. 2008; Dang et al. 2010; Jin et al. 2011). Based on the above analysis, the clusters 13 and 15 sequences could occupy similar niches and both clusters should have the higher adaptability to a wide range of coastal environments as shown by Nitrosomonas sp. Nm143 lineage (Freitag et al. 2006) supported by another study showing no significant correlation with any environmental factors (Dang et al. 2010).
Community structure change of beta-AOB and environmental factors to shape beta-AOB
The beta-AOB diversity decreased with the increase in salinity in some estuaries (de Bie et al. 2001; Francis et al. 2003; Cébron et al. 2004; Urakawa et al. 2006a, 2006b; Ward et al. 2007). However, from the wetland to the open sea in this study, the diversity of beta-AOB was higher in the deep sea of the SCS than in the coastal sediments of the SCS, wetland and estuary, indicating different beta-AOB assemblages in the deep-sea. On the other hand, the community of beta-AOB from mainly Nitrosomonas to Nitrosospira has been proposed to be affected by the salinity in estuarine and coastal environments (Freitag et al. 2006). However, other studies provided a different conclusion about the salinity effect on the community structure of beta-AOB (Bernhard et al. 2007; Dang et al. 2010). In particular at the Jiaozhou Bay of Northern China, the microbe-environment CCA analyses indicated that salinity contributed very little to the distribution and classification of the sediment amoA sequence assemblages (Dang et al. 2010). Current study provides a strong evidence on a much large scale with continuity from the marshes to coastal and then deep-sea to delineate salinity as a factor driving the spatial distribution of beta-AOB from Nitrosomonas- to Nitrosospira-related lineages along the pollution gradient. Other studies also indicated the distinctions of beta-AOB community compositions between freshwater and marine ecosystems (Bollmann and Laanbroek 2002; Coci et al. 2005; Grommen et al. 2005). Microbes flushed from rivers and WWTPs with nutrients could be the main contributors of beta-AOB to the Mai Po Nature Reserve marshes indicating Nitrosomonas-related lineages are the typical beta-AOB in this ecosystem. In this ecosystem, salinity is lower than coastal and the open sea of the SCS for most of the time in a year (Cao et al. 2011b, c, e; He et al. 2009; Jin et al. 2011; Li et al. 2011). Beta-AOB community in the Pearl River Estuary showed a mixture of these two genera (Fig. 2a, b) (Jin et al. 2011) while in the SCS, the Nitrosopira-related lineages are dominant indicating beta-AOB community change along the gradient could be a candidate bio-indicator for environmental pollution and source tracking (Kowalchuk and Stephen 2001; Dang et al. 2010; Cao et al. 2011d).
Although the abundance of AOA was higher than that of beta-AOB in the sampling sites of this study as reported in many other studies, the potential ecological role of AOA still needs further activity confirmation, and the community transition change of beta-AOB along the gradient was apparently a major focus in this study. Sedimentological parameters may influence the bacterial and archaeal community structures responsible for N cycle (Dang et al. 2008, 2009; Jackson and Weeks 2008). In the SCS, based on the CCA analysis, depth and temperature separated all samples into two groups: deep-sea and cold as one, shallow and warm as another indicating the water depth and resulting temperature, and hydro-pressure could shape the community structure of beta-AOB. In addition, NH +4 and NO -2 could be correlated to the samples from the deep-sea in the SCS (Fig. 4). Relationships between the diversity of bet-AOB and depth, temperature and nitrite were weak (Table S2 in the ESM). The oxygen concentration and pressure in the deep-sea sediments should select the beta-AOB to adapt to the unique habitats, resulting in the distinctness of the beta-AOB from others. Although the oxic sediment layers from the SCS sampling stations were known to exhibit higher nitrification activity (Urakawa et al. 2006a, 2006b), the oxygen still could be one limiting factor in the deep-sea sediments. Additionally, at site 201, the most bacterial amoA gene sequences (14 of 21 clones) were grouped into N. oligotropha lineage indicating the impact from the terrestrial environments. This is consistent with the