
Abstract
CD4+FOXP3+ regulatory T cells (Tregs) are a subset of CD4 T cells that play an essential role in maintaining peripheral immune tolerance, controlling acute and chronic inflammation, allergy, autoimmune diseases, and anti-cancer immune responses. Over the past 20 years, a significant progress has been made since Tregs were first characterized in 1995. Many concepts and principles regarding Tregs generation, phenotypic features, subsets (tTregs, pTregs, iTregs, and iTreg35), tissue specificity (central Tregs, effector Tregs, and tissue resident Tregs), homeostasis (highly dynamic and apoptotic), regulation of Tregs by receptors for PAMPs and DAMPs, Treg plasticity (re-differentiation to other CD4 T helper cell subsets, Th1, Th2, Tfh, and Th17), and epigenetic regulation of Tregs phenotypes and functions have been innovated. In this concise review, we want to briefly analyze these eight new progresses in the study of Tregs. We have also proposed for the first time a novel concept that “physiological Tregs” have been re-shaped into “pathological Tregs” in various pathological environments. Continuing of the improvement in our understanding on this important cellular component about the immune tolerance and immune suppression would lead to the future development of novel therapeutics approaches for acute and chronic inflammatory diseases, allergy, allogeneic transplantation-related immunity, sepsis, autoimmune diseases, and cancers.
Similar content being viewed by others


Introduction
CD4+FOXP3+ regulatory T cells are classified as a subpopulation of CD4 T cells specialized in the suppression of immunopathogenic responses from the host immune system against self or foreign antigens and dangerous substances [1,2]. The suppressive function of Tregs in the maintenance of self-tolerance and prevention of the development of autoimmune and chronic inflammatory diseases is mediated by different mechanisms such as Tregs killing of target cells [3], modulation of target cell signaling via cell-cell contact and/or secretion of anti-inflammatory cytokines such as interleukin-10 (IL-10), IL-35 [4,5], and transforming growth factor β (TGF-β) [4,6] as well as modulation of target cells by exosome-carried microRNAs. Currently, several experimental systems are commercially available that simplify the identification, isolation, and characterization of Tregs using fluorescent-conjugated antibodies for CD4, CD25, FOXP3, CD127, cytotoxic T-lymphocyte associated molecule-4 (CTLA-4), glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR), CD39, and CD45RA [7].
Originally termed suppressor T cells, the recognition of regulatory T cells as a cellular mechanism for immune tolerance resulted from experiments performed in the 1960s and 1970s by Gerson and Kondo, which described the induction of suppressor T cells capable of downregulation of antigen-specific T cell responses [8]. Due to the lack of known molecular markers, research on suppressor T cells ceased. However, in 1995, Sagakuchi et al. identified CD25 as a surface phenotypic marker for suppressive CD4 cells in mice [9]. Since then, suppressive T cells have been called regulatory T cells (Tregs). Since the first characterization of Tregs in 1995 [9,10], one of the major milestones in Tregs studies was the identification of FOXP3. FOXP3 is a member of the forkhead/winged-helix family of transcription factors, which acts as a “master regulator” (for multiple pathways) for the development and suppressive function of Tregs [11-13]. The significance of FOXP3 gene was identified by its mutations that cause fatal autoimmune diseases in early life, which is now termed immunodysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome in mice and humans. Since the discovery of the FOXP3 gene, its role and modification have been one of the potential topics in translational medicine field due to the essential function of FOXP3 in maintaining immune tolerance and homeostasis. Similar to T-bet (a T-box gene encoded transcription factor) [14], GATA3 (a trans-acting T cell-specific transcription factor) [15], and RORγt (a RAR-related nuclear orphan receptor family member) [16] identified as the subset-specific transcription factors for the terminal differentiation of type 1 CD4 T helper cells (Th1), Th2, and Th17, respectively, identification of FOXP3 as a Treg-specific transcription factor was a logic approach for the establishment of Tregs as a terminally differentiated and lineage committed subset of CD4 T cells.
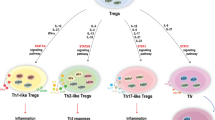
Due to its significant roles in controlling inflammation, innate and adaptive immune responses, and immune tolerance, many unsolved questions remain to be the focus of investigations [17], such as 1) whether Tregs have to be generated in the central lymphoid organ like thymus or Tregs can be “converted” from non-Tregs in peripheral tissues [18,19]. Recently, a special population of Tregs was found in injured muscle, which is distinct from other Treg population and potentiates muscle repair. This report not only revealed a new function of Tregs in modulating tissue repair but also proposed a new pathway for Treg generation [20]; 2) whether there are tissue-resident Tregs, in other words, whether tissue physiological environment modulates Tregs’ genotypes and phenotypes; 3) how do Tregs maintain their own homeostasis. In 2008, we analyzed more than 90 factors involved in regulating homeostasis of Tregs, suggesting that the repertoires of Tregs are undergoing constant adjustment [21]and finally three mechanistic issues were addressed; 4) whether Tregs have functional receptors to sense pathogen-associated molecular patterns (PAMPs) or danger signal-associated molecular patterns (DAMPs) and participate in inflammation [2,22,23]; 5) whether Tregs are a non-changeable lineage committed subset or have differentiation plasticity; and 6) whether epigenetic mechanisms [19] regulate Tregs’ response to changes in physiological tissue environments and pathological conditions such as inflammation. Of note, Treg homeostatic changes have been reported in various diseases [24-66] as shown in Table 1, in which the PMIDs of the papers/reviews are listed. Based on the significant progresses, we have proposed for the first time a novel concept that “physiological Tregs” have been modulated into “pathological Tregs” in various pathological environments. As shown in our novel working model in Figure 1, both physiological and pathological Tregs may undergo changes in differentiation/subset plasticity, homeostasis (survival/death), and proliferation dynamics via molecular mechanisms including pathogen (danger)-associated molecular pattern (PAMPs/DAMPs) receptor signaling, epigenetic mechanisms, microRNAs, and other noncoding RNAs. The issue of how pathological conditions re-shape the subsets of physiological Tregs remains to be poorly addressed. Pathological Tregs can have four different functional status including a) functional suppressors, b) weakened suppressors, c) tumor-enhanced suppressors, and d) malignant Tregs. Improvement of our understanding on these important issues would eventually lead to the future development of Tregs-based novel therapeutics for treating acute and chronic inflammatory diseases, allergy, allogeneic transplantation-related immunity, sepsis, autoimmune diseases, and cancers. For more historical details on the research progress of Tregs, one may refer to others’ excellent reviews as well as ours [2,13,16,18,19,21].

