
Abstract
The Enterobacter cloacae complex is genetically very diverse. The increasing number of complete genomic sequences of E. cloacae is helping to determine the exact relationship among members of the complex. E. cloacae P101 is an endophyte of switchgrass (Panicum virgatum) and is closely related to other E. cloacae strains isolated from plants. The P101 genome consists of a 5,369,929 bp chromosome. The chromosome has 5,164 protein-coding regions, 100 tRNA sequences, and 8 rRNA operons.
Similar content being viewed by others


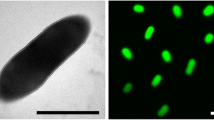
Introduction
Numerous Enterobacter cloacae strains have been associated with plants as agents of disease [1–4], but E. cloacae strains have also been associated with plants as endophytes [5–8], used for biocontrol of fungal pathogens [9–16], and associated with nosocomial infections in hospital settings [17–19]. E. cloacae is in the E. cloacae complex, which also includes the Enterobacter species of E. asburiae, E. hormaechei, E. kobei, E. ludwigii, and E. nimipressuralis. While 16S rRNA sequences are used to initially identify E. cloacae strains, the sequence is not always sufficient for identification at the species and sub-species level [17]. Previous phylogenetic studies with multi-locus sequence analyses of common housekeeping genes demonstrate that there is considerable diversity among the strains designated as E. cloacae due to the formation of multiple clades and the fact that only 3% of the strains group with the type strain E. cloacae subsp. cloacae ATCC 13047 [17,18]. The number of draft and complete E. cloacae genomes has increased recently and there are currently five complete and five draft E. cloacae genomes, with additional registered genome projects [20]. Sequencing and analysis of more E. cloacae genomes may establish a basis for explaining the diversity within the E. cloacae complex and provide new means for more definitive species or sub-species designation.
Classification and features
E. cloacae P101 was isolated from switchgrass (Panicum virgatum) growing on Buena Vista Quarry Prairie near Plover, Wisconsin and is a Gram-negative, rod shaped bacterium of the family Enterobacteriaceae (Table 1). The species within the genus Enterobacter are difficult to identify with biochemical and phylogenetic tests [18], but the increasing number of complete genomes is providing clues as to the relationships among the species. E. cloacae species group separately from other Enterobacter species in a phylogenetic tree using 16s rRNA sequences (Figure 1) with strong support (posterior probability of 100%). In this analysis, P101 is most closely related to E. cloacae EcWSU1 and E. cloacae ENHKU01 which are two other E. cloacae strains that have been isolated from plants. E. cloacae EcWSU1 causes Enterobacter bulb decay on stored onions (Allium cepa) [41] and E. cloacae ENHKU01 was isolated as an endophyte from a pepper (Capsicum annuum) plant infected with Ralstonia solanacearum [42].

Phylogenetic tree of 16S rRNA sequences from Enterobacter sp. with genome sequences. E. cloacae strains grouped separately into a clade from other Enterobacter species using Bayesian phylogenetic analyses of the 16S rRNA region. Analyses were implemented in MRBAYES [39] and the Bayesian Information Criterion (BIC), DT-ModSel [40] was used to determine the nucleotide substitution model best suited for the dataset. To ensure that the average split frequency between runs was less than 1%, the Markov chain Monte Carlo search included two runs with four chains each for 10,000,000 generations. Pectobacterium carotovorum served as the outgroup for the analysis. Numbers in parentheses behind the bacterial names correspond to the GenBank accession numbers for the genome sequences. The scale bar indicates the number of substitutions/site.
Genome sequencing and annotation
Genome project history
The E. cloacae P101 genome project was initiated as part of an undergraduate class at the University of Florida [36]. For the class, whole-genome sequence was obtained using a Genome Sequencer 20 (454 Life Sciences, Branford, CT) and the students used PCR and sequencing to resolve some gaps. Although the project began with these data, little progress was made towards closing the genome. As a result, new next-generation DNA sequencing data for P101 was obtained at the Laboratory for Biotechnology and Bioanalysis at Washington State University using the PacBio RS platform and the PCR products generated to confirm the genome assembly were sequenced at Elim Biopharmaceuticals (Hayward, CA). A BglII cut optical map of P101 was obtained from OpGen (Gaithersburg, MD) in 2009 and was also used in the genome assembly process. The complete chromosome sequence has been deposited in GenBank under the accession number CP006580. Table 2 summarizes the P101 sequencing project.