information on ammonia-oxidizing archaea from this site in which the obvious soil/sediment clade AOA was dominant (Cao et al. 2011c). The highest abundance of beta-AOB existed in the site CF5 where the highest concentration of ammonium was also detected, indicating ammonium as the substrate for nitrification is a controlling factor for the diversity of beta-AOB. Taken all together, sedimentological conditions related to the hydrological regime, for instance, currents, tides, upwelling could affect the sediment composition, inorganic N concentration, redox, pH values, and temperature, so as to shape the microbial community, specifically the beta-AOB assemblages. However, relationships between them still need to be explored in the future studies (Dang et al. 2009, 2010). On the other hand, most bacterial amoA gene sequences from deep-sea sites were represented in the cluster 14 and also grouped with sequences from hydrothermal plum (Lam et al. 2004) without the basal position in the phylogenetic history as that in the AOA (de la Torre et al. 2008; Hatzenpichler et al. 2008; Prosser and Nicol 2008; Zhang et al. 2008; Kato et al. 2009; Dang et al. 2010; Cao et al. 2011c), indicating these two groups of nitrifying microorganisms experienced different evolutionary history. As oxygen limited condition in the deep-sea sediments, the relationships between the aerobic ammonia oxidizers, including AOA and AOB and anaerobic ammonium oxidizing (anammox) bacteria should be much more interesting for the further studies and the anammox bacteria might have a higher contribution to the overall nitrogen cycle.
References
Aakra Å, Utåker JB, Nes IF (1999) RFLP of rRNA genes and sequencing of the 16S–23S rDNA intergenic spacer region of ammonia-oxidizing bacteria: a phylogenetic approach. Int J Syst Bacteriol 49:123–130
Avrahami S, Conrad R (2003) Patterns of community change among ammonia oxidizers in meadow soils upon long-term incubation at different temperatures. Appl Environ Microbiol 69:6152–6164
Beman JM, Francis CA (2006) Diversity of ammonia-oxidizing archaea and bacteria in the sediments of a hypernutrified subtropical estuary: Bahia del Tobari, Mexico. Appl Environ Microbiol 72:7767–7777
Bernhard AE, Donn T, Giblin A, Stahl DA (2005) Loss of diversity of ammonia-oxidizing bacteria correlates with increasing salinity in an estuary system. Environ Microbiol 7:1289–1297
Bernhard AE, Tucker J, Giblin AE, Stahl DA (2007) Functionally distinct communities of ammonia-oxidizing bacteria along an estuarine salinity gradient. Environ Microbiol 9:1439–1447
Bollmann A, Laanbroek HJ (2002) Influence of oxygen partial pressure and salinity on the community composition of ammonia-oxidizing bacteria in the Schelde Estuary. Aquat Microb Ecol 28:239–247
Brochier-Armanet C, Boussau B, Gribaldo S, Forterre P (2008) Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat Rev Microbiol 6:245–252
Caffrey JM, Harrington N, Solem I, Ward BB (2003) Biogeochemical processes in a small California estuary. 2. Nitrification activity, community structure and role in nitrogen budgets. Mar Ecol Prog Ser 248:27–40
Caffrey JM, Bano N, Kalanetra K, Hollibaugh JT (2007) Ammonia oxidation and ammonia-oxidizing bacteria and archaea from estuaries with differing histories of hypoxia. ISME J 1:660–662
Camargo JA, Alonso A (2006) Ecological and toxicological effects of inorganic nitrogen pollution in aquatic ecosystems: a global assessment. Environ Int 32:831–849
Cao H, Hong Y, Li M, Gu J-D (2011a) Lower abundance of ammonia-oxidizing archaea than ammonia-oxidizing bacteria detected in the subsurface sediments of the Northern South China Sea. Geomicrobiol J (in press)
Cao H, Hong Y, Li M, Gu J-D (2011b) Diversity and abundance of ammonia-oxidizing prokaryotes in sediments from the coastal margin to the South China Sea. Antonie van Leeuwenhoek 100:545–556
Cao H, Hong Y, Li M, Gu J-D (2011c) Phylogenetic diversity and ecological pattern of ammonia-oxidizing archaea in the variable surface sediments of the western Pacific. Microb Ecol, doi:10.1007/s00248-011-9901-0
Cao H, Li M, Dang H, Gu J-D (2011d) Responses of aerobic and anaerobic ammonia/ammonium oxidizing microorganisms to anthropogenic pollution in coastal marine environments. In: Klotz MG, Stein LY (eds). Methods in Enzymology, Vol. 496 Research on Nitrification and Related Processes. Part B, pp 35–62.