Our novel working model has been proposed: “Pathological conditions re-shape physiological Tregs into pathological Tregs”.
Review
tTregs, pTregs, iTregs, and iTreg35
FOXP3+CD28+Granzyme B+TGFβ-insensitive naturally occurring thymus-generated Tregs (tTregs or nTregs) [67] are characterized by the expression of CD4, and transcriptional factor FOXP3 and high expression of CD25 [68]. Initially identified for their co-expression of CD4 and CD25 cell surface markers, in subsequent reports tTregs have been recognized by other surface markers such as CD103, CD62L, lymphocyte activation gene 3 protein (LAG 3), C-C chemokine receptor type 5 (CCR5), neurophilin-1 [69-71], the activation antigens glucocorticoid-induced tumor necrosis factor receptor (TNFR) family related gene (GITR), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (also known as CD152), as well as the lack of certain cell surface markers such as CD127 (the α chain of the IL-7 receptor) [72].Of note, Helios, a member of Ikaros transcription factor family is proposed to be a marker of tTregs [73], which can be used to distinguish tTregs from iTregs [67]. Fully matured FOXP3+ tTregs exit the thymus and migrate to the secondary lymphoid organs where they suppress the proliferation of tissue-specific autoimmune T cells, restrain their differentiation into type 1 T helper cells (Th1), Th2, and Th17 lineages in vivo [74], and inhibit polyclonal T cell activation and T-effector cell trafficking [67]. They recognize specific self-antigens and prevent autoimmunity by the inhibition of pathogenic lymphocytes. In addition to adaptive immune cells such as T cells and B cells, tTregs also inhibit the function of innate immune cells including antigen-presenting cells such as macrophages and dendritic cells (DCs) by modulating expression of MARCHI and CD83 on macrophages and DCs [67,74].
So far, the modes of how do tTregs suppress effector cells have been identified: inhibition of cytokine production, prevention of cytotoxic T cells proliferation, and inactivation of antigen-presenting cell function. In addition, Tregs contribute to effector T cells apoptosis via three mechanisms including Fas-FasL-mediated killing, suppressive cytokine-induced impairment [6,75,76], and microRNA transmitted in exosomes [77]. As we reviewed [18], the role of tTregs in suppressing chronic inflammation has been clearly demonstrated in experimental atherosclerosis model in 2006 by Ait-Oufellaet et al., which showed an increase in atherosclerotic lesion size and vulnerability in proatherogenic apolipoprotein E deficient (ApoE−/−) mice after peripheral Tregs were depleted [18,78].
FOXP3+CTLA4+IL-10+TGFβ-sensitive adaptive, inducible, or peripheral Tregs (iTregs/aTregs/pTregs) [79,80] are induced by T cell antigen receptor (TCR) ligation (antigen-specific Tregs) [67] and TGFβ stimulation in periphery from CD4+CD25− T cell precursors [21,79], which acquire the upregulation of CD25 (interleukin-2 receptor α chain (IL-2Rα)). Inducible Tregs are developed from naïve CD4 T cells in the lymphoid tissues in response to specific antigens in the presence of cytokines such as transforming growth factor-β1 (TGF-β1), interleukin-10 (IL-10), and IL-4, while in the absence of pro-inflammatory cytokines such as interferon-γ (IFN-γ), IL-1, IL-6, and IL-12. This antigen presentation in the absence of danger signals is referred as tolerogenic, which is essential for the suppression of undesired immune reactivity against non-harmful materials such as airborne particles, commensal bacteria, and foods. In addition, iTregs depend on IL-2 for development and survival as previously reported [21,81-83], which also explains why iTregs highly express CD25 and probably other IL-2 receptor components. Furthermore, iTregs may be able to redirect macrophage differentiation toward an anti-inflammatory cytokine-producing type 2 macrophage phenotype (M2) rather than pro-inflammatory type 1 macrophages (M1 phenotype) [9].
Different subsets of iTregs have been reported including T regulatory cell type 1 (Tr1) and T helper cell type 3 (Th3) [84]. Tr1 cells are CD25−FOXP3−CD49b+LAG-3+T cells characterized by the secretion of large amounts of IL-10, some IL-5 and IFN-γ with or without TGF-β, IL-2, or IL-4 [85,86]. Tr1 cells control the activation of naïve and memory T cells in vivo and in vitro and also suppress the Th1 and Th2 immune responses to pathogens, tumors, and alloantigen-expressed transplanted tissues [87]. The capacity of DCs to induce T cell proliferation is strongly reduced by the supernatant of activated Tr1 [88], suggesting that Tr1 suppression is mediated by secreted cytokines. Th3 cells have been shown to produce high amounts of TGF-β when induced by oral tolerance in mucosal tissue in an antigen-specific manner [74]. In addition, CD4+LAP+ (latency-associated peptide) Tregs have been identified recently as the third iTregs subtype whose suppression is mediated by TGF-β in immune diseases including experimental autoimmune encephalitis (EAE), type I diabetes mellitus (T1DM), systemic lupus erythematosus (SLE), collagen-induced arthritis, type II diabetes, and atherosclerosis in mice [74,89].
