
Abstract
Roseibacterium elongatum Suzuki et al. 2006 is a pink-pigmented and bacteriochlorophyll a-producing representative of the Roseobacter group within the alphaproteobacterial family Rhodobacteraceae. Representatives of the marine ‘Roseobacter group’ were found to be abundant in the ocean and play an important role in global and biogeochemical processes. In the present study we describe the features of R. elongatum strain OCh 323T together with its genome sequence and annotation. The 3,555,102 bp long genome consists of one circular chromosome with no extrachromosomal elements and is one of the smallest known Roseobacter genomes. It contains 3,540 protein-coding genes and 59 RNA genes. Genome analysis revealed the presence of a photosynthetic gene cluster, which putatively enables a photoheterotrophic lifestyle. Gene sequences associated with quorum sensing, motility, surface attachment, and thiosulfate and carbon monoxide oxidation could be detected. The genome was sequenced as part of the activities of the Transregional Collaborative Research Centre 51 (TRR51) funded by the German Research Foundation (DFG).
Similar content being viewed by others


Introduction
Strain OCh 323T (= DSM 19469T = CIP 107377T = JCM 11220T) is the type strain of Roseibacterium elongatum in the bispecific genus Roseibacterium [1] with R. beibuensis [2] being the second species in the genus. The genus Roseibacterium belongs to the marine Roseobacter group, which was shown to be ubiquitious in the oceans of the world, especially in coastal and polar oceans [3,4]. The strain was isolated from sand located at Monkey Mia, Shark Bay, at the west coast of Australia [1]. The genus Roseibacterium was named after the Latin adjective roseus (‘rose, pink’) and the Greek adjective bakterion (‘rod’); Roseibacterium (‘pink, rod-shaped bacterium’). The species epithet elongatum refers to the Latin adjective elongatum (‘elongated, stretched out’) [1]. Current PubMed records do not indicate any follow-up research with strain OCh 323T after the initial description of R. elongatum [1].
In this study we analyzed the genome sequence of R. elongatum DSM 19469T. We present a description of the genome sequencing and annotation and a summary classification together with a set of features for strain DSM 19469T, including novel aspects of its phenotype and Features of the organism.
Classification and features
16S rRNA gene analysis
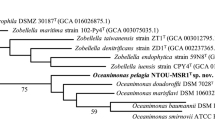
Figure 1 shows the phylogenetic neighborhood of R. elongatum DSM 19469T in a 16S rRNA gene based tree. The sequences of the two identical 16S rRNA gene copies in the genome do not differ from the previously published 16S rRNA gene sequence (AB601471).

Phylogenetic tree highlighting the position of R. elongatum relative to the type strains of the type species of the other genera within the family Rhodobacteraceae. The tree was inferred from 1,331 aligned characters of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion as previously described [5]. Rooting was done initially using the midpoint method [6] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 600 ML bootstrap replicates (left) and from 1,000 maximum-parsimony bootstrap replicates (right) if larger than 60% [5]. Lineages with type strain genome sequencing projects registered in GOLD [7] are labeled with one asterisk, those also listed as ‘Complete and Published’ with two asterisks [8–11].
A representative genomic 16S rRNA gene sequence of R. elongatum DSM 19469T was compared with the Greengenes database [22] for determining the weighted relative frequencies of taxa and (truncated [23]) keywords as previously described [5]. The most frequently occurring genera were Rhodovulum (35.1%), Jannaschia (13.5%), Dinoroseobacter (10.6%), Rhodobacter (9.6%) and Roseobacter (8.5%) (89 hits in total). Regarding the two hits to sequences from members of the species, the average identity within HSPs was 100.0%, whereas the average coverage by HSPs was 99.7%. Among all other species, the one yielding the highest score was Dinoroseobacter shibae (NC_009952), which corresponded to an identity of 95.7% and a HSP coverage of 100.1%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification). The highest-scoring environmental sequence was AF513932 (Greengenes short name ‘Rhodobacter group clone LA4-B3’), which showed an identity of 99.4% and a HSP coverage of 99.9%. The most frequently occurring keywords within the labels of all environmental samples that yielded hits were ‘microbi’ (4.3%), ‘mat’ (2.3%), ‘sea’ (2.0%), ‘marin’ (2.0%) and ‘coral’ (1.9%) (157 hits in total). The most frequently occurring keywords within the labels of those environmental samples that yielded hits of a higher score than the highest scoring species were ‘group, rhodobact’ (33.8%) and ‘rhodobacteracea’ (32.4%) (2 hits in total). These keywords fit well to the known ecology (and phylogenetic relationships) of R. elongatum DSM 19469T.