Cao H, Li M, Hong Y, Gu J-D (2011e) diversity and abundance of ammonia-oxidizing archaea (AOA) and bacteria (AOB) in polluted mangrove sediment. Syst Appl Microbiol 34:513–523
Casciotti KL, Sigman DM, Ward BB (2003) Linking diversity and stable isotope fractionation in ammonia-oxidizing bacteria. Geomicrobiol J 20:335–353
Cébron A, Berthe T, Garnier J (2003) Nitrification and nitrifying bacteria in the lower Seine River and estuary (France). Appl Environ Microbiol 69:7091–7100
Cébron A, Coci M, Garnier J, Laanbroek HJ (2004) Denaturing gradient gel electrophoretic analysis of ammonia-oxidizing bacterial community structure in the lower Seine River: impact of Paris wastewater effluents. Appl Environ Microbiol 70:6726–6737
Chen XP, Zhu YG, Xia Y, Shen JP, He JZ (2008) Ammonia-oxidizing archaea: important players in paddy rhizosphere soil? Environ Microbiol 10:1978–1987
Cheung KC, Poon BHT, Lan CY, Wong MH (2003) Assessment of metal and nutrient concentrations in river water and sediment collected from the cities in the Pearl River Delta, South China. Chemosphere 52:1431–1440
Coci M, Riechmann D, Bodelier PL, Stefani S, Zwart G, Laanbroek HJ (2005) Effect of salinity on temporal and spatial dynamics of ammonia-oxidising bacteria from intertidal freshwater sediment. FEMS Microbiol Ecol 53:359–368
Dai M, Wang L, Guo X, Zhai W, Li Q, He B, Kao SJ (2008) Nitrification and inorganic nitrogen distribution in a large perturbed river/estuarine system: the Pearl River Estuary, China. Biogeosciences Discuss 5:1545–1585
Dang H, Zhang X, Sun J, Li T, Zhang Z, Yang G (2008) Diversity and spatial distribution of sediment ammonia-oxidizing Crenarchaeota in response to estuarine and environmental gradients in the Changjiang Estuary and East China Sea. Microbiology 154:2084–2095
Dang H, Li J, Zhang X, Li T, Tian F, Jin W (2009) Diversity and spatial distribution of amoA-encoding archaea in the deep-sea sediments of the tropical West Pacific Continental Margin. J Appl Microbiol 106:1482–1493
Dang H, Li J, Chen R, Wang L, Luo L, Zhang Z, Klotz MG (2010) Diversity, abundance and spatial distribution of sediment ammonia-oxidizing Betaproteobacteria in response to environmental gradients and coastal eutrophication in Jiaozhou Bay, China. Appl Microbiol Ecol 76:4691–4702
de Bie MJM, Speksnijder AG, Kowalchuk GA, Schuurman T, Zwart G, Stephen JR, Diekmann OE, Laanbroek HJ (2001) Shifts in the dominant populations of ammonia-oxidizing β-subclass Proteobacteria along the eutrophic Schelde estuary. Aquat Microb Ecol 23:225–236
de la Torre JR, Walker CB, Ingalls AE, Könneke M, Stahl DA (2008) Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ Microbiol 10:810–818
Falkowski PG (1997) Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean. Nature 387:272–275
Francis CA, O’Mullan GD, Ward BB (2003) Diversity of ammonia monooxygenase (amoA) genes across environmental gradients in Chesapeake Bay sediments. Geobiology 1:129–140
Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB (2005) Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc Natl Acad Sci U S A 102:14683–14688
Freitag TE, Chang L, Prosser JI (2006) Changes in the community structure and activity of betaproteobacterial ammonia-oxidizing sediment bacteria along a freshwater-marine gradient. Environ Microbiol 8:684–696