Most recently, a new suppressive cytokine IL-35 [6,75,76] has defined a new IL-35-producing iTregs subset, iTreg35 [75], and a subset of IL-35-producing B regulatory cells (Breg) [90,91]. These induced cell subsets are probably responsive to acute and chronic inflammation stimuli since we previously reported that IL-35 is a new category of suppressive cytokine termed responsive cytokine in contrast to the house-keeping suppressive cytokine TGF-β [4].
Tissue-resident Tregs
Similar or parallel to the classification of tTregs, pTregs, iTregs, and iTreg35, it has been proposed that Tregs can also be classified into three new subsets, central Tregs, effector Tregs, and tissue-resident Tregs [17]. Of note, every new classification of cell subsets reflects the improvement of our understanding on the features of the cells. Central Tregs, also termed as resting Tregs or naïve Tregs, make up the majority of Tregs in the circulation and secondary lymphoid organs. Central Tregs have a phenotype of CD62LhighCCR7+ (C-C chemokine receptor type 7) or CD45RAhighCD25low. Effector (memory) Tregs constitute a minor fraction of Tregs in the circulation and secondary lymphoid organs, which have a phenotype of CD62LlowCCR7lowCD44highKLRG1+ (killer cell lectin-like receptor subfamily G member1-positive) CD103+ or CD45RAlowCD25high. Tissue-resident Tregs specify those Tregs that have a long-term residence in non-lymphoid tissues. So far, four types of tissue-resident Tregs have been identified: 1) skin/lung Tregs are specified by two transcription factors FOXP3 and T-bet. The trafficking of skin/lung Tregs are controlled by CXCR3 (receptor for the C-X-C chemokine CXCL9, CXCL10, and CXCL11) and CCR4 (chemokine (C-C motif) receptor 4). The homeostatic mediators for skin/lung Tregs include CD40L, IFN-ɤ, IL-27, and IL-7; 2) gut Tregs are specified by two transcription factors, FOXP3 and Signal transducer and activator of transcription 3 (STAT3). The trafficking of gut Tregs are controlled by CCR6. The homeostatic mediators for gut Tregs include short-chain fatty acid (SCFA), IL-10, IL-6, and IL-1; 3) germinal center Tregs are specified by two transcription factors, FOXP3 and BCL-6. The trafficking of germinal center Tregs are controlled by CXCR5. The homeostatic mediators for germinal center Tregs are still unknown; and 4) adipose tissue Tregs [92] are specified by two transcription factors, FOXP3 and peroxisome proliferator-activated receptor gamma (PPARɤ). The chemokine receptors for trafficking of adipose tissue Tregs are unknown. The homeostatic mediators for adipose tissue Tregs include lipids and long-chain fatty acid (LCFA) [17]. In the near future, we will see more characterizations of tissue-resident Tregs. Identification of tissue-resident Tregs suggests that Tregs express additional transcription factors, chemokine receptors, and receptors for homeostatic mediators in order to respond to the signals of tissue physiological environments.
Highly dynamic and apoptotic populations of Tregs
Our previous reports showed that CD25+/high Tregs are a cell population that are “hungry” for IL-2 for survival and undergo apoptosis easily by upregulating pro-apoptotic protein Bax [81,93] and downregulating anti-apoptotic cytosolic protein translationally controlled tumor protein (TCTP) [82,83,94]. Postulating from our updated understanding on CD25low central Tregs and CD25high effector Tregs, our results suggest that CD25low central Tregs are less likely to undergo acute coronary syndrome- [95], end stage renal disease- [27], and chronic inflammation/infection-related [17], IL-2 decrease-triggered Treg apoptosis [96-98] than CD25high effector Tregs. Based on our results and others’ reports, we proposed that the Tregs apoptotic pathways are novel therapeutic targets for the future development of novel therapy in treating inflammation, autoimmune disease, transplantation-related diseases, allergy, and cancers [21]. Our findings were confirmed by recent excellent reports on high rates of Tregs apoptosis. These reports show that Treg-specific transcription factor FOXP3 is a pro-apoptotic protein [99]; and that mitochondrial anti-apoptosis-regulatory pathway protein Mcl-1 is critical for Tregs survival and niche-filling capacity [100]. However, future work is still needed to determine the apoptotic rates for each subset of Tregs.
High apoptotic rates of Tregs raises the question of how these Tregs maintain their population sizes. Using a special technique to follow up the Thy-1 expressing recent thymic cell emigrants (RTE), investigators found that approximately 30% of CD4+CD25+FOXP3+ Tregs express the markers associated with RTE. Following thymectomy, the numbers of cells expressing these markers fell by 80% within 30 days. In addition, although only ~5% of CD4 single-positive thymocytes express FOXP3 within 24 h after intrathymic injection of fluorescence dye fluorescein isothiocyanate (FITC), more than 30% of the labeled CD4+RTE are FOXP3+, suggesting that some RTE may acquire FOXP3 expression in the periphery. Thus, some RTE may acquire FOXP3 rapidly after emigration from thymus. Tregs are dividing rapidly with apparent half-lives of ~18 days and ~7 days for the CD4+CD25+FOXP3+ and CD4+CD25−FOXP3+ subsets, respectively. The apparent slower turnover of CD4+CD25+FOXP3+ cells is a result of CD4+CD25+FOXP3+ to CD4+CD25−FOXP3+ conversion, with no loss of regulatory function. Therefore, the data suggested that Tregs in adults are relatively short-lived and Tregs numbers are maintained by rapid cell division and continuous replenishment from the thymus [101].