Morphology and physiology
Cells of strain OCh 323T are Gram-negative, non-motile and rod-shaped, 1.6–10.0 µm in length and 0.5–0.8 µm in width (Figure 2). Colonies are circular, smooth, convex and glistening, opaque and pink-pigmented. Optimum growth occurs at a temperature of 27–30°C and a pH of 7.5–8.0. Cells can grow in the presence of 0.5–7.5% NaCl but do not grow in the absence of NaCl. Cells are positive for urease activity but do not show nitrate reductase or phosphate activities. They are negative in the Voges-Prosgauer test but the ONPG reaction is positive. Cells do not produce indole or H2S. Gelatin is hydrolyzed, but alginate, starch and Tween80 are not. Cells do not utilize acetate, citrate, D-glucose, DL-malate, ethanol, pyruvate, succinate. Acid is not produced from D-fructose, D-glucose or lactose (all data from [1]).

Micrograph of R. elongatum DSM 19469T.
In this study the utilization of carbon compounds by R. elongatum DSM 19469T grown at 28°C was also determined using Generation-III microplates in an OmniLog phenotyping device (BIOLOG Inc., Hayward, CA, USA). The microplates were inoculated with a cell suspension at a cell density of 95–96% turbidity and dye IF-A. Further additives were vitamin, micronutrient and sea-salt solutions, which had to be added for dealing with such marine bacteria [24]. The plates were sealed with parafilm to avoid a loss of fluid. The measurement data were exported and further analyzed with the opm package for R [7,25], using its functionality for statistically estimating parameters from the respiration curves such as the maximum height, and automatically translating these values into negative, ambiguous, and positive reactions.
The following substrates were utilized in the Generation-III plates: positive control, pH 6, 1% NaCl, 4% NaCl, D-galactose, D-fucose, L-fucose, L-rhamnose, 1% sodium lactate, D-arabitol, myo-inositol, rifamycin SV, L-aspartic acid, L-glutamic acid, L-histidine, L-serine, D-glucuronic acid, quinic acid, L-lactic acid, citric acid, α-keto-glutaric acid, D-malic acid, L-malic acid, nalidixic acid and sodium formate.
According to Generation-III plates the strain is negative for dextrin, D-maltose, D-trehalose, D-cellobiose, β-gentiobiose, sucrose, D-turanose, stachyose, pH 5, D-raffinose, α-D-lactose, D-melibiose, β-methyl-D-galactoside, D-salicin, N-acetyl-D-glucosamine, N-acetyl-β-D-mannosamine, N-acetyl-D-galactosamine, N-acetyl-neuraminic acid, 8% NaCl, D-glucose, D-mannose, D-fructose, 3-O-methyl-D-glucose, inosine, fusidic acid, D-serine, D-sorbitol, D-mannitol, glycerol, D-glucose-6-phosphate, D-fructose-6-phosphate, D-aspartic acid, D-serine, troleandomycin, minocycline, gelatin, glycyl-L-proline, L-alanine, L-arginine, L-pyroglutamic acid, lincomycin, guanidine hydrochloride, niaproof, pectin, D-galacturonic acid, L-galactonic acid-gamma-lactone, D-gluconic acid, glucuronamide, mucic acid, D-saccharic acid, vancomycin, tetrazolium violet, tetrazolium blue, p-hydroxy-phenylacetic acid, methyl pyruvate, D-lactic acid methyl ester, bromo-succinic acid, lithium chloride, potassium tellurite, tween 40, γ-amino-n-butyric acid, α-hydroxy-butyric acid, β-hydroxy-butyric acid, α-keto-butyric acid, acetoacetic acid, propionic acid, acetic acid, aztreonam, butyric acid and sodium bromate and the negative control.
In a previous study by Suzuki et al. [1], bacterial growth on nine substrates was tested for R. elongatum OCh 323T. According to [1], none of the carbon sources were utilized. In contrast, the OmniLog assay resulted in more than fifteen positive reactions, including sugars, carboxylic and amino acids. This observation can be explained by a higher sensitivity of respiration measurements compared to growth measurements [26]. For instance, the positive reactions detected only in the OmniLog instrument but not by Suzuki et al. [1] might be caused by substrates that were only partially metabolized.
Chemotaxonomy
The principal cellular fatty acids of strain OCh 323T are C18:1 (68%), C16:0 (12%), C18:0 (8%), C19:0 cyclo (4%), C16:0 2-OH (2%), C14:0 3-OH (2%), C15:0 (1%), C17:0 (1%), and C16:1 (1%), whereas C14:0 and C18:2 are only found in traces (all data from [1]).
Genome sequencing and annotation
Genome project history
The genome of strain DSM 19469T was sequenced within the DFG funded project “Ecology, Physiology and Molecular Biology of the Roseobacter group: Towards a Systems Biology Understanding of a Globally Important Clade of Marine Bacteria”. The strain was chosen for genome sequencing according the Genomic Encyclopedia of Bacteria and Archaea (GEBA) criteria [27,28].