Galloway J, Dentener F, Capone D, Boyer E, Howarth R, Seitzinger S, Asner G, Cleveland C, Green P, Holland E, Karl D, Michaels A, Porter J, Townsend A, Vorosmarty C (2004) Nitrogen cycles: past, present, and future. Biogeochemistry 70:153–226
Grommen R, Dauw L, Verstraete W (2005) Elevated salinity selects for a less diverse ammonia-oxidizing population in aquarium biofilters. FEMS Microbiol Ecol 52:1–11
Gruber N, Galloway JN (2008) An Earth-system perspective of the global nitrogen cycle. Nature 451:293–296
Harrison PJ, Yin K, Lee JHW, Gan J, Liu H (2008) Physical–biological coupling in the Pearl River Estuary. Cont Shelf Res 28:1405–1415
Hatzenpichler R, Lebedeva EV, Spieck E, Stoecker K, Richter A, Daims H, Wagner M (2008) A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc Natl Acad Sci U S A 105:2134–2139
Hayashi T, Kaneko R, Tanahashi M, Naganuma T (2007) Molecular diversity of the genes encoding ammonia monooxygenase and particulate methane monooxygenase from deep-sea sediments. Res J Microbiol 2:530–537
He L, Yin K, Yuan X, Li D, Zhang D, Harrison PJ (2009) Spatial distribution of viruses, bacteria and chlorophyII in the northern South China Sea. Aquatic Microbial Ecol 54:153–162
Higgins DG, Thompson JD, Gibson TJ (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Horz HP, Barbrook A, Field CB, Bohannan BJ (2004) Ammonia-oxidizing bacteria respond to multifactorial global change. Proc Natl Acad Sci U S A 101:15136–15141
Huang XP, Huang LM, Yue WZ (2003) The characteristics of nutrients and eutrophication in the Pearl River estuary, South China. Mar Pollut Bull 47:30–36
Hunter EM, Mills HJ, Kostka JE (2006) Microbial community diversity associated with carbon and nitrogen cycling in permeable shelf sediments. Appl Environ Microbiol 72:5689–5701
Jackson CR, Weeks AQ (2008) Influence of particle size on bacterial community structure in aquatic sediments as revealed by 16S rRNA gene sequence analysis. Appl Environ Microb 74:5237–5240
Jin T, Zhang T, Ye L, Lee OO, Wong YH, Qian PY (2011) Diversity and quantity of ammonia-oxidizing archaea and bacteria in sediment of the Pearl River Estuary, China. Appl Microbiol Biotechnol, doi:10.1007/s00253-011-3107-8
Kato S, Kobayashi C, Kakegawa T, Yamagishi A (2009) Microbial communities in iron-silica-rich microbial mats at deep-sea hydrothermal fields of the Southern Mariana Trough. Environ Microbiol 11:2094–2111
Kim OS, Junier P, Imhoff JF, Witzel KP (2008) Comparative analysis of ammonia monooxygenase (amoA) genes in the water column and sediment-water interface of two lakes and the Baltic Sea. FEMS Microbiol Ecol 66:367–378
Klotz MG, Stein LY (2008) Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol Lett 278:146–156
Koops HP, Pommerening-Röser A (2001) Distribution and ecophysiology of the nitrifying bacteria emphasizing cultured species. FEMS Microbiol Ecol 37:1–9
Kowalchuk GA, Stephen JR (2001) Ammonia-oxidizing bacteria: a model for molecular microbial ecology. Annu Rev Microbiol 55:485–529
Laboratory of Environmental Toxicology (2009) Mai Po Inner Deep Bay Ramsar Site Baseline Ecological Monitoring Project 2008–09 Fieldwork and Laboratory Analysis (Bemp 08–09F&LA) Contract Ref. No. AFCD/SQ/96/07. Prepared for and submitted to Agriculture, Fisheries and Conservation Department of the Hong Kong Government. Laboratory of Environmental Toxicology, The University of Hong Kong, Pokfulam Road, Hong Kong