Pathogen-associated molecular pattern receptors on Tregs
After a long time of extensive research, it has been widely accepted that the host innate immune system is equipped with a set of receptors to recognize PAMPs derived from viruses, bacteria, other invasive microorganisms and environmental stimuli or metabolite-related DAMPs [2,22,23]. So far, four types of receptors for PAMPs and DAMPs have been identified; Toll-like receptors (TLRs, 13 members), nucleotide-binding oligomerization domain (NOD) leucine-rich-repeat containing receptors (NLRs, 18 members), C-type lectin receptors (5 members), and retinoic acid-inducible gene I protein (RIG-I) helicase receptors (2 members) [102]. An important question remains whether Tregs are equipped with these sets of receptors for PAMPs and DAMPs. It was reported that stimulation of human Tregs with a mixture of TLR2 ligands Pam2CSK4, Pam3CSK4, and FSL-1 can result in a reversal of suppression on anti-CD3/anti-CD28 stimulated responder T cells. However, the gastric mucosa of the infected TLR2 knock-out mice showed a lower mRNA expression of FOXP3, IL-10, and IL-17A, but a higher expression of IFN-γ compared to the gastric mRNA expression in infected wild-type mice [103]. In addition, TLR5 and TLR8 also modulate the suppressive activity of naturally occurring CD4+CD25high Tregs. The suppressive capacity of Tregs is counter-regulated by TLR ligands indirectly via antigen-presenting cells (APCs) or responder T cells or directly [104]. Similarly, TLR4 stimulation of T effector cells and Tregs co-culture leads to a more pronounced T effector cell activation [105]. TLR ligand lipopolysaccharide (LPS) stimulation of TLR4 on Tregs, similar to T cell antigen receptor ligation, can enhance Tregs suppression on neutrophils [106]. It is obvious that this field is still in the early stage. Further studies are needed to determine the roles of the receptors for PAMPs and DAMPs in regulating Tregs homeostasis and suppressive function.
Tregs plasticity and FOXP3 expression regulated by other transcription factors
Current understandings suggest that naïve CD4 T helper cells can differentiate/polarize into at least six different terminally differentiated/lineage-committed subsets. 1) Naïve CD4 T helper cells can differentiate into type 1 T helper cell subset (Th1) with the expression of Th1-specific transcription factor T-bet in the presence of IL-12 and IFN-γ. 2) Naïve CD4 T cells can differentiate into Th2 subset with the expression of Th2-specific transcription factor GATA3 in the presence of IL-4. 3) Naïve CD4 T cells can differentiate into Th17 subset with the expression of Th17-specific transcription factor RORγt in the presence of IL-6 and TGF-β. 4) Naïve CD4 T cells can differentiate into iTregs subset with the expression of Treg-specific transcription factor FOXP3 in the presence of TGF-β, retinoic acid, and IL-2. 5) Naïve CD4 T cells can also differentiate into follicular T helper cells (Tfh) subset in the presence of IL-21. Naïve CD4+ T cells and Th2 can differentiate into Th9 cells in the presence of TGF-β [107,108].
Among all these CD4 subsets, Th1 and Th2 are relatively stable, but iTregs and Th17 cells can readily switch to other T helper subsets under certain cytokine stimulations. In the presence of B cells and CD40-CD40L interaction, iTregs can switch to follicular T helper (Tfh) cells mediated by transcription factor B-cell lymphoma/leukemia 6 (BCL-6) [109]. In addition, iTregs can also switch to IL-17-producing cells (Th17) mediated by transcription factor STAT3 [109] after stimulation with IL-6 and IL-21. Moreover, iTregs can also convert to Th2 cells mediated by transcription factor Irf4 [109]. Furthermore, iTregs can switch to Th1 cells mediated by T-bet [109]. Finally, Th17 can convert into IFN-γ-producing Th1 cells or IL-4-producing Th2 cells when stimulated by IL-12 or IL-4, respectively. Th2 cells convert to IL-9-producing cells in response to TGF-β stimulation [108]. This conversion of terminally differentiated lineage committed subset to other terminally differentiated lineage committed subsets is termed “plasticity”. Several factors participate in the regulation of T cell conversion including extrinsic and intrinsic ones. The extrinsic factors include accessory immune cells, innate receptors for PAMPs and DAMPs, cytokine microenvironment including the cytokines in polarizing other T helper subsets, cytokine receptor regulation, nutrient availability, and metabolic pathways including long-chain fatty acids and short-chain fatty acids for adipose tissue Tregs. The intrinsic factors are cell cycle and phenotype stability, microRNA mediated control of T cell phenotype, transcription factor dosing and dominance, and epigenetic modifications [109].
In fact, numerous transcription factors have been identified in reshaping Tregs development, function, and homeostasis. These include the first group of transcription factors functional in regulating FOXP3 expression and the second group of transcription factors in forming a complex with FOXP3. Some of the factors are overlapped in these two groups. The first group includes nuclear factor of activated T cells (NFAT), c-Rel (nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB)), activator protein 1 (AP-1), Nr4a (a subfamily of the orphan nuclear receptors), and signal transducer and activator of transcription 5 (STAT5), and FOXP3. In addition to the regulation of FOXP3 gene expression, recent reports indicate that FOXP3 itself is able to form complexes with a number of co-factors to execute cooperative effects during their interaction [110]. The second group of FOXP3 co-factors is composed of as many as 11 sequence specific transcription factors including NFATc2, runt-related transcription factor 1 (RUNX1; also known as acute myeloid leukemia 1 protein (AML1) or core-binding factor subunit alpha-2 (CBFA2)), B-cell lymphoma/leukemia 11B (BCL11b), Foxp1, Foxp4, GATA-3, STAT3, Ikaros (Ikzf1; a lymphoid transcription factor LyF-1), Aiolos (Ikzf3; the Ikaros, Aiolos and Helios are three family members of the hematopoietic specific transcription factors involved in the regulation of lymphocyte development), Ets (E26 transformation-specific family of transcription factors), and Cnot3 (CCR4-NOT transcription complex, subunit 3). The majority of FOXP3 binding sites within the genome lack an identifiable forkhead-binding motif in Tregs, which suggests that a large number of FOXP3 co-factors facilitate the binding of FOXP3 to a given site [111-113]. Since some of these transcription factors, if not all, are also inflammation-regulatory transcription factors [114], it suggests that the inflammation and pathological conditions can regulate the plasticity of Tregs and other T cells. In support of this argument, the expression of Treg-specific transcription factor FOXP3 requires demethylation of FOXP3 promoter [115] whereas proatherogenic stimuli like oxidized low-density lipoprotein (oxLDL) induces increased methylation of FOXP3 promoter and decreased FOXP3 expression [116].