Project information can found in the Genomes OnLine Database [29]. The Whole Genome Shotgun (WGS) sequence is deposited in GenBank and the Integrated Microbial Genomes database (IMG) [30]. A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
A culture of strain DSM 19469T was grown aerobically in DSMZ medium 514 [31] at 28°C. Genomic DNA was isolated using Jetflex Genomic DNA Purification Kit (GENOMED 600100) following the standard protocol provided by the manufacturer but modified by an incubation time of 60 min, incubation on ice over night on a shaker, the use of additional 50 µl proteinase K, and the addition of 100 µl protein precipitation buffer. DNA is available from the DSMZ through the DNA Network [32].
Genome sequencing and assembly
The genome was sequenced using a combination of two libraries (Table 2). Illumina sequencing was performed on a GA IIx platform with 150 cycles. The paired-end library contained inserts of an average of 441 bp in length. The first run delivered 2.7 million reads. To increase the sequencing depth, a second Illumina run was performed, providing another 1.2 million reads. After error correction and clipping by fastq-mcf [33] and quake [34], the data was assembled using Velvet [35]. The first draft assembly from 1,753,098 filtered reads with an average read length of 89 bp resulted in 97 contigs.
To gain information on the contig arrangement an additional 454 run was performed. The paired-end jumping library of 3kb insert size was sequenced on a 1/8 lane. Pyrosequencing resulted in 174,493 reads, with an average read length of 360 bp, assembled with Newbler (Roche Diagnostics). The resulting draft assembly consisted of 22 scaffolds. Both draft assemblies (Illumina and 454 sequences) were fractionated into artificial Sanger reads 1,000 bp in length plus 75 bp overlap on each site. These artificial reads served as an input for the phred/phrap/consed package [36]. In combination the assembly resulted in 39 contigs organized in four scaffolds. Subsequently, small unlocalized contigs were mapped to the scaffolds using both minimus2 [37] and NUCmer [38]. By manual editing, the number of contigs could be reduced to 21, organized in one chromosomal scaffold. The remaining ordered gaps were closed by bridging PCR fragments and primer walking. A total of 50 reactions were required to conclude the assembly process. The genome was sequenced with a 93× coverage.
Genome annotation
Genes were identified using Prodigal [39] as part of the JGI genome annotation pipeline. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Identification of RNA genes was carried out by using HMMER 3.0rc1 [40] (rRNAs) and tRNAscan-SE 1.23 [41] (tRNAs). Other non-coding genes were predicted using INFERNAL 1.0.2 [42] Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [43] CRISPR elements were detected using CRT [44] and PILERCR [45].
Genome properties
The genome statistics are provided in Table 3 and Figure 3. The genome has a total length of 3,555,109 bp and a G+C content of 65.7%. Of the 3,599 genes predicted, 3,540 were identified as protein-coding, and 59 as RNAs. The majority of the protein-coding genes were assigned a putative function (79.6%) while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COG functional categories is presented in Table 4.

Graphical map of the chromosome. From outside to center: Genes on forward strand (colored by COG categories), Genes on reverse strand (colored by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content (black), GC skew (purple/olive).
Genomic insights
Whole genome sequencing of strain R. elongatum DSM 19469T revealed a complete and finished genome size of 3,555,109 bp, which seems to be the smallest completed genome of representatives of the Roseobacter group up to date [46]. The two other isolates Loktanella vestfoldensis SKA53 and Sulfitobacter sp. EE-36 both reveal a genome length shorter than that of strain DSM 19469T, but remain still in draft state. Whereas many members of the Roseobacter group contain plasmids [47], no extrachromosomal elements could be detected in strain DSM 19469T.
The fraction of shared genes between strain R. elongatum DSM 19469T and the neighboring strains D. shibae DFL-12 (DSM 16493T) [11,48] (Figure 1) and Jannaschia sp. CSS1 (which turned out to have similar genomic characteristics in the course of this study, too), both members of the Roseobacter group are shown in a Venn diagram (Figure 4). The number of pairwise genes was inferred from the phylogenetic profiler of the IMG-ER platform [43]. Homologous genes were detected with an E-value cutoff of 10−5 and a minimum identity of 30%.

Venn diagram (total numbers in parentheses) of R. elongatum DSM 19469T, D. shibae DSM 16493T and Jannaschia sp. CSS1.
A total of 2,287 genes are shared by all three genomes, corresponding to 54.3% and 53.4% of the gene count in D. shibae DSM 16493T [11,48] and Jannaschia sp. CCS1, respectively. With only 3.5 Mbp in length, the genome of R. elongatum DSM 19469T shares more than 64.6% of genes with the other two genomes. A number of 645 genes that have no homologs in the other genomes were detected, including a sensor protein of blue-light using FAD (BLUF, roselon_02123) and the Phn gene cluster (roselon_02168-79) involved in the uptake and degradation of phosphonates.