Growth conditions and DNA isolation
E. cloacae P101 was cultured overnight in LB broth [45] on a rotary shaker at 200 rpm at 28°C. To remove excess exopolysaccharides prior to genomic DNA isolation, the cells were washed twice with equal volumes of sterile, distilled water. Genomic DNA was then isolated from the washed cells using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI) following the kit protocol for Gram-negative bacteria.
Genome sequencing and assembly
Genome sequencing was performed at the Laboratory for Biotechnology and Bioanalysis at Washington State University on a PacBio RS instrument (Pacific Biosciences, Menlo Park, CA). A small insert library for circular consensus reads was prepared from 5 µg of P101 genomic DNA. The genomic DNA was first fragmented to 1 Kb pieces using 20 shearing cycles at speed code 6 through the small shearing assembly of a Hydroshear Plus (Digilab, Marlborough, MA). The library was then constructed using the DNA Template Prep Kit 2.0 (250 bp-<3 kb) (Pacific Biosciences, Menlo Park, CA). Two large insert (10 Kb) libraries for continuous long reads (CLR) were also prepared. For one library, 10 µg of genomic DNA was sheared using 20 shearing cycles at speed code 11 through the large shearing assembly of a Hydroshear Plus. The second library was prepared with 5 µg of genomic DNA that was fragmented by passing the DNA twice through a g-TUBE (Covaris, Woburn, MA) at 6,000 × g in a microcentrifuge. Both large libraries were prepared using DNA Template Prep Kit 2.0 (3–10 Kb) (Pacific Biosciences). The resulting libraries were bound to the C2 DNA polymerase (Pacific Biosciences) and loaded into the SMRT cell (Pacific Biosciences) zero mode waveguides by diffusion (small libraries and first large library) or with mag-bead assistance (second large library). The prepared libraries were loaded on a total of 16 SMRT cells. The four SMRT cells that contained the small insert libraries were observed with two 55 minute movies while the 12 SMRT cells with large libraries were observed with a single 120 minute movie. Pre-filtering, there was 1.5 Gbp of data in 1.2 million reads with an average read length of 1,244 bp and read quality of 0.284. After filtering to remove any reads shorter than 100 bp or below the minimum accuracy of 0.8, 0.96 Gbp of data remained and consisted of 287,709 reads with an average quality of 0.857 and an average read length of 3,323 bp.
The raw data from the 16 SMRT cells were assembled using the HGAP protocol of the SMRT Analysis v2.0.0 software (Pacific Biosciences). The standard bacterial HGAP assembly protocol with an expected genome size of 5.0 Mb was used. The same protocol was also used to assemble the data from 12 SMRT cells, which excluded four CLR SMRT cells run under instrument software v1.3.0, due to concerns of artifacts in the assembly based on how the quality scores were handled by that version of the software. The 20 contigs from the 16 SMRT cell assembly were used as the base set of contigs. The largest contig was 1.7 Mbp in length and the average coverage for all the contigs was 131× with an N50 of 591,864 bp. The 12 SMRT cell contig set was essentially the same, but there were 28 contigs with an N50 of 3,479,841 bp (also the length of the longest contig). The contigs were mapped to the P101 optical map. This allowed the contigs to be ordered and for overlapping regions to be joined together. Primer pairs for regions throughout the genome assembly were generated and used to verify the assembly using GoTaq Polymerase (Promega) according to the manufacturer’s protocol and 50 ng of P101 genomic DNA, which had an annealing temperature of 52°C and an extension of 1 m. Sequencing was completed for both strands of the PCR amplicons using the same primers used for amplification of the fragments. The assembled chromosome and sequences from the PCR products were aligned with Bioedit (Ibis Biosciences, Carlsbad, CA).