Lam P, Cowen JP, Jones RD (2004) Autotrophic ammonia oxidation in a deep-sea hydrothermal plume. FEMS Microbiol Ecol 47:191–206
Lam P, Jensen MM, Lavik G, McGinnis DF, Muller B, Schubert CJ, Amann R, Thamdrup B, Kuypers MM (2007) Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proc Natl Acad Sci U S A 104:7104–7109
Li M, Cao H, Hong Y, Gu J-D (2011) Spatial distribution and abundance of ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB) in mangrove sediments. Appl Microbiol Biotechnol 89:1243–1254
Liang Y, Wong MH (2003) Spatial and temporal organic and heavy metal pollution at Mai Po Marshes Nature Reserve, Hong Kong. Chemosphere 52:1647–1658
Limpiyakorn T, Shinohara Y, Kurisu F, Yagi O (2005) Communities of ammonia-oxidizing bacteria in activated sludge of various sewage treatment plants in Tokyo. FEMS Microbiol Ecol 54:205–217
Lozupone C, Hamady M, Knight R (2006) UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinforma 7:371
Lydmark P, Almstrand R, Samuelsson K, Mattsson A, Sorensson F, Lindgren PE, Hermansson M (2007) Effects of environmental conditions on the nitrifying population dynamics in a pilot wastewater treatment plant. Environ Microbiol 9:2220–2233
Magalhães CM, Joye SB, Moreira RM, Wiebe WJ, Bordalo AA (2005) Effect of salinity and inorganic nitrogen concentrations on nitrification and denitrification rates in intertidal sediments and rocky biofilms of the Douro River estuary, Portugal. Water Res 39:1783–1794
Meyer B, Kuever J (2008) Phylogenetic diversity and spatial distribution of the microbial community associated with the Caribbean deep-water sponge Polymastia cf. corticata by 16S rRNA, aprA, and amoA gene analysis. Microb Ecol 56:306–321
Mosier AC, Francis CA (2008) Relative abundance and diversity of ammonia-oxidizing archaea and bacteria in the San Francisco Bay estuary. Environ Microbiol 10:3002–3016
Nakagawa T, Mori K, Kato C, Takahashi R, Tokuyama T (2007) Distribution of cold-adapted ammonia-oxidizing microorganisms in the deep-ocean of the northeastern Japan Sea. Microbes Environ 22:365–372
Nakamura Y, Satoh H, Kindaichi T, Okabe S (2006) Community structure, abundance, and in situ activity of nitrifying bacteria in river sediments as determined by the combined use of molecular techniques and microelectrodes. Environ Sci Technol 40:1532–1539
Nicolaisen MH, Ramsing NB (2002) Denaturing gradient gel electrophoresis (DGGE) approaches to study the diversity of ammonia-oxidizing bacteria. J Microbiol Methods 50:189–203
Nold SC, Zhou J, Devol AH, Tiedje JM (2000) Pacific Northwest marine sediments contain ammonia-oxidizing bacteria in the beta subdivision of the Proteobacteria. Appl Environ Microbiol 66:4532–4535
O’Mullan GD, Ward BB (2005) Relationship of temporal and spatial variabilities of ammonia-oxidizing bacteria to nitrification rates in Monterey Bay, California. Appl Environ Microbiol 71:697–705
Ottosen LDM, Risgaard-Petersen N, Nielsen LP, Dalsgaard T (2001) Denitrification in exposed intertidal mud-flats, measured with a new 15N-ammonium spray technique. Mar Ecol Prog Ser 209:35–42