Epigenetic regulation and microRNA regulation of Tregs
Under inflammatory stimulations, the suppressive function of Tregs is decreased and TGF-β-induced Tregs development is attenuated by an epigenetic manner [117]. It suggests that epigenetic regulation of Tregs function, development, and homeostasis is patho-physiologically relevant. In correlation with this finding, Tregs suppression is also reported to be attenuated in autoimmune type 1 diabetes, in which epigenetics is one of the pathological mechanisms involved [118]. What is epigenetics? Epigenetics refers to heritable changes that occur in gene expression without modification in the DNA sequence of the genome. These epigenetics mechanisms, which briefly include DNA methylation/demethylation, histones modifications, and micro-RNAs (miRNAs) are the principal mechanisms involved in regulating chromosomal organization, chromosomal remodeling and then gene expression via different dynamic levels. More specifically, it has been demonstrated that epigenetics mechanisms play a critical role in regulating FOXP3 expression and lead to further regulations in Tregs functions and homeostasis [119-121]. Emerging epigenetics therapies are providing new therapeutic agents for the control of various diseases [13,16,18,21,81-83,115,122].
The term epigenetics was first introduced by Conrad Waddington in 1942 [123]. Epigenetics [123] integrates organism genotypes by the influence and response of environmental stimuli on their phenotype, which can take place in chromosomal DNA or in the proteins linked with the chromosomal DNA such as histones. In recent years, many epigenetic proteins have been investigated in laboratories and in clinic, while inhibitor development for modification enzymes is the frontier for drug discovery. So far, epigenetic modifications have been grouped into four main categories: DNA methylation, histone modification including histone methylation/demethylation, histone acetylation/deacetylation, histone phosphorylation, histone SUMOylation [124], small and long noncoding RNAs, as well as chromatin remodeling [124,125].
Noncoding RNAs (ncRNAs) are a type of functional RNA molecule, which are not translated into proteins. More functional groups of ncRNAs have been categorized by the following: four short noncoding RNAs (17–31 base pairs (bp)) (microRNAs (miRNAs), Piwi-interacting RNAs (piRNAs), small interfering RNAs (siRNAs), and transcription initiation RNAs), mid-size noncoding RNAs (<200 bp) (small nucleolar RNAs, promoter-associated small RNAs (PASRs), TSS-associated RNAs (TSSa-RNAs), and promoter upstream transcripts (PROMPTs)), long noncoding RNAs (lncRNAs, >200 bp), and its subgroups such as long intergenic noncoding RNAs (lincRNAs), enhancer RNAs (eRNAs), transcribed ultraconserved regions (T-UCRs) and other lncRNAs [126]. It has been widely shown that ncRNAs not only regulate gene expression at the transcriptional and post-transcriptional levels but also play a role in the control of epigenetic pathways [127]. Of note, we recently published a comprehensive review on epigenetics enzymes as new therapeutic targets for Treg-based therapy, which one may refer to for the details [19].
Conclusions
The roles of Tregs in inhibiting inflammation, autoimmune diseases, allergy, transplantation, and modulating anti-cancer immune responses have been well established. Due to extensive studies, several new concepts and principles regarding Tregs in physiological and pathological status have emerged, which are concisely discussed in this brief review. Continuation in improving our understanding on the molecular mechanisms underlying Tregs development, homeostasis, and functions will eventually lead to the future development of novel Tregs-based therapeutics to treat inflammation, chronic cardiovascular diseases, allergy, allogeneic transplantation-related immune responses, sepsis, autoimmune diseases, and cancers.
References
Sakaguchi S, Setoguchi R, Yagi H, Nomura T. Naturally arising Foxp3-expressing CD25 + CD4+ regulatory T cells in self-tolerance and autoimmune disease. Curr Top Microbiol Immunol. 2006;305:51–66. Epub 2006/05/27.
Yang XF, Yin Y, Wang H. Vascular inflammation and atherogenesis are activated via receptors for pamps and suppressed by regulatory t cells. Drug Discov Today Ther Strat. 2008;5(2):125–42. Epub 2008/01/01.
Askenasy N. Enhanced killing activity of regulatory T cells ameliorates inflammation and autoimmunity. Autoimmun Rev. 2013;12(10):972–5. Epub 2013/05/21.
Li X, Mai J, Virtue A, Yin Y, Gong R, Sha X, et al. IL-35 is a novel responsive anti-inflammatory cytokine–a new system of categorizing anti-inflammatory cytokines. PLoS One. 2012;7(3):e33628. Epub 2012/03/23.
Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–8. Epub 2012/07/21.
Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, et al. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. 2012;13(3):290–9. Epub 2012/02/07.
Simonetta F, Chiali A, Cordier C, Urrutia A, Girault I, Bloquet S, et al. Increased CD127 expression on activated FOXP3 + CD4+ regulatory T cells. Eur J Immunol. 2010;40(9):2528–38. Epub 2010/08/07.
Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18(5):723–37. Epub 1970/05/01.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64.
Sakaguchi S. Naturally arising Foxp3-expressing CD25 + CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6(4):345–52.