Phages
Phages are widely distributed and common in marine environments [49–51]. Horizontal gene transfer of the phage genome and its integration in the host genome are known to drive the bacterial diversity [51,52]. In the genome sequence of R. elongatum DSM 19469T several putative phage-associated gene sequences were detected, particularly organized in gene clusters (e.g., roselon_02355 – 02370).
Quorum sensing
Quorum sensing (QS) is a cell-to-cell communication system, where bacteria interact with each other in dependence of their population density. Gram-negative bacteria use small signal molecules called autoinducers, which are produced, excreted through the bacterial membrane and detected by conspecific bacteria. Consequently, when the concentration of those membrane-diffusible autoinducers reaches a specific threshold value, the population responds with an activation of gene expression to coordinate a population-wide behavior [53–58]. QS was first detected in the marine gammaproteobacterium Vibrio fischerii, a species often found to live in symbiosis with squids or fishes. Here, the autoinducer accumulation and the activation of certain genes result in biolumescence [59,60]. Other examples for QS-induced bacterial physiological aspects are biofilm formation, exopolysaccharide production and virulence [53,61]. Interestingly, many representatives of the Roseobacter group were shown to encode and/or express gene sequences associated with QS [e.g., 62–65].
Genome analysis of strain R. elongatum DSM 19469T revealed the presence of genes putatively associated with QS like a N-acyl-L-homoserine lactone synthetase (LuxI homolog; roselon_01555) and a regulator of the LuxR family (roselon_3097).
Photosynthetic gene cluster
Light is used as energy source by many bacteria in the ocean. An increasing number of representatives belonging to the Roseobacter group have been found to be aerobic anoxygenic photoheterotrophs, containing bacteriochlorophyll a (Bchl a) [3,4,66–69]. They transform light energy into a proton motive force (pmf) across the membrane that is used for the generation of ATP, which could have an importance for marine environments and global cycles [66–68]. Aerobic anoxygenic photoheterotrophs represent a significant fraction of the microbial population depending on the location [69–73]. It was further shown that aerobic anoxygenic photoheterotrophs synthesize Bchl a only in the presence of oxygen [66,74] and that the photosynthetic pigments of aerobic alphaproteobacteria are synthesized under dark conditions [75–77], whereas some members of the gammaproteobacterial OM60/NOR5 clade also synthesize pigments in the light [78]. Furthermore, Elsen and colleagues reported that genes encoding the photosynthetic apparatus and related genes are mainly organized in a large gene cluster [79].
In the description of strain OCh 323T, the authors showed that the absorption spectrum of the membranes of ultrasonically disrupted cells exhibit a significant photosynthetic reaction center absorption peak (at 800 nm) and a light-harvesting complex I absorption peak (at 879 nm) [1].
The genome sequence of strain R. elongatum DSM 19469T encodes a functional photosynthetic gene cluster (roselon_01064 – 01096) containing a set of bch genes, puf genes, crt genes, hem genes and genes for proteins with sensory activity (Figure 5).

Motility and flagellar genes
Strain R. elongatum DSM 19469T was originally described as non-flagellated [1]. In the genome a flagella gene cluster was found flanking the chromosome-partitioning gene dnaA (roselon_1273). Flagella formation depends on external stimuli such as incubation temperature or composition of the media [81]. Thus, strain DSM 19469T might exhibit a motile phenotype under certain, as yet unknown, conditions. Flagellar genes of strain DSM 19469T involved in flagellar assembly and function were analyzed to assess potential motility behavior. The cluster consists of 28 genes (roselon_01279 – 01316). Three further motor switch proteins, including fliG were detected upstream of roselon_03222. Together with fliM (roselon_03295) and fliN (roselon_01309) fliG forms a protein that controls rotation behavior of flagella. This dissociation of flagellar operons has been seen in two groups of alphaproteobacteria [82]. No master regulator genes operon (flhDC) [83] could be detected. Whereas genes controlling the early flagellum assembly were not detected, several proteins necessary for the formation of the basal body were found, including flgDEFGHIKL and fliF. Genome analysis of strain DSM 19469T revealed further the presence of genes involved in the formation of the export apparatus: the previously mentioned C-ring forming complex fliGMN and the protein-encoding sequences flhA, flhB, fliP, fliQ and fliR, which are involved in pore-forming through the membrane [84]. Whereas two motor protein-encoding gene sequences motAB were found (roselon_01316, roselon_01313), a homolog of the fliO gene as part of the channel-forming apparatus was absent. Additionally, the genome of strain R. elongatum DSM 19469T revealed the presence of regulatory genes controlling the late phase, such as the hook capping protein (roselon_01279), the flagellar hook-length control protein (roselon_01280) and the flagellin-encoding gene sequence fliC (roselon_01284). Methyl-accepting chemotaxis proteins that sense external stimuli, and therefore direct flagella-induced motility of strain DSM 19469T, could not be detected.
