
Abstract
Since their ‘re-discovery’ more than two decades ago, FOXP3+ regulatory T cells (Tregs) have been an important subject of investigation in the biomedical field and our understanding of the mechanisms that drive their phenotype and function in health and disease has advanced tremendously. During the past few years it has become clear that Tregs are not a terminally differentiated population but show some degree of plasticity, and can, under specific environmental conditions, acquire the phenotype of effector T cells. In particular, recent works have highlighted the acquisition of a Th1-like phenotype by Tregs in several pathological environments. In this review we give an update on the concept of Treg plasticity and the advances in defining the molecular mechanisms that underlie the generation of Th1-like Tregs during an immune response and in different disease settings.
Similar content being viewed by others

Regulatory T cells control the immune system
The immune system is armed with a variety of effector mechanisms to recognize and destroy foreign pathogens as well as with several peripheral tolerance processes to maintain tolerance to self. In this context, cells with regulatory capacity are crucial for maintaining immune homeostasis and peripheral tolerance, and they play an essential role in controlling autoimmune responses, allergies and limiting immunopathology [1,2,3,4,5,6]. While the spectrum of regulatory cells is wide and comprises many different cell types, naturally occurring regulatory T cells (Tregs) is the best studied population of cells with suppressive capacity [6]. They are generated in the thymus at the stage of CD4 single-positive thymocytes [7] as a separate cell lineage, and are thought to be enriched in self-reactive T-cell receptors (TCR), which is essential for maintenance of self-tolerance. As with the development of other cell lineages in the thymus [8, 9], a specific requirement for TCR signaling is essential for the induction of its lineage specification transcription factor FOXP3 and Treg cell lineage commitment, and TCR specificity plays a critical role in this differentiation. Studies with TCR transgenic mice [10,11,12,13] and sequence analysis of polyclonal TCR repertoires from Tregs as compared to conventional T cells bearing a single transgene-encoded TCRβ chain [14,15,16] have led to the conclusion that Treg cell selection is probably instructed by TCRs with affinities for self-peptide–MHC complexes that are of an intermediate affinity between those that induce positive selection of non-Treg cells and those that mediate negative selection of self-reactive T cells. Medullary thymic epithelial cells (mTECs) are the major antigen-presenting cells in the thymus, and their phenotype and function are mediated in part by the transcription factor Aire, which regulates the expression of a multitude of genes, including antigens characteristic of fully differentiated cells in peripheral tissues, shaping the array of self-peptides presented by MHC and thus, the negative selection of effector T cells [17,18,19] and the positive selection of Tregs [20,21,22,23]. Additional signals needed for Treg differentiation in the thymus include cytokines such as IL-2 [24] and to a lesser extent IL-7 and IL-15 [25], and costimulatory molecules, among which, CD28 plays an important role in promoting natural Treg differentiation. Mice studies have shown that mice deficient in CD28 or its ligands CD80 and CD86 have significantly impaired Tregs [26, 27], whereas CTLA-4 ablation results in a higher frequency of natural Tregs [28]. Both TCR and CD28 signaling trigger a myriad of intracellular signaling events that lead to the activation of transcription factors, including NFκB, which has been shown to be essential for natural Treg generation [29,30,31]. Lineage specificity is imprinted in early phases during development by the expression of FOXP3 [32] and by the induction of stable epigenetic changes [33,34,35].
Tregs are characterized by the expression of the master transcription factor FOXP3 [36, 37], the IL-2 receptor alpha chain, CD25 [6, 38], and the low expression of the IL-7 receptor alpha chain, CD127 [39]. Many other markers have been described to be expressed in subpopulations of Tregs, arguing for heterogeneity within this population. IL-2 is an essential cytokine for Treg homeostasis and function. Tregs constitutively express high levels of CD25, and IL-2 is essential to preserve tolerance by influencing Treg homeostasis and activation [40, 41]. In mice, knocking out or blocking IL-2 or CD25 results in Treg deficiency, impaired Treg development and reduced Treg function [42]. Furthermore, IL-2-driven signals through the JAK/STAT signaling pathway directly stabilize FOXP3 expression through STAT5 activation, subsequently driving their suppressive function [43, 44]. Due to the absence of IL-2R signaling, IL-2 and CD25 knock out mice exhibit lethal autoimmunity caused by uncontrolled CD4+ T cell activation and proliferation [45, 46]. Treg numbers are reduced in these mice [47] and co-transfer of functional Tregs can prevent autoimmunity [48], highlighting the importance of IL-2 signaling on Tregs for their role in controlling immune responses.
In the setting of an immune response, CD4+ naïve T cells produce massive amounts of IL-2 and upregulate CD25, resulting in a self-enhancing loop that favors augmented CD4+ T cell activation, proliferation and polarization into T helper effector CD4+ T cells [49]. To counteract increased effector T cell activation, Tregs respond to IL-2 and activate mechanisms to regulate effector T cells to prevent uncontrolled pro-inflammatory and potentially harmful responses.
FOXP3 as a master regulator of the Treg lineage
FOXP3 is crucial for Treg development [37, 50], function [51, 52] and maintenance [53]. Thus, forced expression of FOXP3 in CD4+ T cells results in the acquisition of a regulatory phenotype, although it does not completely recapitulate Treg gene-specific signature [54, 55], and experimental deletion of the FOXP3 gene in Tregs results in the loss of their suppressive capabilities [32, 37, 53]. Mutations in the FOXP3 gene leads to the human autoimmune immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX), characterized by a loss of Treg function and severe autoimmunity. Patients with IPEX suffer from early-onset insulin-dependent diabetes mellitus, thyroiditis, massive lymphoproliferation, eczema, entheropathy and other autoimmune pathologies that are usually fatal during the first years of life [56, 57]. Due to its essential role in maintaining Treg function and stability, it is not surprising that Foxp3 expression is tightly regulated. Transcription of Foxp3 gene has been shown to be modulated at the epigenetic level [58], and FOXP3 protein expression and stability may be controlled by post-translational modifications such as phosphorylation [59,60,61], acetylation [62, 63] and ubiquitination [64, 65], among others. Experiments with genetically engineered mouse models have shown that the genomic region of the Foxp3 locus has several conserved non-coding sequences (CNS1, CNS2, CNS3), which perform diverse functions in the regulation of Foxp3 transcription. CNS1 region contains binding sites for NFAT and AP-1, being important for peripheral generation of adaptive Tregs [58, 66], while CNS3 plays a role in both natural and adaptive Treg generation and contains binding sites for transcription factors such as c-Rel [58]. Runx1-CBFβ complexes bind to CNS2 region to control Foxp3 expression and stability [67]. Moreover, epigenetic modifications of highly conserved regions within CNS in the Foxp3 locus are involved in the transcription of Foxp3. Thus, CNS2 contains a conserved CpG island (TSDR region) that is highly demethylated in natural Tregs and hypermethylated in conventional CD4+ T cells [34, 68, 69], which determines Foxp3 expression and the stability of the Treg lineage [33, 69, 70]. This TSDR region has been widely used to distinguish bona fide Tregs from T cell populations that can transiently upregulate FOXP3 upon activation [71]. Lastly, although FOXP3 is an essential transcription factor required by Tregs to maintain their phenotype and function, over the last few years several works in the literature have demonstrated that FOXP3 does not function alone but forms protein complexes with more than 300 potential partners [72]. Many of these partners are transcription factors such as, among others, NFAT, Gata-3, Smad, Runx1 and FOXO [66, 72,73,74,75]. These transcription factors have been shown to be required to define the Treg cell phenotype and to establish their unique transcriptional program [76].
Functionally, Tregs utilize cell–cell contact mechanisms and soluble factors to inhibit the activation of many different cell types. Thus, Tregs can suppress not only CD4+ and CD8+ T cells [77] but also other immune cells such as B lymphocytes [78,79,80,81], dendritic cells [82,83,84], monocytes [85, 86], and NK cells [87, 88], as well as non-immune cell types such as osteoclasts [89, 90], underscoring the importance of this population to maintain immune homeostasis.
FOXP3−CD4+ T cells in the periphery can also acquire FOXP3 expression and suppressive function when they encounter their cognate antigen in the presence of TFGβ and IL-2 under certain environmental conditions. These Tregs are termed adaptive or induced Tregs (iTregs), and they show important epigenetic differences as compared to natural Tregs; however, we currently lack specific markers that distinguish both populations [91].
Finally, FOXP3 expression also defines a population of CD8+ T cells with regulatory capacity both in mice and humans that seems to play a role in autoimmune, infectious and transplantation settings [92, 93], although their origin and their function in the immune response in these disease scenarios is less studied than those of CD4+ Tregs. Interestingly, some early reports suggested that their suppressive function mainly depends on HLA-E recognition [94, 95] and is mediated by IFNγ secretion [96, 97], although the molecular mechanisms underlying this observation have not been examined in depth.