Katoh H, Zheng P, Liu Y. FOXP3: genetic and epigenetic implications for autoimmunity. J Autoimmun. 2013;41:72–8. Epub 2013/01/15.
Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182(1):259–73. Epub 2008/12/26.
Yang XF, Fang P, Meng S, Jan M, Xiong X, Yin Y, et al. The FOX transcription factors regulate vascular pathology, diabetes and Tregs. Front Biosci (Schol Ed). 2009;1:420–36. Epub 2009/06/02.
Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13(11):777–89. Epub 2013/10/12.
Kanhere A, Hertweck A, Bhatia U, Gokmen MR, Perucha E, Jackson I, et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun. 2012;3:1268. Epub 2012/12/13.
Mai J, Wang H, Yang XF. Th 17 cells interplay with Foxp3+ Tregs in regulation of inflammation and autoimmunity. Front Biosci (Landmark Ed). 2010;15:986–1006. Epub 2010/06/03.
Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–65. Epub 2014/02/01.
Pastrana JL, Sha X, Virtue A, Mai J, Cueto R, Lee IA, et al. Regulatory T cells and atherosclerosis. J Clin Exp Cardiol. 2012;2012 Suppl 12:2. Epub 2013/09/03.
Pastrana JL, Shao Y, Chernaya V, Wang H, Yang XF. Epigenetic enzymes are the therapeutic targets for CD4CD25Foxp3 regulatory T cells. Transl Res. 2014. Epub 2014/09/07.
Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–95. Epub 2013/12/10.
Yang XF. Factors regulating apoptosis and homeostasis of CD4 + CD25highFOXP3+ regulatory T cells are new therapeutic targets. Front Biosci. 2008;13:1472–99.
Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, et al. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol. 2009;22(2):311–22. Epub 2009/06/10.
Yin Y, Pastrana JL, Li X, Huang X, Mallilankaraman K, Choi ET, et al. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Front Biosci. 2013;18:638–49. Epub 2013/01/02.
Ammirati E, Cianflone D, Banfi M, Vecchio V, Palini A, De Metrio M, et al. Circulating CD4 + CD25hiCD127lo regulatory T-Cell levels do not reflect the extent or severity of carotid and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(9):1832–41. Epub 2010/06/12.
Cheng X, Yu X, Ding YJ, Fu QQ, Xie JJ, Tang TT, et al. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol. 2008;127(1):89–97. Epub 2008/02/26.
Han SF, Liu P, Zhang W, Bu L, Shen M, Li H, et al. The opposite-direction modulation of CD4 + CD25+ Tregs and T helper 1 cells in acute coronary syndromes. Clin Immunol. 2007;124(1):90–7. Epub 2007/05/22.
Meier P. FOXP3+ regulatory T-cells in chronic kidney disease: molecular pathways and clinical implications. Adv Exp Med Biol. 2009;665:163–70. Epub 2009/01/01.
Zeng C, Shi X, Zhang B, Liu H, Zhang L, Ding W, et al. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med (Berl). 2012;90(2):175–86. Epub 2011/10/04.
Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. 2011;186(2):1162–72. Epub 2010/12/21.
Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. 2011;60(11):2954–62. Epub 2011/09/14.
Wagner NM, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S, et al. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity (Silver Spring, Md. 2013;21(3):461-8. Epub 2013/04/18.
Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS One. 2011;6(1):e16376. Epub 2011/02/08.
Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, et al. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J Exp Med. 2004;199(11):1567–75. Epub 2004/06/03.
Ling EM, Smith T, Nguyen XD, Pridgeon C, Dallman M, Arbery J, et al. Relation of CD4 + CD25+ regulatory T-cell suppression of allergen-driven T-cell activation to atopic status and expression of allergic disease. Lancet. 2004;363(9409):608–15. Epub 2004/02/28.
Bellinghausen I, Klostermann B, Knop J, Saloga J. Human CD4 + CD25+ T cells derived from the majority of atopic donors are able to suppress TH1 and TH2 cytokine production. J Allergy Clin Immunol. 2003;111(4):862–8. Epub 2003/04/22.
Crispin JC, Martinez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2003;21(3):273–6. Epub 2003/11/06.
Alvarado-Sanchez B, Hernandez-Castro B, Portales-Perez D, Baranda L, Layseca-Espinosa E, Abud-Mendoza C, et al. Regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2006;27(2):110–8. Epub 2006/08/08.
Barath S, Aleksza M, Tarr T, Sipka S, Szegedi G, Kiss E. Measurement of natural (CD4 + CD25high) and inducible (CD4 + IL-10+) regulatory T cells in patients with systemic lupus erythematosus. Lupus. 2007;16(7):489–96. Epub 2007/08/03.
Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol. 2005;140(2):360–7. Epub 2005/04/06.
Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmstrom V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther. 2004;6(4):R335–46. Epub 2004/07/01.
Lawson CA, Brown AK, Bejarano V, Douglas SH, Burgoyne CH, Greenstein AS, et al. Early rheumatoid arthritis is associated with a deficit in the CD4 + CD25high regulatory T cell population in peripheral blood. Rheumatology (Oxford). 2006;45(10):1210–7. Epub 2006/03/31.
Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200(3):277–85. Epub 2004/07/29.
de Kleer IM, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, et al. CD4 + CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol. 2004;172(10):6435–43.
Monneret G, Debard AL, Venet F, Bohe J, Hequet O, Bienvenu J, et al. Marked elevation of human circulating CD4 + CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med. 2003;31(7):2068–71. Epub 2003/07/09.
Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Increased percentage of CD4 + CD25+ regulatory T cells during septic shock is due to the decrease of CD4 + CD25- lymphocytes. Crit Care Med. 2004;32(11):2329–31.
Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F, et al. Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. 2009;35(4):678–86. Epub 2008/10/24.