Prosser JI, Nicol GW (2008) Relative contributions of archaea and bacteria to aerobic ammonia oxidation in the environment. Environ Microbiol 10:2931–2941
Purkhold U, Pommerening-Roser A, Juretschko S, Schmid MC, Koops H-P, Wagner M (2000) Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl Environ Microbiol 66:5368–5382
Purkhold U, Wagner M, Timmermann G, Pommerening-Röser A, Koops HP (2003) 16S rRNA and amoA-based phylogeny of 12 novel betaproteobacterial ammonia-oxidizing isolates: extension of the dataset and proposal of a new lineage within the nitrosomonads. Int J Syst Evol Microbiol 53:1485–1494
Rotthauwe J-H, Witzel K-P, Liesack W (1997) The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl Environ Microbiol 63:4704–4712
Rysgaard S, Thastum P, Dalsgaard T, Christensen PB, Sloth NP (1999) Effects of salinity on NH4 adsorption capacity, nitrification, and denitrification in Danish estuarine sediments. Estuaries 22:21–30
Sahan E, Muyzer G (2008) Diversity and spatio-temporal distribution of ammonia-oxidizing archaea and bacteria in sediments of the Westerschelde estuary. FEMS Microbiol Ecol 64:175–186
Santoro AE, Francis CA, de Sieyes NR, Boehm AB (2008) Shifts in the relative abundance of ammonia-oxidizing bacteria and archaea across physicochemical gradients in a subterranean estuary. Environ Microbiol 10:1068–1079
Satoh H, Nakamura Y, Okabe S (2007) Influences of infaunal burrows on the community structure and activity of ammonia-oxidizing bacteria in intertidal sediments. Appl Environ Microbiol 73:1341–1348
Schleper C, Nicol GW (2010) Ammonia-oxidising archaea—physiology, ecology and evolution. Adv Microb Physiol 57:1–41
Schleper C, Jurgens G, Jonuscheit M (2005) Genomic studies of uncultivated archaea. Nat Rev Microbiol 3:479–488
Schloss PD, Handelsman J (2006) Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72:6773–6779
Shaw LJ, Nicol GW, Smith Z, Fear J, Prosser JI, Baggs EM (2006) Nitrosospira spp. can produce nitrous oxide via a nitrifier denitrification pathway. Environ Microbiol 8:214–222
Shen PP, Zhou H, Gu J-D (2010) Patterns of polychaete communities in relation to environmental perturbations in a subtropical wetland of Hong Kong. J Mar Biol Assoc UK 90:923–932
Shi JP, Ip CC, Zhang G, Jiang GB, Li XD (2010) Mercury profiles in sediments of the Pearl River Estuary and the surrounding coastal area of South China. Environ Pollut 158:1974–1979
Spang A, Hatzenpichler R, Brochler-Armanet C, Rattei T, Tischler P, Spieck E, Streit W, Stahl DA, Wagner M, Schleper C (2010) Distinct gene set in two different lineages of ammonia-oxidizing archaea supports the phylum Thaumarchaeota. Trends Microbiol 18:331–340
Stehr G, Biittcher B, Dittberner P, Rath G, Koops H-P (1995) The ammonia-oxidizing nitrifying population of the River Elbe estuary. FEMS Microbiol Ecol 17:177–186
Stein LY, Arp DJ, Berube PM, Chain PS, Hauser L, Jetten MS, Klotz MG, Larimer FW, Norton JM, Op den Camp HJ, Shin M, Wei X (2007) Whole-genome analysis of the ammonia-oxidizing bacterium, Nitrosomonas eutropha C91: implications for niche adaptation. Environ Microbiol 9:2993–3007
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular Evolutionary Genetics Analysis using Likelihood, Distance, and Parsimony methods. Mol Biol Evol (in press)