Regulatory T cell plasticity
Traditionally, Tregs have been considered as a stable cell lineage with strong suppressive capabilities and a terminally differentiated phenotype. But the idea of phenotype irreversibility has been recently challenged by a body of work demonstrating that Tregs are not a completely committed cell lineage, but can retain some degree of plasticity. This observation is not surprising in the context of an immune response, as multitude of works have clearly demonstrated that cell plasticity is an inherent property of most, if not all, immune cells that helps them adapt their phenotype and function to the changing environment [98,99,100,101,102]. In this regard, it is important to distinguish between functional plasticity and lineage instability. For the purpose of this review, we will consider functional plasticity as the capacity of Tregs to acquire a different phenotype due to environmental cues, anatomical location, among other factors, but maintaining either FOXP3 expression or Treg-specific epigenetic patterns. As such, three major plasticity events have been described with regards to Treg phenotype and/or function:
‘ex-FOXP3’ cells
Cell-fate reporter mice have revealed that under certain inflammatory conditions, a small number of Tregs can lose FOXP3 expression and acquire effector-like phenotypes (‘ex-FOXP3’ cells), producing pro-inflammatory cytokines such as IL-2 and TNF and contributing to inflammation [103,104,105,106]. In some experimental conditions, these ex-FOXP3 cells appear to retain the Treg-specific epigenetic signature, potentially being able to be reconverted to FOXP3+ Tregs in the absence of the environmental cues that induced loss of FOXP3 expression [107]. The de-differentiation of Tregs into effector-like cells has also been observed in humans under several pathological settings [108,109,110].
In relation to the loss of FOXP3 expression on Tregs, other studies have shown that FOXP3 degradation favors the secretion of cytokines such as IL-2, TNF and IFNγ and the decrease in suppressive function, especially in type I pathogenic settings [64]. The E3 ubiquitin ligases Stub1 and USP21 seem to play antagonistic roles in modulating the degradation of FOXP3 with Stub1 promoting degradation while USP21 stabilizing FOXP3 expression [64, 65]. Pro-inflammatory cues such as cytokines and LPS signaling induce K48-linked polyubiquitination of FOXP3 by its interaction with Stub1, resulting in FOXP3 degradation, increased expression of IFNγ and reduced expression of characteristic Treg genes like CD25 and CTLA-4 [64] and subsequently disrupting Treg function. On the contrary, specific deletion of the ubiquitin ligase USP21 on mice Tregs induces an immune disorder characterized by increased expression of IFNγ by effector cells, and a Th1-like phenotype by Tregs. USP21 prevents FOXP3 degradation through deubiquination, thus stabilizing Treg phenotype and antagonizing the development of Th1-like Tregs [65]. While USP21 and Stub1 directly interact with FOXP3, the E3 ubiquitin ligase VHL indirectly regulates Th1-like Treg generation by increasing the expression of HIF-1α, which binds to the IFNG promoter, increasing IFNγ production [111].
Treg plasticity as a means of controlling immune responses and/or adapting to the tissue where they reside
Several studies have demonstrated that Tregs utilize the transcription factor program of the population they are suppressing. Thus, Tregs that express T-BET efficiently suppress type 1 inflammation [112], IRF4 expression on Tregs is essential for controlling Th2 responses [113], and STAT3 is utilized by Tregs to control Th17 responses [114] in mouse models of inflammation.
Recent studies in mice have also indicated that tissue-resident Tregs show a distinct gene expression pattern and TCR usage as compared to circulating Tregs [115,116,117]. For instance, the peroxisome proliferator-activated receptor gamma (PPARγ) was identified as the characteristic transcription factor utilized by fat tissue-resident Tregs to maintain their unique phenotype [115]. Interestingly, PPARγ is predominantly expressed on adipocytes, where its function is to regulate adipocyte differentiation and to mediate glucose metabolism [118]. This suggests that tissue-specific Treg phenotypes are also driven by tissue-specific transcription factors, introducing an additional type of plasticity. Similarly, other studies have identified roles for Treg cells in muscle repair [119, 120], regulation of skin homeostasis, and prevention of skin infections [121]. Both muscle- and skin-derived Tregs show a differential gene expression pattern as compared to blood- and fat-derived Tregs, indicating that Tregs might adapt their phenotype and function to the tissue they populate, a process that is probably mediated by the specific tissue microenvironment.
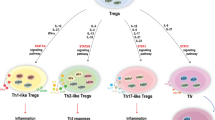
Th-like Tregs
Lastly, under certain inflammatory settings, some Tregs can acquire an effector T helper (Th)-like phenotype with the capacity to express pro-inflammatory cytokines, mainly IFNγ (Th1-like Tregs), IL-17 (Th17-like Tregs) and IL-13 (Th2-like Tregs), and lose suppressive capacities while maintaining FOXP3 expression. In this regard, little is known about the phenotype and function of Th17-like Tregs under inflammatory conditions. For instance, when Tregs are stimulated in vitro in the presence of dectin-1-activated dendritic cells (DC), they upregulate RORγt and express IL-17 [122]. Interestingly, Th17-like Tregs have been observed in vivo in humans under physiological conditions [123, 124] and in mice, preferentially located in the intestine [125]. Human Th17-like Tregs seem to maintain their suppressive capacity despite IL-17 expression in healthy individuals [123, 124], and they can be induced in vitro by stimulation of Tregs in the presence of IL-6 and IL-1β [123]. These cells could have pathogenic potential, contributing to mucosal disease and being involved in the development of colon cancer [110, 126, 127] and inflammatory bowel disease [128]. In this respect, Saito et al. have recently observed that some tumors from patients with colorectal cancer contain an increased frequency of Foxp3low Tregs with increased IL-17 secretion and decreased suppressive function as compared to peripheral blood, and this group of patients seems to have a better prognosis than those who do not display the increase in Th17-like Tregs [110]. Other recent reports have described the presence of Th17-like Tregs in the skin of psoriasis [129] as well as arthritis [130] patients.
Th2-like Tregs have been recently observed in a mouse model of food allergy, and they are increased in frequency in children with milk allergy as compared to healthy donors or children with other food allergies. These Th2-like Tregs are characterized by an increased expression of the transcription factors GATA3 and IRF4 and increased secretion of IL-4 and IL-13 [131, 132]. The production of both cytokines by Tregs directly contributes to disease, as specific disease amelioration is observed upon Treg cell-specific deletion of IL-4 and IL-13 [132]. Moreover, viral infection with the respiratory syncytial virus (RSV), which is known to increase the risk for asthma in adults when infection occurs early in life, induces a Th2-like inflammation in the lung, which promotes a Th2-like effector phenotype in Treg cells and a loss of suppressive function [131]. In vitro, Th2-like Tregs can be polarized by the stimulation of Tregs in the presence of IL-4 and IL-13 [131, 132].
Similarly, we and others have shown that Tregs from healthy individuals stimulated in vitro in the presence of IL-12 acquire a Th1-like phenotype characterized by the secretion of IFNγ, upregulation of T-BET and other Th1-related markers, such as CXCR3 and CCR5, both in mice and humans [68, 133]. Despite the maintained expression of FOXP3, these Th1-like Tregs are defective in suppressive capacity as compared to Tregs, although they retain some degree of inhibitory function in most cases [68, 133, 134]. Several reports have described the in vivo generation of Th1-like Tregs in inflammatory environments such as Toxoplasma gondii infection [135], neurotropic hepatitis virus [136] and in patients with autoimmune diseases such as relapsing-remitting Multiple sclerosis [133, 137] and type 1 diabetes [134].
In this review we give an update on the molecular mechanisms responsible for Th1-like Treg generation focusing on two Treg plasticity aspects: the acquisition of a Th1-suppressing phenotype to control type 1 immune responses, and the acquisition of an effector-like phenotype characterized by their inflammatory nature. We also review recent literature on the role of Th1-like Tregs in several pathological settings such as autoimmune diseases, infections and cancer, and the potential modulation of Treg plasticity as a therapeutic strategy in human disease.
Suppressive Th1-like Tregs: a method to control specific immune responses
During the past years, it has become clear that Treg function is a finely modulated process during the extent of an immune response and is very much dependent on the anatomical location of the occurring reaction, as well as the type of immune response they are controlling. In this regard, it has been observed that Tregs acquire the expression of the master transcription factor that is similar to the effector T cell population or type of immune response that they are suppressing. Thus, Tregs co-opt for IRF4 to inhibit Th2 responses [113] and STAT3 to control Th17 pathology [114], while T-BET expression acquisition by Tregs is necessary for the control of type 1 inflammation in vivo [112].
In mice, Koch et al. demonstrated that a Treg subpopulation expressing CXCR3 (a surface marker preferentially expressed on Th1 CD4+ T cells) is present in the circulation of wild-type animals [112]. CXCR3 expression is regulated by the transcription factor T-BET [138]. T-BET expression in turn is further induced in Tregs during type-I inflammation, subsequently resulting in an enrichment of T-BET+ Tregs that is necessary to control T cell responses during Mycobacterium sp. infection [112]. Although T-BET is classically defined as a Th1-driving transcription factor controlling the expression of IFNγ, T-BET+ Tregs do not seem to express IFNγ in this model; this is likely to be due to the absence of IL-12 receptor beta 2 subunit (IL12RB2) expression and the lack of IL-12-induced signaling pathways [139]. The differentiation of Tregs into Th1-suppressing Tregs is likely to be tightly controlled by cytokines in the microenvironment. In the case of a type 1 immune response, antigen presenting cells (APCs) and other cells from the innate immune system, such as NK cells and macrophages, release Th1-associated cytokines, IL-12, TNFα and IFNγ, which induce a Th1 phenotype on CD4+ effector T cells. These cytokines also shape the phenotype and function of Tregs.