Oberholzer C, Oberholzer A, Clare-Salzler M, Moldawer LL. Apoptosis in sepsis: a new target for therapeutic exploration. Faseb J. 2001;15(6):879–92. Epub 2001/04/09.
Wesche DE, Lomas-Neira JL, Perl M, Chung CS, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. 2005;78(2):325–37. Epub 2005/04/09.
Ni Choileain N, MacConmara M, Zang Y, Murphy TJ, Mannick JA, Lederer JA. Enhanced regulatory T cell activity is an element of the host response to injury. J Immunol. 2006;176(1):225–36.
D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4 + CD25 + Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119(10):2898–913. Epub 2009/09/23.
Chauhan SK, Saban DR, Lee HK, Dana R. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol. 2009;182(1):148–53. Epub 2008/12/26.
Taylor PA, Noelle RJ, Blazar BR. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J Exp Med. 2001;193(11):1311–8. Epub 2001/06/08.
Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25 + CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. J Immunol. 2004;172(2):923–8. Epub 2004/01/07.
Zhang ZX, Yang L, Young KJ, DuTemple B, Zhang L. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat Med. 2000;6(7):782–9. Epub 2000/07/11.
Bestard O, Cruzado JM, Rama I, Torras J, Goma M, Seron D, et al. Presence of FoxP3+ regulatory T Cells predicts outcome of subclinical rejection of renal allografts. J Am Soc Nephrol. 2008;19(10):2020–6. Epub 2008/05/23.
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4 + CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167(3):1245–53.
Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169(5):2756–61. Epub 2002/08/24.
Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003;9(12):4404–8. Epub 2003/10/14.
Kawaida H, Kono K, Takahashi A, Sugai H, Mimura K, Miyagawa N, et al. Distribution of CD4 + CD25high regulatory T-cells in tumor-draining lymph nodes in patients with gastric cancer. J Surg Res. 2005;124(1):151–7.
Kono K, Kawaida H, Takahashi A, Sugai H, Mimura K, Miyagawa N, et al. CD4(+)CD25high regulatory T cells increase with tumor stage in patients with gastric and esophageal cancers. Cancer Immunol Immunother. 2006;55(9):1064–71. Epub 2005/12/06.
Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4 + CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer. 2003;98(5):1089–99.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9.
Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, et al. Reduced frequencies and suppressive function of CD4 + CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106(6):2018–25.
Yang ZZ, Novak AJ, Stenson MJ, Witzig TE, Ansell SM. Intratumoral CD4 + CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood. 2006;107(9):3639–46.
Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Attenuation of CD8(+) T-cell function by CD4(+)CD25(+) regulatory T cells in B-cell non-Hodgkin's lymphoma. Cancer Res. 2006;66(20):10145–52.
Wang X, Zheng J, Liu J, Yao J, He Y, Li X, et al. Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur J Haematol. 2005;75(6):468–76.
Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. 2014;259(1):88–102. Epub 2014/04/10.
Stephens GL, Shevach EM. Foxp3+ regulatory T cells: selfishness under scrutiny. Immunity. 2007;27(3):417–9. Epub 2007/09/26.
Tordjman R, Lepelletier Y, Lemarchandel V, Cambot M, Gaulard P, Hermine O, et al. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat Immunol. 2002;3(5):477–82. Epub 2002/04/16.
Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209(10):1713–22. 10. Epub 2012/09/12.
Weiss JM, Bilate AM, Gobert M, Ding Y, de Lafaille MA C, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209(10):1723–42. 10. Epub 2012/09/12.
Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62.
Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184(7):3433–41. Epub 2010/02/26.
Sasaki N, Yamashita T, Takeda M, Hirata K. Regulatory T cells in atherogenesis. Journal of atherosclerosis and thrombosis. 2012;19(6):503-15. Epub 2012/04/14.
Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010;11(12):1093–101. Epub 2010/10/19.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450(7169):566–9. Epub 2007/11/23.
Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, et al. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity. 2014;41(1):89–103. Epub 2014/07/19.
Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12(2):178–80. Epub 2006/02/08.
de Lafaille MA C, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30(5):626–35. Epub 2009/05/26.
Okada M, Hibino S, Someya K, Yoshmura A. Regulation of regulatory T cells: epigenetics and plasticity. Adv Immunol. 2014;124:249–73. Epub 2014/09/02.
Xiong Z, Song J, Yan Y, Huang Y, Cowan A, Wang H, et al. Higher expression of Bax in regulatory T cells increases vascular inflammation. Front Biosci. 2008;13:7143–55. Epub 2008/05/30.
Xiong Z, Yan Y, Song J, Fang P, Yin Y, Yang Y, et al. Expression of TCTP antisense in CD25(high) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis. 2009;203(2):401–8. Epub 2008/09/16.
Yan Y, Xiong Z, Zhang S, Song J, Huang Y, Thornton AM, et al. CD25high T cells with a prolonged survival inhibit development of diabetes. Int J Immunopathol Pharmacol. 2008;21(4):767–80.
Jonuleit H, Schmitt E. The regulatory T cell family: distinct subsets and their interrelations. J Immunol. 2003;171(12):6323–7. Epub 2003/12/10.
Hippen KL, Riley JL, June CH, Blazar BR. Clinical perspectives for regulatory T cells in transplantation tolerance. Semin Immunol. 2011;23(6):462–8. Epub 2011/08/09.
Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. 2013;19(6):739–46. Epub 2013/04/30.
Groux H. Type 1 T-regulatory cells: their role in the control of immune responses. Transplantation. 2003;75(9 Suppl):8S–12S. Epub 2003/06/24.
Roncarolo MG, Levings MK, Traversari C. Differentiation of T regulatory cells by immature dendritic cells. J Exp Med. 2001;193(2):F5–9. Epub 2001/02/24.