To compare the flagellar gene clusters of neighboring species (Figure 6), homologs of flgG coding for a protein mainly involved in the formation of the basal body in R. sphaeroides ATCC 17029 [85] were identified using the IMG/ER platform [43]. All compared genomes show a similar gene cluster structure, but have variations such as differences in gene length for fliK, which controls the completion of previous flagellum-assembly steps. The fliK protein in R. sphaeroides is 700 amino-acid residues (AA) in length [85]. A genome BLAST search (minimal similarity 30%, maximal e-value 10−5) against putative fliK proteins revealed that the gene-encoding sequence length of fliK varies from 102 AA in R. sphaeroides strains WS8N and 2.4.1 to 937 AA in Citreicella sp. SE45. The genomes of the three species Salipiger mucosus, Sagittulla stellata and Pelagibaca bermudensis each encode a truncated fliK-encoding gene sequence, but those strains do not form flagella [86–88]. These truncations could be the reason for inactive proteins resulting in a non-motile phenotype. In contrast, the genome of Jannaschia sp. CCS1 codes for a fliK protein of 612 AA (Jann_4206) and, interestingly, this strain was reported to be motile.

Map of the flagella cluster of R. elongatum DSM 19469T (roselon_Rosei_p5_w02) and homologous ORFs in the genomes of the four comparable strains Jannaschia sp. CCS1 (NC_007802), S. stellata E-37 (NZ_AAYA01000005), R. sphaeroides ATCC 17029 (NC_009049) and Citreicella sp. E45 (NZ_GG704601). Prediction of homologs was conducted using the conserved-neighborhood tool of the IMG-ER platform [43]. The colored areas represent differences in the genomic structure within the flagella cluster.
The second marked region (Figure 6) is well conserved in the first four genomes, but is missing in strain R. sphaeroides ATCC 17029. This cluster consists of the rod-forming gene flgJ and three proteins involved in the regulation of the flagella assembly. Homologs of the R. elongatum DSM 19469T flagellin gene (roselon_01284) are absent in R. sphaeroides. Thus, the regulation of the flagella operon might be conducted by other genes: one of the genes coding for the flagellin-forming FliC in R. sphaeroides is located on the chromosome within the flagellar cluster. An additional set of three regulation genes is detected on the 120 kb plasmid (NC_009040) of the genome. In area 3 of Figure 6 the genomes of both S. stellata and Citreicella sp. lack three flagellar genes: fliL and fliF, which are both involved in the formation of the basal body, and fliP (export apparatus). An additional PAS/PAC sensor hybrid histidine kinase (Rsph17029_2967) is found in the R. sphaeroides genome.
Morphological traits
The genome sequence of strain R. elongatum DSM 19469T was found to have specific genes associated with the putative biosynthesis and export of exopolysaccharides (roselon_01150, roselon_01343 – 01343) and the putative export of capsule polysaccharides (e.g., roselon_00513, roselon_01783 – 01785).
Additionally, the genome of strain R. elongatum DSM 19469T encodes several gene sequences associated with flp-type pili biogenesis and formation (e.g., roselon_01843 – 01852). Hence, the formed pili might play a role in adhesion or switching-type motility on solid surfaces.
Further, strain R. elongatum DSM 19469T seems to accumulate polyhydroxyalkanoates as storage compounds (e.g., roselon_00211 – 00214).
Metabolic plasticity
The genome sequence of strain R. elongatum DSM 19469T encodes a gene cluster associated with a Sox multienzyme complex (roselon_02191 – 02202) that could be utilized for the oxidation of thiosulfate to sulfate. Carbon monoxide could be putatively oxidized by aerobic-type carbon monoxide dehydrogenases (roselon_01738, roselon_01976 – 01977, roselon_02472, roselon_02474).
Several genes play a role in the electron transport chain, such as those associated with the NADH dehydrogenase (e.g., roselon_00011 – 00023), succinate dehydrogenase (roselon_01681 – 01684) and cytochrome bd ubiquinol oxidase (roselon_00027 – 00028). In addition two different cytochrome c oxidases (caa3-type [e.g. roselon_02733 – 02734] or cbb3-type [roselon_00626 – 00628]) were detected.
References
Suzuki T, Mori Y, Nishimura Y. Roseibacterium elongatum gen. nov., sp. nov., an aerobic, bacteriochlorophyll-containing bacterium isolated from the westcoast of Australia. Int J Syst Evol Microbiol 2006; 56:417–421.
Mao Y, Wei J, Zeng Q, Xiao N, Li Q, Fu Y, Wang Y, Jiao N. Roseibacterium beibuensis sp. nov., a novel member of Roseobacter clade isolated from Beibu Gulf in the South China Sea. Curr Microbiol 2012; 65:568–574.