TNFα can trigger pro- and anti-inflammatory pathways in Tregs. A subset of both murine and human Tregs express the TNF receptor 2 (TNFR2) [140, 141]. TNFα stimulation induces, synergistically with IL-2, the expression of other members of the TNFR superfamily such as 4-1BB and OX40 [142]. Interestingly, OX40 (CD134; TNFRSF4) has recently been shown to induce Treg activation and suppressive function [143]. Furthermore, in vitro experiments have demonstrated the induction of Treg proliferation and promotion of Treg stability upon OX40 engagement via APC-mediated activation and stimulation with OX40 ligand (OX40L). TNFα stimulation increases OX40 expression and promotes Treg suppressive function [143, 144]. During type I inflammation, Th1 effector T cells as well as macrophages release a large amount of TNFα. TNFα in turn is essential to stabilize Treg function in a mouse model of colitis [145] as well as murine autoimmune diabetes [146]. Mediating the accumulation of suppressive Tregs in a TNFα-dependent manner poses a potential feedback mechanism to regulate strong type I inflammatory responses. On the other hand, TNFα can inflict negative effects on Treg stability and function. For instance, Valencia et al. demonstrated in vitro that the addition of TNFα in concentrations of 50 ng/ml inhibits Treg suppression and it is accompanied by decreased FOXP3 expression in a TNFR2-dependent manner [147]. In rheumatoid arthritis, TNFα negatively modulates Treg suppressive function [147] and dephosphorylates FOXP3 [61]. Additionally, anti-TNFα therapy induced Tregs in rheumatoid arthritis patients [148, 149]. However, Zhong et al. observed inhibitory effects of TNFα mainly on Helioslow Tregs [150], suggesting that the observed differential effects of TNFα might be highly dependent on the specific Treg subpopulation and/or the inflammatory milieu.
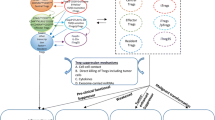
IFNγ is mainly released by NK cells and Th1 effector cells in the context of type-I inflammation. In Tregs, IFNγ increases TBET expression in a STAT1-dependent manner resulting in induction of IL12RB2 expression [139]. The impact of IFNγ on Tregs is thus dichotomic as the upregulation of IL12RB2 renders Tregs susceptible to IL-12 signaling and subsequent polarization into Th1-like Tregs. However, during type-I immune responses, IFNγ fosters the polarization of Th1-suppressing Tregs [112]. As IFNγ signaling induces TBET expression, IFNγ released by Th1 effector CD4+ T cells can act as a feedback mechanism to control type-I immune responses through expansion of Th1-suppressing Tregs and maintenance of high levels of T-BET expression. In regards to IL-12 signaling on Tregs, some reports in the literature suggested that in steady state conditions, Tregs expressed much lower levels of the IL12RB2 subunit mRNA as compared to FOXP3− T cells [139], suggesting that these cells would not be responsive to IL-12 ex vivo. IFNγ induces IL12RB2 in a STAT4-dependent manner in both murine and human Tregs in vitro [139, 143]. Subsequent exposure of IFNγ-primed Tregs to IL-12 renders Tregs dysfunctional and favors the polarization of Th1-like IFNγ-producing Tregs leading to a disrupted regulation of type-I inflammatory responses [151]. However, the upregulation of IFNγ at RNA and protein levels by human and mice Tregs upon ex vivo stimulation in the presence of IL-12 has been widely demonstrated [68, 133,134,135]. IL-12 induces IFNγ-producing Th1-like Tregs that in addition show inhibited proliferative capacities, reduced suppressive function and expression of CD25 [113, 133, 134]. In agreement with these results, recent data suggest that human Tregs do express IL12RB2 at the RNA level in the steady state [152], which would explain the rapid increase in IFNG expression upon IL-12 stimulation. Thus, the balance between differentiation into Th1-suppressing or Th1-like Tregs might be somewhat different between mice and humans and might not be controlled by the expression of IL12RB2 and sensitivity to IL-12, but rather by the amount and availability of IL-12 in the microenvironment and/or the duration of IL-12 signaling (Fig. 1). In this regard, a subpopulation of Tregs expressing OX40, which can be induced by TNFα, has been found to differentiate into Th1-suppressing Tregs and OX40 to compete for IL-12 [143], unraveling another possible mechanism that drives Tregs into different Th1-associated phenotype and functions.

Functional differentiation of Treg cells into Th1-suppressing and Th1-like Tregs. FOXP3+ Tregs upregulate T-BET expression upon type I inflammatory stimuli such as IFNγ in a STAT1-dependent manner. T-BET+FOXP3+ Tregs retain their suppressive function and contribute to resolution of type I inflammation. Additional or subsequent exposure to IL-12 drives Tregs to express T-BET and release IFNγ. IFNγ-producing Tregs either contribute to resolution of type I inflammation (Th1-suppressing) or lose suppressive function and fail to efficiently control autoimmune responses (Th1-like Tregs)
Th1-like Treg signaling
In humans, in vitro stimulation of Tregs with IL-12 induces a Th1-like Treg phenotype with upregulation of T-BET, CXCR3, CCR5 and IFNγ expression [133, 151]. In contrast to mice studies [139], human Treg stimulation in the presence of IFNγ does not induce a Th1-like phenotype, with IL-12 efficiently inducing IFNG gene expression as soon as 24 h after stimulation in vitro ([133, 153] and unpublished data). While FOXP3 expression and the methylation status of the TSDR locus remain unaffected by IL-12 exposure, Th1-like Tregs diminish their suppressive function in in vitro co-cultures with Treg-depleted CD4+ T cells. In relation to the signaling pathways that drive the generation of Th1-like Tregs in humans, we recently defined the PI3K/AKT/FOXO pathway as a major axis involved in IFNγ production by Tregs [153]. PI3K/AKT is a critical signaling node in all eukaryotic cells and the AKT family of proteins is one of the most versatile and important kinases in human physiology and disease. The AKT family of proteins comprises 3 mammalian isoforms (AKT1, AKT2 and AKT3), encoded by separate genes [154]. AKT is activated downstream of PI3K, which can be triggered by multiple stimuli such as T cell receptor (TCR) engagement [155], costimulatory molecules such as CD28 [156], cytokine receptors [153, 157], G protein-coupled receptors [158] and insulin [159], among others. AKT is fully activated by phosphorylation at both Ser 473 by mTORC2 [160], and at Thr 308 by PI3K-activated PDK1 [161]. Although phosphorylation at Ser 473 is necessary for full activation, once triggered by growth factors, phosphorylation at this residue by mTORC2 targets AKT for degradation by the ubiquitin system [162]. Once activated, AKT is able to phosphorylate and activate a myriad of downstream substrates, influencing diverse cellular and physiological processes such as cell cycle progression, cell growth, cell differentiation, cell survival, metabolism, angiogenesis and motility [163,164,165]. One of these multiple AKT targets is the FOXO family of transcription factors, which has been widely involved in Treg development and function in mice studies [73, 166, 167]. The PI3K/AKT/FOXO axis is tightly regulated by the function of several phosphatases that act at different levels of the pathway, among which, PTEN inhibits PI3K by dephosphorylating PI(3,4,5)P3 to PI(4,5)P2 and PHLPP phosphatases dephosphorylate AKT kinases [168].
IFNγ+ Tregs isolated from healthy individuals show an increased expression of AKT1 and decreased expression of FOXO3, AKT3 and PTEN. Interestingly, using IL-12 as an in vitro model for Th1-like Treg generation, it is observed that IL-12 directly induces Th1 polarization by activation of the PI3K/AKT/FOXO1/3 pathway as measured by phosphorylation of AKT at residue Thr 308 and FOXO1/3 at Ser 319 [133, 153]. Despite the well known involvement of AKT in cell survival, in vitro experiments did not show increased Th1-like Treg survival as compared to control Tregs [153], although the in vivo role of this pathway in promoting Th1-like Treg cell survival remains to be elucidated. Previous studies had highlighted the importance of the PI3K/AKT axis in Treg development [169, 170] and function [171], demonstrating that diminished AKT activation is necessary for human Tregs to foster suppressive capacities [171]. The importance of the PI3K/AKT/FOXO1/3 pathway is further highlighted by a series of in vitro experiments where interference with the pathway by either pharmacological activation of PI3K or AKT1, or inhibition of PTEN or FOXO1/3 resulted in increased expression of IFNγ and T-BET as well as reduced suppressive function [153]. These data are in agreement with some recent works demonstrating that PI3K and PTEN are essential for Treg stability in vivo in mouse Tregs [172, 173]. PTEN stabilizes the metabolic balance between glycolysis and mitochondrial fitness, and PTEN-deficient Tregs show increased phosphorylation of AKT and higher expression of activation markers. In these mice, activated memory-effector T cells produce high amounts of IFNγ and upregulate CXCR3, underscoring the importance of the PTEN/AKT axis to stabilize Treg phenotype and modulate Treg-mediated control of type 1 immune responses [172]. Tregs deficient in PTEN show increased PI3K activity and downregulate the expression of CD25 and FOXP3 which leads to reduced suppressive capacity further demonstrating the relevance of the PI3K/AKT/FOXO1/3 pathway for Treg homeostasis and function [173]. FOXO1 has also been involved in the stability and function of Tregs, as FOXO−/− Tregs display an increase in IFNγ expression and a pro-inflammatory phenotype, and mice with Treg-specific deletion of FOXO1 succumb to a fatal autoimmune disorder similar in severity to that observed in FOXP3 deficient mice [73] (Fig. 2).