Chen Y, Chen Q, Lu G, Fan Z, Zhong S. The autocrine regulatory effect of vasoactive intestinal peptide on the growth of human pancreatic carcinoma cells. Chin Med Sci J. 1994;9(4):215–9.
Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20(6):633–41. Epub 2014/04/20.
Shen P, Roch T, Lampropoulou V, O'Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366–70. Epub 2014/02/28.
Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013;17(6):851–9. Epub 2013/06/12.
Yang XF, Weber GF, Cantor H. A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity. 1997;7(5):629–39.
Yang Y, Yang F, Xiong Z, Yan Y, Wang X, Nishino M, et al. An N-terminal region of translationally controlled tumor protein is required for its antiapoptotic activity. Oncogene. 2005;24(30):4778–88.
Mor A, Luboshits G, Planer D, Keren G, George J. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur Heart J. 2006;27(21):2530–7. Epub 2006/09/07.
Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol. 2004;4(9):665–74.
Dietrich T, Hucko T, Schneemann C, Neumann M, Menrad A, Willuda J, et al. Local delivery of IL-2 reduces atherosclerosis via expansion of regulatory T cells. Atherosclerosis. 2012;220(2):329–36. Epub 2011/11/09.
Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea 3rd EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055–66. Epub 2011/12/02.
Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, et al. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity. 2013;38(6):1116–28. Epub 2013/06/12.
Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14(9):959–65. Epub 2013/07/16.
Mabarrack NH, Turner NL, Mayrhofer G. Recent thymic origin, differentiation, and turnover of regulatory T cells. J Leukoc Biol. 2008;84(5):1287–97. Epub 2008/08/07.
Netea MG, van der Meer JW. Immunodeficiency and genetic defects of pattern-recognition receptors. N Engl J Med. 2011;364(1):60–70. Epub 2011/01/07.
Sun X, Zhang M, El-Zataari M, Owyang SY, Eaton KA, Liu M, et al. TLR2 mediates Helicobacter pylori-induced tolerogenic immune response in mice. PLoS One. 2013;8(9):e74595. Epub 2013/09/24.
Oberg HH, Juricke M, Kabelitz D, Wesch D. Regulation of T cell activation by TLR ligands. Eur J Cell Biol. 2011;90(6-7):582–92. Epub 2011/02/05.
Cordiglieri C, Marolda R, Franzi S, Cappelletti C, Giardina C, Motta T, et al. Innate immunity in myasthenia gravis thymus: pathogenic effects of Toll-like receptor 4 signaling on autoimmunity. J Autoimmun. 2014;52:74–89. Epub 2014/01/09.
Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. Lipopolysaccharide-activated CD4 + CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol. 2006;177(10):7155–63.
Luckheeram RV, Zhou R, Verma AD, Xia B. CD4(+)T cells: differentiation and functions. Clin Dev Immunol. 2012;2012:925135. Epub 2012/04/05.
Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30(5):646–55. Epub 2009/05/26.
Coomes SM, Pelly VS, Wilson MS. Plasticity within the alphabeta(+)CD4(+) T-cell lineage: when, how and what for? Open Biol. 2013;3(1):120157. Epub 2013/01/25.
Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. 2012;13(10):1010–9. Epub 2012/08/28.
Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, Macisaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445(7130):931–5.
Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445(7130):936–40.
Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151(1):153–66. Epub 2012/10/02.
Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9(10):692–703. Epub 2009/10/28.
Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. 2009;114(18):3727–35. Epub 2009/07/31.
Jia L, Zhu L, Wang JZ, Wang XJ, Chen JZ, Song L, et al. Methylation of FOXP3 in regulatory T cells is related to the severity of coronary artery disease. Atherosclerosis. 2013;228(2):346–52. Epub 2013/04/10.
Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36(5):717–30. Epub 2012/05/15.
Jeker LT, Bour-Jordan H, Bluestone JA. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harbor Perspect Med. 2012;2(3):a007807. Epub 2012/03/07.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. Epub 2003/01/11.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4 + CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6. Epub 2003/03/04.
Vahedi G, CP A, Hand TW, Laurence A, Kanno Y, O'Shea JJ, et al. Helper T-cell identity and evolution of differential transcriptomes and epigenomes. Immunol Rev. 2013;252(1):24–40. Epub 2013/02/15.
Yang XF, Fang P, Meng S, Jan M, Xiong X, Yin Y, et al. The forkhead transcription factors play important roles in vascular pathology and immunology. Adv Exp Med Biol. 2009;665:90–105. Epub 2009/01/01.
Dvir A, Conaway JW, Conaway RC. Mechanism of transcription initiation and promoter escape by RNA polymerase II. Curr Opin Genet Dev. 2001;11(2):209–14. Epub 2001/03/16.
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. Epub 2012/04/14.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68. Epub 2010/10/15.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74. Epub 2011/11/19.
Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010;24(20):2264–9. Epub 2010/10/19.
Acknowledgements
This work is partially supported by NIH grants to Drs. XF. Yang and H. Wang.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WY and YS carried out the primary literature search and drafted the manuscript. JL and JM provided material input, and helped revising the manuscript. HW and XFY conceived the study, provided field expertise, and finalized the manuscript. All authors read and approved the final manuscript.
Authors’ information
WY is a Research Assistant. YS, MS, is an Assistant Scientist. JL, MD, is a Postdoc Fellow. JM, PHD, is a Postdoc Fellow. HW, MD, PhD, EMBA, FAHA, is a Professor. XFY, MD, PhD, EMBA, FAHA, is a Professor.
William Y Yang and Ying Shao contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Yang, W.Y., Shao, Y., Lopez-Pastrana, J. et al. Pathological conditions re-shape physiological Tregs into pathological Tregs. Burn Trauma 3, 1 (2015). https://doi.org/10.1186/s41038-015-0001-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41038-015-0001-0