Buchan A, Gonzalez JM, Moran MA. Overview of the marine Roseobacter lineage. Appl Environ Microbiol 2005; 71:5665–5677.
Wagner-Döbler I, Biebl H. Environmental biology of the Roseobacter lineage. Annu Rev Microbiol 2006; 60:255–280.
Göker M, Cleland D, Saunders E, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, Tapia R, et al. Complete genome sequence of Isosphaera pallida type strain (IS1BT). Stand Genomic Sci 2011; 4:63–71.
Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669–674.
Vaas LA, Sikorski J, Hofner B, Fiebig A, Buddruhs N, Klenk HP, Göker M. opm: an R package for analysing OmniLog phenotype microarray data. Bioinformatics 2013; 29:1823–1824.
Riedel T, Fiebig A, Spring S, Petersen J, Göker M, Klenk HP. Genome sequence of the Wenxinia marina type strain (DSM 24838T), a representative of the Roseobacter clade isolated from oilfield sediments. Stand Genomic Sci (Submitted).
Trash JC, Cho JC, Vergin KL, Giovannoni SJ. Genome sequences of Oceanicola granulosus HTCC2516T and Oceanicola batsensis HTCC2597T. J Bacteriol 2010; 192:3549–3550.
Vollmers J, Voget S, Dietrich S, Gollnow K, Smits M, Meyer K, Brinkhoff T, Simon M, Daniel R. Poles apart: Arctic and Antarctic Octadecabacter strains share high genome plasticity and a new type of xanthorhodopsin. PLoS ONE 2013; 8:e63422.
Wagner-Döbler I, Ballhausen B, Berger M, Brinkhoff T, Bunk B, Cypionka H, Daniel R, Drepper T, Gerdts G, Hahnke S, et al. The complete genome sequence of the algal symbiont Dinoroseobacter shibae: a hitchhiker’s guide to life in the sea. ISME J 2010; 4:61–77.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547.
Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, Gilbert J, Glöckner FO, Hirschman L, Karsch-Mzrachi I, et al. Clarifying Concepts and Terms in Biodiversity Informatics. PLoS Biol 2013; 9:e1001088.
Woese CR, Kandler O, Weelis ML. Towards a natural system of organisms. Proposal for the domains Archaea, Bacteria and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579.
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl nov. In: Brenner DJ, Krieg NR, Stanley JT, Garrity GM (eds), Bergey’s Manual of Sytematic Bacteriology, second edition. Vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria), Springer, New York, 2005, p. 1.
Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov. In: Brenner DJ, Krieg NR, Stanley JT, Garrity GM (eds), Bergey’s Manual of Sytematic Bacteriology, second edition. Vol. 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), Springer, New York, 2005, p. 1.
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6.
Garrity GM, Bell JA, Lilburn T. Order III. Rhodobacterales ord. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, second edition. vol. 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), Springer, New York, 2005, p. 161.
Garrity GM, Bell JA, Lilburn T. Family I. Rhodobacteraceae fam. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds), Bergey’s Manual of Systematic Bacteriology, second edition. vol. 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), Springer, New York, 2005, p. 161.
BAuA. Classification of Bacteria and Archaea in risk groups. TRBA 2010; 466:93.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072.
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130–137.
Buddruhs N, Pradella S, Göker M, Päuker O, Michael V, Pukall R, Spröer C, Schumann P, Petersen J, Brinkhoff T. Molecular and phenotypic analyses reveal the non-identity of the Phaeobacter gallaeciensis type strain deposits CIP 105210T and DSM 17395. Int J Syst Evol Microbiol 2013; 63:4340–4349.
Vaas LAI, Sikorski J, Michael V, Göker M, Klenk HP. Visualization and curve-parameter estimation strategies for efficient exploration of phenotype microarray kinetics. PLoS ONE 2012; 7:e34846.
Vaas LAI, Marheine M, Sikorski J, Göker M, Schumacher M. Impacts of pr-10a overexpression at the molecular and the phenotypic level. Int J Mol Sci 2013; 14:15141–15166.
Göker M, Klenk HP. Phylogeny-driven target selection for large-scale genome-sequencing (and other) projects. Stand Genomic Sci 2013; 8:360–374.
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven Genomic Encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40:D571–D579.
Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Pati A, Huntemann M, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res 2012; 40:D115–D122.
List of growth media used at the DSMZ: http://www.dmsz.de/catalogues/cataloque-microorganisms/culture-technology/list-of-media-for-microorganisms.html.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011; 9:51–55.
Aronesty E. ea-utils: Command-line tools for processing biological sequencing data. 2011; http://code.google.com/p/ea-utils.
Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biol 2010; 11:R116.
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829.
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998; 8:195–202.
Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 2007; 8:64.
Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast Algorithms for Large-scale Genome Alignment and Comparision. Nucleic Acids Res 2002; 30:2478–2483.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119.