The PI3K/AKT/FOXO pathway is a key regulator of Th1-like Treg differentiation. Upon T cell activation via the T cell receptor or cytokine receptors, the phosphatidylinositol 3-kinase (PI3K) phosphorylates PI(4,5)P2 to PI(3,4,5)P3 which in turn recruits PDK1. PTEN counteracts PI3K activity by dephosphorylating PI(3,4,5)P3 to PI(4,5)P2. PDK1 phosphorylates AKT at Thr 308. Additional phosphorylation of AKT at Ser 473 by mTORC2, activates AKT to phosphorylate FOXO transcription factors, which promotes their nuclear export. FOXO1/3 are essential to stabilize FOXP3 expression in Tregs. In the case of Th1-like Tregs, AKT phosphorylation is enhanced resulting in increased FOXO1/3 nuclear export, which enables other signaling events to trigger TBET and IFNG gene expression
Interestingly, AKT isoforms exhibit differential functions on the context of Th1-like Treg polarization [153]. While AKT1 is upregulated in IFNγ-producing Th1-like Tregs and AKT1 blockade can prevent Th1-like Treg polarization [153, 171], AKT3 silencing was sufficient to induce IFNγ production by human Tregs [153]. Other studies have suggested non-overlapping functions of AKT isoforms in the context of cancer biology and vascular disease [154, 165, 174]. Moreover, in the context of autoimmunity, a recent study has attributed AKT3 a protective role in EAE development in mice [175].
Besides the PI3K/AKT/FOXO pathway, gene expression analysis of human IFNγ+ and IFNγ− Tregs has shown that other signaling pathways are differentially expressed in Th1-like Tregs, suggesting that they could also be involved in Th1-like Treg generation, although their functional relevance remains to be determined [153].
Th1-like Tregs in autoimmunity
In regards to autoimmune pathologies, several works in the literature have defined the presence of Th1-like Tregs in patients with various diseases, such as Multiple Sclerosis (MS) [133, 153], type 1 diabetes (T1D) [134], de novo autoimmune hepatitis in patients with liver transplant [176] and in inflammatory bowel disease [177,178,179] (IBD). We described some years ago that untreated relapsing-remitting (RR) MS patients display an increased frequency of Th1-like Tregs in peripheral blood. These Tregs express increased levels of T-BET, CXCR3, CCR5, and IFNγ and decreased levels of TGFβ, and CTLA-4, being defective in function [133]. Interestingly, IFNγ is involved in the decreased suppressive capacity observed in Tregs from RRMS patients, as IFNγ blockade in ex vivo co-cultures of Tregs and Treg-depleted CD4+ T cells from patients with MS, significantly increased their suppressive function. Moreover, the elevated frequency of Th1-like Tregs in RRMS patients is due, at least in part, to the in vivo activated status of the AKT/FOXO pathway as compared to Tregs from healthy individuals, with increased expression of phosphorylated AKT (Thr 308) and FOXO1/3 (Ser 319) [153]. Therefore, blockade of PI3K activation in ex vivo stimulated Tregs from MS patients decreased the frequency of IFNγ-producing Tregs and increased their suppressive capacity. The presence of Th1-like Tregs has also been observed in Tregs infiltrating the CNS in vivo, during EAE development, utilizing a FOXP3 knock-in mouse model. In this model, antigen-specific Tregs and effector T cells were traced by MOG-specific tetramers at different points after EAE induction [137]. Upon EAE induction, MOG-specific Tregs were not capable of suppressing CNS-infiltrating MOG-specific T cells in vivo and in vitro and prevent disease onset, and secreted IFNγ at the onset and peak of the disease, decreasing the amount during the recovery phase and increasing IL-10 secretion [137].
Similarly, patients with T1D display an increased frequency of Th1-like Tregs as compared to healthy individuals [134] that can be observed in in vitro expanded or in ex vivo isolated Tregs. Similar to the observation made with MS patients, Th1-like Tregs in T1D patients contained both Helios+ and Helios− Tregs. Although initially thought to be a marker of thymus-derived Tregs [180], contradictory works have occluded the use of this marker to differentiate natural versus adaptive Tregs [181,182,183,184]. In vivo, a recent work has demonstrated that in the pre-diabetes phase in the NOD mouse model there is an increased frequency of Th1-like Tregs in the draining lymph nodes, characterized by the expression of T-BET, CXCR3, ICOS and IFNγ. These CXCR3+ICOS+ Th1-like Tregs, however, retain some degree of suppressive capacity in vivo in adoptive transfer experiments [185].
Tregs are increased in frequency in inflamed intestinal tissue in animal models and patients with IBD [178, 179] and most of them express IFNγ and IL-17, potentially enhancing inflammation or inhibiting regulation. But contradictory results have been found in in vivo models of colitis with regards to the protective or pathogenic role of Th1-like Tregs in this setting [177,178,179]. While antigen-specific IFNγ+ Tregs were able to prevent colitis in an adoptive transfer model with flagellin-specific Tregs [178], FOXO−/− Tregs, which show a Th1-like phenotype, we unable to prevent diseases in a colitis model, with IFNγ being involved on the Treg defect in function, as FOXO−/− IFNG−/− double knock-out mice were able to partially recover from the wasting syndrome [73].
Th1-like Tregs in infections and tumor environments
Both Th1-suppressing and Th1-like Tregs have been described in several models of infection and tumor environments, with each of the two populations contributing to disease manifestations and outcome. Th1-suppressing Tregs, characterized by elevated levels of T-BET and CXCR3 but not IFNγ, are induced in a model of type I inflammation with Leishmania major and Mycobacterium tuberculosis infections in mice [112]. T-BET-expressing Tregs accumulated at the site of infection, balanced Th1 inflammatory responses and maintained their homeostasis and function. In other infection models, Th1-like Tregs gained the ability to express IFNγ resulting in differential clinical outcomes of the infection models. Thus, during T. gondii infection, T-BET and IFNγ expression were triggered in Tregs while FOXP3 expression declined coinciding with strong immunopathology and subsequent lethal disease progression [135]. Although IFNγ is considered to be a highly pro-inflammatory cytokine, Hall et al. reported the rise of IFNγ+ Tregs that were able to suppress Th1 effector T cells and limit effector T cell responses to T. gondii infection [186]. Along this line, IFNγ+ Tregs in a colitis model retained suppressive function in vitro and inhibited the induction of colitis by microbiota antigen-specific T cells in vivo [113]. IFNγ+ Th1-suppressing Tregs were also observed in a chronic corona-virus-induced encephalomyelitis model during both acute and chronic phases of infection [136]. Again, Th1-suppressing Tregs produced IFNγ and retained their function in this model, being more likely to contribute to diminishing immunopathology. Koenecke et al., further demonstrated the protective role of IFNγ-producing Tregs as these cells appear in both Listeria monocytogenes infection and in acute graft-versus-host disease (GVHD). Ablation of IFNγ in Tregs resulted in the development of lethal GVHD undermining the importance of IFNγ-production by Tregs to prevent GVHD [187].
A recent study identified differential surface expression of OX40 to coincide with IFNγ-expression and Treg function in Tregs isolated from liver tissue from human hepatocellular carcinoma and cirrhosis patients [143]. These data suggest that OX40 expression efficiently separates Th1-like (OX40−) Tregs with IFNγ expression and reduced suppressive function from Th1-suppressing (OX40+) Tregs. In this study, cirrhosis tissue and tumor microenvironments favor the accumulation of OX40+ Th1-suppressing Tregs, whereas OX40− Th1-like Tregs preferentially accumulate in non-cirrhotic chronic HCV-associated liver tissue. OX40 stimulation can abolish Treg function and thus it has been investigated as a potential antitumor target [188]. Furthermore, recent data in patients with colorectal cancer have shown that a small percentage of Tregs from either peripheral blood or infiltrating the tumor express IFNγ at similar levels, but the study lacks healthy individuals for absolute comparison [110].
Taken together, these data demonstrate that Th1-suppressive and Th1-like regulatory T cells appear in a variety of infections and tumor environments. Whereas the expression of the transcription factor T-BET seems to be a characteristic feature in all models, additional differentiation and expression of IFNγ can either be connected with protective or pro-inflammatory function in these disease settings.
Treg plasticity as a potential therapeutic strategy
Tregs represent a major barrier to effective immune responses in antitumor immunity, as well as in chronic viral infections. The identification of pathways that maintain Treg cell stability or that induce Th-like effector functions on Tregs could potentially present important novel therapeutic approaches to undermine intratumoral Tregs or Tregs in chronic infections and to enhance disease clearance.
Modulation of Treg differentiation might be a potential therapeutic target but also harbors some pitfalls. Engagement of certain surface molecules and activation of subsequent signaling pathways that might drive Tregs into either Th1-like or Th1-suppressing subtypes can dictate the beneficial outcome in settings of infection or tumor development. Several clinical studies have been undertaken to exploit antibody therapy against CTLA-4 and PD-1/PD-L1 thus dampening intratumoral Treg responses and enabling tumor clearance by effector T cells [189, 190]. However, systemic administration of antibodies or other reagents that interfere with Treg signaling pathways harbor the danger of shifting the subpopulation balance in disfavor when considering the performance of other immune responses.
Furthermore, the underlying pathways that drive Treg plasticity into either direction seem to be critically dependent on the microenvironment. MS patients display an increased number of IFNγ+ Th1-like Tregs that lost suppressive function in peripheral blood and are thus believed to be a crucial factor as to why autoimmune reactions in these patients are not controlled efficiently. The PI3K/AKT/FOXO1/3 pathway has been identified as one of the key pathways involved in Th1-like Treg generation and is an interesting prospective target for immunomodulation [153]. However, IFNγ+ Th1-like Tregs in other disease settings do not lose their suppressive function which might indicate the involvement of different signaling pathways ultimately resulting in similar but yet different phenotypic outcomes.