ter Braak CJF, Smilauer P (2002) CANOCO Reference Manual and CanoDraw for Windows User's Guide: Software for Canonical Community Ordination (Version 4.5). Microcomputer Power, Ithaca, NY, USA
Urakawa H, Maki H, Kawabata S, Fujiwara T, Ando H, Kawai T, Hiwatari T, Kohata K, Watanabe M (2006a) Abundance and population structure of ammonia-oxidizing bacteria that inhabit canal sediments receiving effluents from municipal wastewater treatment plants. Appl Environ Microbiol 72:6845–6850
Urakawa H, Kurata S, Fujiwara T, Kuroiwa D, Maki H, Kawabata S, Hiwatari T, Ando H, Kawai T, Watanabe M, Kohata K (2006b) Characterization and quantification of ammonia-oxidizing bacteria in eutrophic coastal marine sediments using polyphasic molecular approaches and immunofluorescence staining. Environ Microbiol 8:787–803
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66–74
Wankel SD, Mosier AC, Hansel CM, Paytan A, Francis CA (2011) Spatial variability in nitrification rates and ammonia-oxidizing microbial communities in the agriculturally-impacted Elkhorn slough estuary. Appl Environ Microbiol 77:269–280
Ward BB (2005) Molecular approaches to marine microbial ecology and the marine nitrogen cycle. Annu Rev Earth Planet Sci 33:301–333
Ward BB, O'Mullan GD (2002) Worldwide distribution of Nitrosococcus oceani, a marine ammonia-oxidizing gamma-proteobacterium, detected by PCR and sequencing of 16S rRNA and amoA genes. Appl Environ Microbiol 68:4153–4157
Ward BB, Martino DP, Diaz MC, Joye SB (2000) Analysis of ammonia-oxidizing bacteria from hypersaline Mono Lake, California, on the basis of 16S rRNA sequences. Appl Environ Microbiol 66:2873–2881
Ward BB, Eveillard D, Kirshtein JD, Nelson JD, Voytek MA, Jackson GA (2007) Ammonia-oxidizing bacterial community composition in estuarine and oceanic environments assessed using a functional gene microarray. Environ Microbiol 9:2522–2538
Zhang CL, Ye Q, Huang Z, Li W, Chen J, Song Z, Zhao W, Bagwell C, Inskeep WP, Ross C, Gao L, Wiegel J, Romanek CS, Shock EL, Hedlund BP (2008) Global occurrence of archaeal amoA genes in terrestrial hot springs. Appl Environ Microbiol 74:6417–6426
Zhou W, Long A, Jiang T, Chen S, Huang L, Huang H, Cai C, Yan Y (2011) Bacterioplankton dynamics along the gradient from highly eutrophic Pearl River Estuary to oligotrophic northern South China Sea in wet season: implication for anthropogenic inputs. Mar Pollut Bull. doi:10.1016/j.marpolbul.2011.01.018
Acknowledgments
The research was supported by a Ph.D. studentship (H-LC) from The University of Hong Kong and in part by the grants from Agriculture, Fisheries and Conservation Department of the Hong Kong Government (J-DG). Ms. Jessie Lai was thanked for the kindly laboratory assistance during the research conducted at The University of Hong Kong.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplemental materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 307 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cao, H., Hong, Y., Li, M. et al. Community shift of ammonia-oxidizing bacteria along an anthropogenic pollution gradient from the Pearl River Delta to the South China Sea. Appl Microbiol Biotechnol 94, 247–259 (2012). https://doi.org/10.1007/s00253-011-3636-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3636-1