Finn DR, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Research 2011, Web Server Issue 39:W29–W37.
Lowe TM, Eddy SR. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res 1997; 25:955–964.
Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009; 25:1335–1337.
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278.
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 2007; 8:209.
Anonymous. PILER Genomic repeat analysis software. 2009.
Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE, Howard EC, King E, Oakley CA, Reisch CR, et al. Genome characteristics of a generalist marine bacterial lineage. ISME J 2010; 4:784–798.
Petersen J, Frank O, Göker M, Pradella S. Extrachromosomal, extraordinary and essential—the plasmids of the Roseobacter clade. Appl Microbiol Biotechnol 2013; 97:2805–2815.
Biebl H, Allgaier M, Tindall BJ, Koblizek M, Lunsdorf H, Pukall R, Wagner-Döbler I. Dinoroseobacter shibae gen. nov., sp. nov., a new aerobic phototrophic bacterium isolated from dinoflagellates. Int J Syst Evol Microbiol 2005; 55:1089–1096.
Wommack KE, Colwell RR. Viroplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev 2000; 64:69–114.
Proctor LM, Fuhrman JA, Ledbetter MC. Marine bacteriophages and bacterial mortality. Eos 1988; 69:1111–1112.
Paul JH. Prophages in marine bacteria: dangerous molecular bombs or the key to survival in the seas? ISME J 2008; 2:579–589.
Canchaya C, Proux C, Fournous G, Bruttin A. Prophage genomics. Microbiol Mol Biol Rev 2003; 67:238–276.
Bassler BL. How bacteria talk to each other: regulation of gene expression by quorum sensing. Curr Opin Microbiol 1999; 2:582–587.
Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol 2001; 55:165–199.
Fuqua C, Parsek MR, Greenberg EP. Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annu Rev Genet 2001; 35:439–468.
Waters CM, Bassler BL. Quorum Sensing: Cell-to-Cell Communication in Bacteria. Annu Rev Cell Dev Biol 2005; 21:319–346.
Case RJ, Labbate M, Kjelleberg S. AHL-driven quorum-sensing circuits: their frequency and function among the Proteobacteria. ISME J 2008; 2:345–349.
Ng WL, Bassler BL. Bacterial quorum-sensing network architectures. Annu Rev Genet 2009; 43:197–222.
Nealson KH, Hastings JW. Bacterial Bioluminescence: Its Control and Ecological Significance. Microbiol Rev 1979; 43:496–518.
Visick KL, Foster J, Doino J, McFall-Ngai M, Ruby EG. Vibrio fischeri lux genes play an important role in colonization and development of the host light organ. J Bacteriol 2000; 182:4578–4586.
Sauer K, Camper AK, Ehrlich GD, Costerton JW, Davies DG. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J Bacteriol 2002; 184:1140–1152.
Cude WN, Buchan A. Acyl-homoserine lactone-based quorum sensing in the Roseobacter clade: complex cell-to-cell communication controls multiple physiologies. Front Microbiol 2013; 4:336.
Riedel T, Hazuki T, Petersen J, Fiebig A, Davenport K, Daligault H, Erkkila T, Gu W, Munk C, Xu Y, et al. Genome sequence of the Leisingera aquimarina type strain (DSM 24565T), a member of the marine Roseobacter clade rich in extrachromosomal elements. Stand Genomic Sci 2013; 8:389–402.
Riedel T, Fiebig A. Petersen, Gronow S, Kyrpides NC, Göker M, Klenk HP. Genome sequence of the Litoreibacter arenae type strain (DSM 19593T), a member of the Roseobacter clade isolated from sea sand. Stand Genomic Sci 2013; 9:117–127.
Patzelt D, Wang H, Buchholz I, Rohde M, Gröbe L, Pradella S, Neumann A, Schulz S, Heyber S, Münch K, et al. You are what you talk: quorum sensing induces individual morphologies and cell division modes in Dinoroseobacter shibae. ISME J 2013; 7:2274–2286.
Yurkov VV, Beatty JT. Aerobic anoxygenic phototrophic bacteria. Microbiol Mol Biol Rev 1998; 62:695–724.
Allgaier M, Uphoff A, Wagner-Döbler I. Aerobic anoxygenic photosynthesis in Roseobacter clade bacteria from diverse marine habitats. Appl Environ Microbiol 2003; 69:5051–5059.
Brinkhoff T, Giebel HA, Simon M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol 2008; 189:531–539.
Béjà O, Suzuki MT, Heidelberg JF, Nelson WC, Preston CM, Hamada T, Eisen JA, Fraser CM, DeLong EF. Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature 2002; 415:630–633.
Oz A, Sabehi G, Koblizek M, Massana R, Beja O. Roseobacter-like bacteria in Red and Mediterranean Sea aerobic anoxygenic photosynthetic populations. Appl Environ Microbiol 2005; 71:344–353.