Clinically, Tregs have recently been beneficially used for therapy against graft versus host disease following allogeneic bone marrow or stem cell transplantation [191, 192] and type 1 diabetes patients [193]. In these settings, autologous polyclonal Tregs are expanded in vitro and re-transferred into the patient. The use of in vitro Treg expansion protocols gives the opportunity to analyze Treg phenotype and function as well as specifically control cell numbers necessary to re-transfer for a beneficial outcome. Several clinical trial studies have reported successful application of Treg transfer therapy in T1D and GVHD [193, 194]. As antigen-specific Tregs are more efficient at regulating disease-specific immunological processes, the generation of antigen-specific Tregs using chimeric antigen receptors (CAR) has been explored as well, as an improvement to Treg therapy. CAR Tregs have been shown to prevent GVHD in a humanized mouse model highlighting their improved therapeutic potential [195].
Combination of adoptive transfer of antigen-specific Tregs with modulation of Treg plasticity proposes strong therapeutic potential. In vitro expansion of Tregs provides the opportunity to also include cytokine treatments to reprogram Tregs into phenotypic subtypes most beneficial for the respective disease. Furthermore, defective Treg phenotypes could be corrected by either interfering with involved pathways by using pharmacological agents or by introducing recent gene editing technologies such as the CRISPR/Cas9 system.
Conclusions and future perspectives
During the past few years it has become clear that Tregs possess some degree of plasticity and can adapt their phenotype to the microenvironment where they exert their functions. Furthermore, aberrant reprogramming of Tregs into Th1-like Tregs has been observed in several human autoimmune diseases, suggesting their contribution to disease. The relative importance of Th1-like Tregs in the pathophysiology of the diseases where they have been observed, their role in promoting or protecting from inflammation-derived damage and their influence in disease outcome are fundamental open questions that remain to be answered and that will undoubtedly provide valuable information for the potential manipulation of Treg plasticity with therapeutic purposes for these diseases. In order to design better therapeutic options targeted to Tregs in cancer, infectious and autoimmune diseases, it is imperative to understand the signaling pathways that govern the acquisition of specific effector characteristics by Tregs in different disease settings. In this regard, most of the current literature on Th1-like Tregs defines IFNγ and IL-12 as the major inducers of Th1- reprogramming in Treg cells. However, the discovery of the PI3K/AKT axis as a major signaling pathway that regulates Th1-like Treg generation, and the variety of upstream ligands/receptors that can activate it, makes the generation of Th1-like Tregs a plausible event in many disease settings, and it strongly suggests that there are likely other environmental cues apart from Th1 cytokines that induce the generation of dysfunctional Th1-Tregs or Th1-suppressing Tregs. Moreover, there are likely many other signaling pathways involved in Th1-like Treg generation in specific disease settings yet to be defined [93], that will further improve our knowledge on the molecular mechanisms that regulate human Treg plasticity with potential therapeutic applications.
In conclusion, it is crucial to fully understand the underlying pathways and mechanisms that regulate Treg plasticity and the environmental cues that induce such phenotypes in specific disease settings in order to be able to take advantage of Treg plasticity for therapeutic purposes. Furthermore, it will be important to perform investigations that focus on understanding the differences between Th1-suppressive Th1-Tregs and dysfunctional Th1-Tregs observed in autoimmune disease settings and acute phases of infection as well as how to transition from one state to another, with potential important applications in therapy.
References
Rosser EC, Mauri C (2015) Regulatory B cells: origin, phenotype, and function. Immunity 42(4):607–612. doi:10.1016/j.immuni.2015.04.005
Miyara M, Sakaguchi S (2011) Human FoxP3(+)CD4(+) regulatory T cells: their knowns and unknowns. Immunol Cell Biol 89(3):346–351. doi:10.1038/icb.2010.137
Wang YM, Alexander SI (2009) CD8 regulatory T cells: what’s old is now new. Immunol Cell Biol 87(3):192–193. doi:10.1038/icb.2009.8
Fu B, Tian Z, Wei H (2014) Subsets of human natural killer cells and their regulatory effects. Immunology 141(4):483–489. doi:10.1111/imm.12224
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9(3):162–174. doi:10.1038/nri2506
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10(7):490–500. doi:10.1038/nri2785
Fontenot JD, Dooley JL, Farr AG, Rudensky AY (2005) Developmental regulation of Foxp3 expression during ontogeny. J Exp Med 202(7):901–906. doi:10.1084/jem.20050784
Germain RN (2002) T-cell development and the CD4–CD8 lineage decision. Nat Rev Immunol 2(5):309–322. doi:10.1038/nri798
Singer A, Adoro S, Park JH (2008) Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat Rev Immunol 8(10):788–801. doi:10.1038/nri2416
Apostolou I, Sarukhan A, Klein L, von Boehmer H (2002) Origin of regulatory T cells with known specificity for antigen. Nat Immunol 3(8):756–763. doi:10.1038/ni816
Kawahata K, Misaki Y, Yamauchi M, Tsunekawa S, Setoguchi K, Miyazaki J, Yamamoto K (2002) Generation of CD4(+)CD25(+) regulatory T cells from autoreactive T cells simultaneously with their negative selection in the thymus and from nonautoreactive T cells by endogenous TCR expression. J Immunol 168(9):4399–4405
Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S (1994) High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell 78(3):399–408
Olivares-Villagomez D, Wang Y, Lafaille JJ (1998) Regulatory CD4(+) T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med 188(10):1883–1894
Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY (2004) Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21(2):267–277. doi:10.1016/j.immuni.2004.07.009
Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L (2006) Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity 25(2):249–259. doi:10.1016/j.immuni.2006.05.016
Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C (2007) Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol 178(11):7032–7041
Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D (2005) The cellular mechanism of Aire control of T cell tolerance. Immunity 23(2):227–239. doi:10.1016/j.immuni.2005.07.005
Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC (2003) Aire regulates negative selection of organ-specific T cells. Nat Immunol 4(4):350–354. doi:10.1038/ni906
Taniguchi RT, DeVoss JJ, Moon JJ, Sidney J, Sette A, Jenkins MK, Anderson MS (2012) Detection of an autoreactive T-cell population within the polyclonal repertoire that undergoes distinct autoimmune regulator (Aire)-mediated selection. Proc Natl Acad Sci USA 109(20):7847–7852. doi:10.1073/pnas.1120607109
Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, Bosl MR, Hollander GA, Hayashi Y, Malefyt Rde W, Nitta T, Takahama Y (2011) Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J Exp Med 208(2):383–394. doi:10.1084/jem.20102327
Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP, Socci ND, Savage PA (2013) Aire-dependent thymic development of tumor-associated regulatory T cells. Science 339(6124):1219–1224. doi:10.1126/science.1233913
Perry JS, Lio CW, Kau AL, Nutsch K, Yang Z, Gordon JI, Murphy KM, Hsieh CS (2014) Distinct contributions of Aire and antigen-presenting-cell subsets to the generation of self-tolerance in the thymus. Immunity 41(3):414–426. doi:10.1016/j.immuni.2014.08.007
Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D (2015) Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348(6234):589–594. doi:10.1126/science.aaa7017
Burchill MA, Yang J, Vang KB, Farrar MA (2007) Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol Lett 114(1):1–8. doi:10.1016/j.imlet.2007.08.005
Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA (2008) IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol 181(5):3285–3290
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12(4):431–440
Tai X, Cowan M, Feigenbaum L, Singer A (2005) CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol 6(2):152–162. doi:10.1038/ni1160
Verhagen J, Gabrysova L, Minaee S, Sabatos CA, Anderson G, Sharpe AH, Wraith DC (2009) Enhanced selection of FoxP3+ T-regulatory cells protects CTLA-4-deficient mice from CNS autoimmune disease. Proc Natl Acad Sci USA 106(9):3306–3311. doi:10.1073/pnas.0803186106
Barnes MJ, Krebs P, Harris N, Eidenschenk C, Gonzalez-Quintial R, Arnold CN, Crozat K, Sovath S, Moresco EM, Theofilopoulos AN, Beutler B, Hoebe K (2009) Commitment to the regulatory T cell lineage requires CARMA1 in the thymus but not in the periphery. PLoS Biol 7(3):e51. doi:10.1371/journal.pbio.1000051
Schmidt-Supprian M, Tian J, Grant EP, Pasparakis M, Maehr R, Ovaa H, Ploegh HL, Coyle AJ, Rajewsky K (2004) Differential dependence of CD4+CD25+ regulatory and natural killer-like T cells on signals leading to NF-kappaB activation. Proc Natl Acad Sci USA 101(13):4566–4571. doi:10.1073/pnas.0400885101
Wan YY, Chi H, Xie M, Schneider MD, Flavell RA (2006) The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol 7(8):851–858. doi:10.1038/ni1355
Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY (2005) Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22(3):329–341. doi:10.1016/j.immuni.2005.01.016
Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J, Thiel A, Boeld TJ, Hoffmann P, Edinger M, Turbachova I, Hamann A, Olek S, Huehn J (2007) DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol 37(9):2378–2389. doi:10.1002/eji.200737594
Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, Osaki M, Tanaka Y, Yamashita R, Nakano N, Huehn J, Fehling HJ, Sparwasser T, Nakai K, Sakaguchi S (2012) T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 37(5):785–799. doi:10.1016/j.immuni.2012.09.010
Ohkura N, Kitagawa Y, Sakaguchi S (2013) Development and maintenance of regulatory T cells. Immunity 38(3):414–423. doi:10.1016/j.immuni.2013.03.002
Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY (2007) Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445(7129):771–775. doi:10.1038/nature05543
Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299(5609):1057–1061. doi:10.1126/science.1079490
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA (2001) CD4+CD25high regulatory cells in human peripheral blood. J Immunol 167(3):1245–1253
Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, de St. Groth BF, Clayberger C, Soper DM, Ziegler SF, Bluestone JA (2006) CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 203(7):1701–1711. doi:10.1084/jem.20060772
Boyman O, Sprent J (2012) The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 12(3):180–190. doi:10.1038/nri3156
Malek TR, Bayer AL (2004) Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 4(9):665–674. doi:10.1038/nri1435
Cheng G, Yu A, Malek TR (2011) T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev 241(1):63–76. doi:10.1111/j.1600-065X.2011.01004.x
Passerini L, Allan SE, Battaglia M, Di Nunzio S, Alstad AN, Levings MK, Roncarolo MG, Bacchetta R (2008) STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25− effector T cells. Int Immunol 20(3):421–431. doi:10.1093/intimm/dxn002
Murawski MR, Litherland SA, Clare-Salzler MJ, Davoodi-Semiromi A (2006) Upregulation of Foxp3 expression in mouse and human Treg is IL-2/STAT5 dependent: implications for the NOD STAT5B mutation in diabetes pathogenesis. Ann N Y Acad Sci 1079:198–204. doi:10.1196/annals.1375.031
Sadlack B, Lohler J, Schorle H, Klebb G, Haber H, Sickel E, Noelle RJ, Horak I (1995) Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol 25(11):3053–3059. doi:10.1002/eji.1830251111
Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW (1995) Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3(4):521–530
Almeida AR, Legrand N, Papiernik M, Freitas AA (2002) Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol 169(9):4850–4860
Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ (2002) Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J Exp Med 196(6):851–857
Chaplin DD (2010) Overview of the immune response. J Allergy Clin Immunol 125(2 Suppl 2):S3–23. doi:10.1016/j.jaci.2009.12.980
Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4(4):330–336
Khattri R, Cox T, Yasayko SA, Ramsdell F (2003) An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 4(4):337–342. doi:10.1038/ni909
Wan YY, Flavell RA (2007) Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 445(7129):766–770
Williams LM, Rudensky AY (2007) Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol 8(3):277–284. doi:10.1038/ni1437
Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C (2007) Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27(5):786–800. doi:10.1016/j.immuni.2007.09.010
Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, Sakaguchi S (2006) Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol 18(8):1197–1209. doi:10.1093/intimm/dxl060
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27(1):20–21
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME (2001) X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27(1):18–20. doi:10.1038/83707
Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY (2010) Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463(7282):808–812. doi:10.1038/nature08750
Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S, Gao Z, Piccioni M, Tsun A, Cai S, Zheng SG, Zhang Y, Li B (2014) PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem 289(39):26872–26881. doi:10.1074/jbc.M114.586651
Morawski PA, Mehra P, Chen C, Bhatti T, Wells AD (2013) Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem 288(34):24494–24502. doi:10.1074/jbc.M113.467704
Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, Liu X, Xiao L, Chen X, Wan B, Chin YE, Zhang JZ (2013) Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med 19(3):322–328. doi:10.1038/nm.3085
Kwon HS, Lim HW, Wu J, Schnolzer M, Verdin E, Ott M (2012) Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J Immunol 188(6):2712–2721. doi:10.4049/jimmunol.1100903
van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, Brenkman AB, Hijnen DJ, Mutis T, Kalkhoven E, Prakken BJ, Coffer PJ (2010) Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 115(5):965–974. doi:10.1182/blood-2009-02-207118
Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, Jinasena D, Fu J, Lin F, Chen C, Zhang J, Yu N, Li X, Shan Z, Nie J, Gao Z, Tian H, Li Y, Yao Z, Zheng Y, Park BV, Pan Z, Zhang J, Dang E, Li Z, Wang H, Luo W, Li L, Semenza GL, Zheng SG, Loser K, Tsun A, Greene MI, Pardoll DM, Pan F, Li B (2013) The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 39(2):272–285. doi:10.1016/j.immuni.2013.08.006
Li Y, Lu Y, Wang S, Han Z, Zhu F, Ni Y, Liang R, Zhang Y, Leng Q, Wei G, Shi G, Zhu R, Li D, Wang H, Zheng SG, Xu H, Tsun A, Li B (2016) USP21 prevents the generation of T-helper-1-like Treg cells. Nat Commun 7:13559. doi:10.1038/ncomms13559
Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M (2008) Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol 9(2):194–202. doi:10.1038/ni1549
Kitoh A, Ono M, Naoe Y, Ohkura N, Yamaguchi T, Yaguchi H, Kitabayashi I, Tsukada T, Nomura T, Miyachi Y, Taniuchi I, Sakaguchi S (2009) Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity 31(4):609–620. doi:10.1016/j.immuni.2009.09.003
Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O’Shea JJ, Zhao K (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30(1):155–167. doi:10.1016/j.immuni.2008.12.009
Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J (2007) Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 5(2):e38
Nagar M, Vernitsky H, Cohen Y, Dominissini D, Berkun Y, Rechavi G, Amariglio N, Goldstein I (2008) Epigenetic inheritance of DNA methylation limits activation-induced expression of FOXP3 in conventional human CD25−CD4+ T cells. Int Immunol 20(8):1041–1055. doi:10.1093/intimm/dxn062
Huehn J, Polansky JK, Hamann A (2009) Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol 9(2):83–89. doi:10.1038/nri2474
Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, Rudensky AY (2012) Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 13(10):1010–1019. doi:10.1038/ni.2402
Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, Meijer D, Zhao K, Rudensky AY, Atwal G, Zhang MQ, Li MO (2012) Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491(7425):554–559. doi:10.1038/nature11581
Hu H, Djuretic I, Sundrud MS, Rao A (2007) Transcriptional partners in regulatory T cells: Foxp3, Runx and NFAT. Trends Immunol 28(8):329–332. doi:10.1016/j.it.2007.06.006
Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S (2007) Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446(7136):685–689. doi:10.1038/nature05673
Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, Gazit R, Adoro S, Glimcher L, Chan S, Kastner P, Rossi D, Collins JJ, Mathis D, Benoist C (2012) A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol 13(10):972–980. doi:10.1038/ni.2420
Thornton AM, Shevach EM (1998) CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med 188(2):287–296
Iikuni N, Lourenco EV, Hahn BH, La Cava A (2009) Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol 183(3):1518–1522. doi:10.4049/jimmunol.0901163
Lim HW, Hillsamer P, Banham AH, Kim CH (2005) Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol 175(7):4180–4183
Sage PT, Ron-Harel N, Juneja VR, Sen DR, Maleri S, Sungnak W, Kuchroo VK, Haining WN, Chevrier N, Haigis M, Sharpe AH (2016) Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat Immunol 17(12):1436–1446. doi:10.1038/ni.3578
Gotot J, Gottschalk C, Leopold S, Knolle PA, Yagita H, Kurts C, Ludwig-Portugall I (2012) Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proc Natl Acad Sci USA 109(26):10468–10473. doi:10.1073/pnas.1201131109
Navarrete AM, Meslier Y, Teyssandier M, Andre S, Delignat S, Triebel F, Kaveri SV, Lacroix-Desmazes S, Bayry J et al (2009) CD4+CD25+ regulatory T cells modulate human dendritic cell chemokines via multiple mechanisms: comment on the article by Kolar et al. Arthritis Rheum 60(9):2848–2849. doi:10.1002/art.24784 (author reply 2849–2851)
Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, Dustin ML (2006) Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med 203(3):505–511. doi:10.1084/jem.20050783
Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, Flores M, Li N, Schweighoffer E, Greenberg S, Tybulewicz V, Vignali D, Clynes R (2008) Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol 180(9):5916–5926
Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, Bijlsma JW, Lafeber FP (2005) Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol 66(3):222–230. doi:10.1016/j.humimm.2004.12.006
Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS (2007) CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci USA 104(49):19446–19451. doi:10.1073/pnas.0706832104
Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, Puig PE, Novault S, Escudier B, Vivier E, Lecesne A, Robert C, Blay JY, Bernard J, Caillat-Zucman S, Freitas A, Tursz T, Wagner-Ballon O, Capron C, Vainchencker W, Martin F, Zitvogel L (2005) CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med 202(8):1075–1085. doi:10.1084/jem.20051511
Ralainirina N, Poli A, Michel T, Poos L, Andres E, Hentges F, Zimmer J (2007) Control of NK cell functions by CD4+CD25+ regulatory T cells. J Leukoc Biol 81(1):144–153. doi:10.1189/jlb.0606409
Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB (2007) Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun 357(4):1046–1052. doi:10.1016/j.bbrc.2007.04.042
Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G (2007) Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum 56(12):4104–4112. doi:10.1002/art.23138
Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A (2016) Induced regulatory T cells: their development, stability, and applications. Trends Immunol 37(11):803–811. doi:10.1016/j.it.2016.08.012