Cottrell MT, Mannino A, Kirchman DL. Aerobic anoxygenic phototrophic bacteria in the Mid-Atlantic Bight and the North Pacific Gyre. Appl Environ Microbiol 2006; 72:557–564.
Lami R, Cottrell MT, Ras J, Ulloa O, Obernosterer I, Claustre H, Kirchman DL, Lebaron P. High abundances of aerobic anoxygenic photosynthetic bacteria in the South Pacific Ocean. Appl Environ Microbiol 2007; 73:4198–4205.
Jiao N, Zhang F, Hong N. Significant roles of bacteriochlorophylla supplemental to chlorophylla in the ocean. ISME J 2010; 4:595–597.
Koblizek M, Mlcouskova J, Kolber Z, Kopecky J. On the photosynthetic properties of marine bacterium COL2P belonging to Roseobacter clade. Arch Microbiol 2010; 192:41–49.
Yurkov VV, van Gemerden H. Impact of light/dark regime on growth rate, biomass formation and bacteriochlorophyll synthesis in Erythromicrobium hydrolyticum. Arch Microbiol 1993; 159:84–89.
Biebl H, Wagner-Döbler I. Growth and bacteriochlorophyll a formation in taxonomically diverse aerobic anoxygenic phototrophic bacteria in chemostat culture: influence of light regimen and starvation. Process Biochem 2006; 41:2153–2159.
Tomasch J, Gohl R, Bunk B, Suarez-Diez M, Wagner-Döbler I. Transcriptional response of the photoheterotrophic marine bacterium Dinoroseobacter shibae to changing light regimes. ISME J 2011; 5:1957–1968.
Spring S, Riedel T. Mixotrophic growth of bacteriochlorophyll a-containing members of the OM60/NOR5 clade of marine gammaproteobacteria is carbon-starvation independent and correlates with the cellular redox state. BMC Microbiol 2013; 13:117.
Elsen S, Jaubert M, Pignol D, Giraud E. PpsR: a multifaceted regulator of photosynthesis gene expression in purple bacteria. Mol Microbiol 2005; 57:17–26.
Spring S, Lünsdorf H, Fuch BM, Tindall BJ. The photosynthetic apparatus and its regulation in the aerobic gammaproteobacterium Congregibacter litoralis gen. nov., sp. nov. PLoS ONE 2009; 4:e4866.
Holt PS, Chaubal PH. Detection of motility and putative synthesis of flagellar proteins in Salmonella pullorum cultures. J Clin Microbiol 1997; 35:1016–1020.
Liu R, Ochman H. Origins of flagellar gene operons and secondary flagellar systems. J Bacteriol 2007; 189:7098–7104.
Smith TG, Hoover TR. Deciphering bacterial flagellar gene regulatory networks in the genomic era. Adv Appl Microbiol 2009; 67:257–295.
Terashima H, Kojima S, Homma M. Flagellar motility in bacteria structure and function of flagellar motor. Int Rev Cell Mol Biol 2008; 270:39–85.
González-Pedrajo B, de la Mora J, Ballado T, Camarena L, Dreyfus G. Characterization of the flgG operon of Rhodobacter sphaeroides WS8 and its role in flagellum biosynthesis. Biochim Biophys Acta 2002; 1579:55–63.
Martinez-Cánovas MJ, Quesada E, Martinez-Checa F, del Moral A, Béjar V. Salipiger mucescens gen.nov., sp.nov., a moderately halophilic, exopolysaccharide-producing bacterium isolated from hypersaline soil, belonging to the alpha-Proteobacteria. Int J Syst Evol Microbiol 2004; 54:1725–1740.
González JM, Mayer F, Moran MA, Hodson RE, Whitman WB. Sagittula stellata gen. nov., sp. nov., a lignin-transforming bacterium from a coastal environment. Int J Syst Bacteriol 1997; 47:773–780.
Cho JC, Giovannoni SJ. Pelagibaca bermudensis gen. nov., sp. nov., a novel marine bacterium within the Roseobacter clade in the order Rhodobacterales. Int J Syst Evol Microbiol 2006; 56:855–859.
Acknowledgements
The authors gratefully acknowledge Evelyne Brambilla for DNA extraction and quality control as well as Stefan Spring for his great support (both at the DSMZ). The work was performed under the auspices of the German Research Foundation (DFG) Transregio-SFB 51 Roseobacter grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Riedel, T., Fiebig, A., Göker, M. et al. Complete genome sequence of the bacteriochlorophyll a-containing Roseibacterium elongatum type strain (DSM 19469T), a representative of the Roseobacter group isolated from Australian coast sand. Stand in Genomic Sci 9, 840–854 (2014). https://doi.org/10.4056/sigs.5541028
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.5541028