Lu L, Kim HJ, Werneck MB, Cantor H (2008) Regulation of CD8+ regulatory T cells: interruption of the NKG2A-Qa-1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc Natl Acad Sci USA 105(49):19420–19425. doi:10.1073/pnas.0810383105
Panoutsakopoulou V, Huster KM, McCarty N, Feinberg E, Wang R, Wucherpfennig KW, Cantor H (2004) Suppression of autoimmune disease after vaccination with autoreactive T cells that express Qa-1 peptide complexes. J Clin Investig 113(8):1218–1224. doi:10.1172/JCI20772
Hu D, Ikizawa K, Lu L, Sanchirico ME, Shinohara ML, Cantor H (2004) Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol 5(5):516–523. doi:10.1038/ni1063
Sarantopoulos S, Lu L, Cantor H (2004) Qa-1 restriction of CD8+ suppressor T cells. J Clin Investig 114(9):1218–1221. doi:10.1172/JCI23152
Balashov KE, Khoury SJ, Hafler DA, Weiner HL (1995) Inhibition of T cell responses by activated human CD8+ T cells is mediated by interferon-gamma and is defective in chronic progressive multiple sclerosis. J Clin Investig 95(6):2711–2719. doi:10.1172/JCI117973
Long X, Cheng Q, Liang H, Zhao J, Wang J, Wang W, Tomlinson S, Chen L, Atkinson C, Zhang B, Chen X, Zhu P (2017) Memory CD4+ T cells are suppressed by CD8+ regulatory T cells in vitro and in vivo. Am J Transl Res 9(1):63–78
Bonelli M, Shih HY, Hirahara K, Singelton K, Laurence A, Poholek A, Hand T, Mikami Y, Vahedi G, Kanno Y, O’Shea JJ (2014) Helper T cell plasticity: impact of extrinsic and intrinsic signals on transcriptomes and epigenomes. Curr Topics Microbiol Immunol 381:279–326. doi:10.1007/82_2014_371
DuPage M, Bluestone JA (2016) Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol 16(3):149–163. doi:10.1038/nri.2015.18
Galli SJ, Borregaard N, Wynn TA (2011) Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 12(11):1035–1044. doi:10.1038/ni.2109
Perez-Shibayama C, Gil-Cruz C, Ludewig B (2014) Plasticity and complexity of B cell responses against persisting pathogens. Immunol Lett 162(1 Pt A):53–58. doi:10.1016/j.imlet.2014.07.003
Takashima A, Yao Y (2015) Neutrophil plasticity: acquisition of phenotype and functionality of antigen-presenting cell. J Leukoc Biol 98(4):489–496. doi:10.1189/jlb.1MR1014-502R
Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA (2009) Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 10(9):1000–1007. doi:10.1038/ni.1774
Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J (2009) Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol 39(4):948–955. doi:10.1002/eji.200839196
Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S (2009) Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA 106(6):1903–1908. doi:10.1073/pnas.0811556106
Yurchenko E, Shio MT, Huang TC, Da Silva Martins M, Szyf M, Levings MK, Olivier M, Piccirillo CA (2012) Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS One 7(4):e35572. doi:10.1371/journal.pone.0035572
Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S (2012) Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 36(2):262–275. doi:10.1016/j.immuni.2011.12.012
Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, Olek S, Dietmaier W, Andreesen R, Edinger M (2009) Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol 39(4):1088–1097. doi:10.1002/eji.200838904
Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I (2008) Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 112(6):2340–2352. doi:10.1182/blood-2008-01-133967
Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, Nagase H, Nishimura J, Yamamoto H, Takiguchi S, Tanoue T, Suda W, Morita H, Hattori M, Honda K, Mori M, Doki Y, Sakaguchi S (2016) Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 22(6):679–684. doi:10.1038/nm.4086
Lee JH, Elly C, Park Y, Liu YC (2015) E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell Stability and suppressive capacity. Immunity 42(6):1062–1074. doi:10.1016/j.immuni.2015.05.016
Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ (2009) The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10(6):595–602. doi:10.1038/ni.1731
Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, Rudensky AY (2009) Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458(7236):351–356. doi:10.1038/nature07674
Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, Rudensky AY (2009) CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326(5955):986–991. doi:10.1126/science.1172702
Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D (2012) PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486(7404):549–553. doi:10.1038/nature11132
Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D (2009) Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15(8):930–939. doi:10.1038/nm.2002
Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, Wagers A, Germain RN, Benoist C, Mathis D (2015) Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab 21(4):543–557. doi:10.1016/j.cmet.2015.03.005
Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM (2000) Transcriptional regulation of adipogenesis. Genes Dev 14(11):1293–1307
Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D (2013) A special population of regulatory T cells potentiates muscle repair. Cell 155(6):1282–1295. doi:10.1016/j.cell.2013.10.054
Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, Mathis D (2016) Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44(2):355–367. doi:10.1016/j.immuni.2016.01.009
Xia M, Hu S, Fu Y, Jin W, Yi Q, Matsui Y, Yang J, McDowell MA, Sarkar S, Kalia V, Xiong N (2014) CCR10 regulates balanced maintenance and function of resident regulatory and effector T cells to promote immune homeostasis in the skin. J Allergy Clin Immunol 134(3):634–644. doi:10.1016/j.jaci.2014.03.010
Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, Reis e Sousa C (2008) DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol 38(12):3274–3281. doi:10.1002/eji.200838950
Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, Hafler DA (2009) IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood 113(18):4240–4249. doi:10.1182/blood-2008-10-183251
Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, Bover L, Hanabuchi S, Khalili J, Marinova E, Zheng B, Littman DR, Liu YJ (2009) Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci USA 106(12):4793–4798
Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR (2008) TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453(7192):236–240. doi:10.1038/nature06878
Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, Ham S, Sandall BP, Khan MW, Mahvi DM, Halverson AL, Stryker SJ, Boller AM, Singal A, Sneed RK, Sarraj B, Ansari MJ, Oft M, Iwakura Y, Zhou L, Bonertz A, Beckhove P, Gounari F, Khazaie K (2012) Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci Transl Med 4(164):164ra159. doi:10.1126/scitranslmed.3004566
Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, Huang E, Greenson J, Chang A, Rolinski J, Radwan P, Fang J, Wang G, Zou W (2011) IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol 186(7):4388–4395. doi:10.4049/jimmunol.1003251
Hovhannisyan Z, Treatman J, Littman DR, Mayer L (2011) Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 140(3):957–965. doi:10.1053/j.gastro.2010.12.002
Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ (2011) Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J Investig Dermatol 131(9):1853–1860. doi:10.1038/jid.2011.139
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20(1):62–68. doi:10.1038/nm.3432
Krishnamoorthy N, Khare A, Oriss TB, Raundhal M, Morse C, Yarlagadda M, Wenzel SE, Moore ML, Peebles RS Jr, Ray A, Ray P (2012) Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat Med 18(10):1525–1530. doi:10.1038/nm.2896
Noval Rivas M, Burton OT, Wise P, Charbonnier LM, Georgiev P, Oettgen HC, Rachid R, Chatila TA (2015) Regulatory T cell reprogramming toward a Th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity 42(3):512–523. doi:10.1016/j.immuni.2015.02.004
Dominguez-Villar M, Baecher-Allan CM, Hafler DA (2011) Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med 17(6):673–675. doi:10.1038/nm.2389
McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, Hoffmuller U, Baron U, Olek S, Bluestone JA, Brusko TM (2011) Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol 186(7):3918–3926. doi:10.4049/jimmunol.1003099
Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O’Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y (2009) Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31(5):772–786. doi:10.1016/j.immuni.2009.10.001
Zhao J, Zhao J, Fett C, Trandem K, Fleming E, Perlman S (2011) IFN-gamma- and IL-10-expressing virus epitope-specific Foxp3(+) T reg cells in the central nervous system during encephalomyelitis. J Exp Med 208(8):1571–1577. doi:10.1084/jem.20110236
Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK (2007) Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med 13(4):423–431. doi:10.1038/nm1564
Beima KM, Miazgowicz MM, Lewis MD, Yan PS, Huang TH, Weinmann AS (2006) T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J Biol Chem 281(17):11992–12000. doi:10.1074/jbc.M513613200
Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ (2012) T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity 37(3):501–510. doi:10.1016/j.immuni.2012.05.031
Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ (2010) Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur J Immunol 40(4):1099–1106. doi:10.1002/eji.200940022
Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ (2008) Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol 180(10):6467–6471
Hamano R, Huang J, Yoshimura T, Oppenheim JJ, Chen X (2011) TNF optimally activatives regulatory T cells by inducing TNF receptor superfamily members TNFR2, 4-1BB and OX40. Eur J Immunol 41(7):2010–2020. doi:10.1002/eji.201041205
Piconese S, Timperi E, Pacella I, Schinzari V, Tripodo C, Rossi M, Guglielmo N, Mennini G, Grazi GL, Di Filippo S, Brozzetti S, Fazzi K, Antonelli G, Lozzi MA, Sanchez M, Barnaba V (2014) Human OX40 tunes the function of regulatory T cells in tumor and nontumor areas of hepatitis C virus-infected liver tissue. Hepatology 60(5):1494–1507. doi:10.1002/hep.27188
Piconese S, Timperi E, Barnaba V (2014) ‘Hardcore’ OX40+ immunosuppressive regulatory T cells in hepatic cirrhosis and cancer. Oncoimmunology 3:e29257. doi:10.4161/onci.29257
Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ (2013) TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol 190(3):1076–1084. doi:10.4049/jimmunol.1202659
Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, Martin GH, Elhage R, Derian N, Carpentier W, Marodon G, Klatzmann D, Piaggio E, Salomon BL (2010) Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J Clin Investig 120(12):4558–4568. doi:10.1172/JCI42945
Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE (2006) TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 108(1):253–261. doi:10.1182/blood-2005-11-4567
Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C (2004) Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med 200(3):277–285. doi:10.1084/jem.20040165
Nadkarni S, Mauri C, Ehrenstein MR (2007) Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med 204(1):33–39. doi:10.1084/jem.20061531
Zhong H, Yazdanbakhsh K (2013) Differential control of Helios(+/−) Treg development by monocyte subsets through disparate inflammatory cytokines. Blood 121(13):2494–2502. doi:10.1182/blood-2012-11-469122
Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY (2010) Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 142(6):914–929. doi:10.1016/j.cell.2010.08.012
Bhairavabhotla R, Kim YC, Glass DD, Escobar TM, Patel MC, Zahr R, Nguyen CK, Kilaru GK, Muljo SA, Shevach EM (2016) Transcriptome profiling of human FoxP3+ regulatory T cells. Hum Immunol 77(2):201–213. doi:10.1016/j.humimm.2015.12.004
Kitz A, de Marcken M, Gautron AS, Mitrovic M, Hafler DA, Dominguez-Villar M (2016) AKT isoforms modulate Th1-like Treg generation and function in human autoimmune disease. EMBO Rep 17(8):1169–1183. doi:10.15252/embr.201541905
Gonzalez E, McGraw TE (2009) The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8(16):2502–2508. doi:10.4161/cc.8.16.9335
Ward SG, Ley SC, MacPhee C, Cantrell DA (1992) Regulation of D-3 phosphoinositides during T cell activation via the T cell antigen receptor/CD3 complex and CD2 antigens. Eur J Immunol 22(1):45–49. doi:10.1002/eji.1830220108
Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP (1999) Impaired Fas response and autoimmunity in Pten+/− mice. Science 285(5436):2122–2125
Migone TS, Rodig S, Cacalano NA, Berg M, Schreiber RD, Leonard WJ (1998) Functional cooperation of the interleukin-2 receptor beta chain and Jak1 in phosphatidylinositol 3-kinase recruitment and phosphorylation. Mol Cell Biol 18(11):6416–6422
Schwindinger WF, Robishaw JD (2001) Heterotrimeric G-protein betagamma-dimers in growth and differentiation. Oncogene 20(13):1653–1660. doi:10.1038/sj.onc.1204181
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM (2006) A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125(4):733–747. doi:10.1016/j.cell.2006.03.035
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307(5712):1098–1101. doi:10.1126/science.1106148
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7(4):261–269
Wu YT, Ouyang W, Lazorchak AS, Liu D, Shen HM, Su B (2011) mTOR complex 2 targets Akt for proteasomal degradation via phosphorylation at the hydrophobic motif. J Biol Chem 286(16):14190–14198. doi:10.1074/jbc.M111.219923
Buzzi F, Xu L, Zuellig RA, Boller SB, Spinas GA, Hynx D, Chang Z, Yang Z, Hemmings BA, Tschopp O, Niessen M (2010) Differential effects of protein kinase B/Akt isoforms on glucose homeostasis and islet mass. Mol Cell Biol 30(3):601–612. doi:10.1128/MCB.00719-09
Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114(Pt 16):2903–2910
Yu H, Littlewood T, Bennett M (2015) Akt isoforms in vascular disease. Vascul Pharmacol 71:57–64. doi:10.1016/j.vph.2015.03.003
Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch’en IL, Stockmann C, Katayama CD, Hedrick SM (2010) Foxo transcription factors control regulatory T cell development and function. Immunity 33(6):890–904. doi:10.1016/j.immuni.2010.12.002
Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO (2010) Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol 11(7):618–627. doi:10.1038/ni.1884
Chen M, Nowak DG, Trotman LC (2014) Molecular pathways: PI3K pathway phosphatases as biomarkers for cancer prognosis and therapy. Clin Cancer Res 20(12):3057–3063. doi:10.1158/1078-0432.CCR-12-3680
Merkenschlager M, von Boehmer H (2010) PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med 207(7):1347–1350. doi:10.1084/jem.20101156
Haxhinasto S, Mathis D, Benoist C (2008) The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 205(3):565–574. doi:10.1084/jem.20071477
Crellin NK, Garcia RV, Levings MK (2007) Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood 109(5):2014–2022. doi:10.1182/blood-2006-07-035279
Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H (2015) Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol 16(2):178–187. doi:10.1038/ni.3076
Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, Bluestone JA, Turka LA (2015) Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol 16(2):188–196. doi:10.1038/ni.3077
Linnerth-Petrik NM, Santry LA, Petrik JJ, Wootton SK (2014) Opposing functions of Akt isoforms in lung tumor initiation and progression. PLoS One 9(4):e94595. doi:10.1371/journal.pone.0094595
Tsiperson V, Gruber RC, Goldberg MF, Jordan A, Weinger JG, Macian F, Shafit-Zagardo B (2013) Suppression of inflammatory responses during myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis is regulated by AKT3 signaling. J Immunol 190(4):1528–1539. doi:10.4049/jimmunol.1201387
Arterbery AS, Osafo-Addo A, Avitzur Y, Ciarleglio M, Deng Y, Lobritto SJ, Martinez M, Hafler DA, Kleinewietfeld M, Ekong UD (2016) Production of proinflammatory cytokines by monocytes in liver-transplanted recipients with de novo autoimmune hepatitis is enhanced and induces TH1-like regulatory T cells. J Immunol 196(10):4040–4051. doi:10.4049/jimmunol.1502276
Cao AT, Yao S, Stefka AT, Liu Z, Qin H, Liu H, Evans-Marin HL, Elson CO, Nagler CR, Cong Y (2014) TLR4 regulates IFN-gamma and IL-17 production by both thymic and induced Foxp3+ Tregs during intestinal inflammation. J Leukoc Biol 96(5):895–905. doi:10.1189/jlb.3A0114-056RR
Feng T, Cao AT, Weaver CT, Elson CO, Cong Y (2011) Interleukin-12 converts Foxp3+ regulatory T cells to interferon-gamma-producing Foxp3+ T cells that inhibit colitis. Gastroenterology 140(7):2031–2043. doi:10.1053/j.gastro.2011.03.009
Holmen N, Lundgren A, Lundin S, Bergin AM, Rudin A, Sjovall H, Ohman L (2006) Functional CD4+CD25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflamm Bowel Dis 12(6):447–456
Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM (2010) Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol 184(7):3433–3441. doi:10.4049/jimmunol.0904028
Elkord E (2016) Helios should not be cited as a marker of human thymus-derived Tregs. Commentary: helios(+) and helios(−) cells coexist within the natural FOXP3(+) T regulatory cell subset in humans. Front Immunol 7:276. doi:10.3389/fimmu.2016.00276
Gottschalk RA, Corse E, Allison JP (2012) Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol 188(3):976–980. doi:10.4049/jimmunol.1102964
Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK (2013) Helios+ and Helios− cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol 190(5):2001–2008. doi:10.4049/jimmunol.1201379
Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, Ignatowicz L (2015) Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ regulatory T cells. PLoS One 10(10):e0141161. doi:10.1371/journal.pone.0141161
Kornete M, Mason ES, Girouard J, Lafferty EI, Qureshi S, Piccirillo CA (2015) Th1-Like ICOS+ Foxp3+ Treg cells preferentially express CXCR3 and home to beta-islets during pre-diabetes in BDC2.5 NOD mice. PLoS One 10(5):e0126311. doi:10.1371/journal.pone.0126311
Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno ED, Wagage S, Roos DS, Scott P, Turka LA, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, Hunter CA (2012) The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37(3):511–523. doi:10.1016/j.immuni.2012.06.014
Koenecke C, Lee CW, Thamm K, Fohse L, Schafferus M, Mittrucker HW, Floess S, Huehn J, Ganser A, Forster R, Prinz I (2012) IFN-gamma production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol 189(6):2890–2896. doi:10.4049/jimmunol.1200413
Weinberg AD, Morris NP, Kovacsovics-Bankowski M, Urba WJ, Curti BD (2011) Science gone translational: the OX40 agonist story. Immunol Rev 244(1):218–231. doi:10.1111/j.1600-065X.2011.01069.x
Philips GK, Atkins M (2015) Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol 27(1):39–46. doi:10.1093/intimm/dxu095
Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE (2015) Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA 112(19):6140–6145. doi:10.1073/pnas.1417320112
Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, Curtsinger J, Verneris MR, MacMillan ML, Levine BL, Riley JL, June CH, Le C, Weisdorf DJ, McGlave PB, Blazar BR, Wagner JE (2016) Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood 127(8):1044–1051. doi:10.1182/blood-2015-06-653667
June CH, Blazar BR (2006) Clinical application of expanded CD4+25+ cells. Semin Immunol 18(2):78–88. doi:10.1016/j.smim.2006.01.006
Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7(315):315ra189. doi:10.1126/scitranslmed.aad4134
Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, Owczuk R, Szadkowska A, Witkowski P, Mlynarski W, Jarosz-Chobot P, Bossowski A, Siebert J, Trzonkowski P (2014) Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets—results of one year follow-up. Clin Immunol 153(1):23–30. doi:10.1016/j.clim.2014.03.016
MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, Broady R, Levings MK (2016) Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Investig 126(4):1413–1424. doi:10.1172/JCI82771
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kitz, A., Dominguez-Villar, M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell. Mol. Life Sci. 74, 4059–4075 (2017). https://doi.org/10.1007/s00018-017-2569-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2569-y