Genome annotation
The submission file for GenBank was prepared using Sequin [46]. The genome sequence was submitted to GenBank and annotated with the NCBI Prokaryotic Genome Annotation Pipeline [44].
Genome properties
The genome of E. cloacae P101 has one circular chromosome of 5,369,929 bp (Table 3). The average G+C content for the genome is 54.4% (Table 3). There are 100 tRNA genes and 8 rRNA operons, each consisting of a 16S, 23S, and 5S rRNA gene. There are 5,164 predicted protein-coding regions and 29 pseudogenes in the genome. A total of 4,419 genes (83.6%) have been assigned a predicted function while the remainders have been designated as hypothetical proteins (Table 3). The numbers of genes assigned to each COG functional category are listed in Table 4. Of the annotated genes, 19.6% were not assigned to a COG or are of unknown function.
References
Bishop AL. Internal decay of onions caused by Enterobacter cloacae. Plant Dis 1990; 74:692–694. http://dx.doi.org/10.1094/PD-74-0692
Nishijima KA, Alvarez AM, Hepperly PR, Shintaku MH, Keith LM, Sato DM, Bushe BC, Armstrong JW, Zee FT. Association of Enterobacter cloacae with rhizome rot of edible ginger in Hawaii. Plant Dis 2004; 88:1318–1327. http://dx.doi.org/10.1094/PDIS.2004.88.12.1318
Nishijima KA, Couey HM, Alvarez AM. Internal yellowing, a bacterial disease of papaya fruits caused by Enterbacter cloacae. Plant Dis 1987; 71:1029–1034. http://dx.doi.org/10.1094/PD-71-1029
Nishijima KA, Wall MM, Siderhurst MS. Demonstrating pathogenicity of Enterobacter cloacae on macadamia and identifying associated volatiles of gray kernel of macadamia in Hawaii. Plant Dis 2007; 91:1221–1228. http://dx.doi.org/10.1094/PDIS-91-10-1221
Madmony A, Chernin L, Pleban S, Peleg E, Riov J. Enterobacter cloacae, an obligatory endophyte of pollen grains of Mediterranean pines. Folia Microbiol (Praha) 2005; 50:209–216. PubMed http://dx.doi.org/10.1007/BF02931568
McInroy JA, Kloepper JW. Survey of indigenous bacterial endophytes from cotton and sweet corn. Plant Soil 1995; 173:337–342. http://dx.doi.org/10.1007/BF00011472
Hinton DM, Bacon CW. Enterobacter cloacae is an endophytic symbiont of corn. Mycopathologia 1995; 129:117–125. PubMed http://dx.doi.org/10.1007/BF01103471
Mukhopadhyay K, Garrison NK, Hinton DM, Bacon CW, Khush GS, Peck HD, Datta N. Identification and characterization of bacterial endophytes of rice. Mycopathologia 1996; 134:151–159. PubMed http://dx.doi.org/10.1007/BF00436723
Van Dijk K, Nelson EB. Inactivation of seed exudate stimulants of Pythium ultimum sporangium germination by biocontrol strains of Enterobacter cloacae and other seed-associated bacteria. Soil Biol Biochem 1998; 30:183–192. http://dx.doi.org/10.1016/S0038-0717(97)00106-5
Nelson EB. Biological control of Pythium seed rot and preemergence damping-off of cotton with Enterobacter cloacae and Erwinia herbicola applied as seed treatments. Plant Dis 1988; 72:140–142. http://dx.doi.org/10.1094/PD-72-0140
Nelson EB, Chao WL, Norton JM, Nash GT, Harman GE. Attachment of Enterobacter cloacae to hyphae of Pythium ultimum: Possible role in the biological control of Pythium preemergence damping-off. Phytopathology 1986; 76:327–335. http://dx.doi.org/10.1094/Phyto-76-327
Nelson EB, Craft CM. Introduction and establishment of strains of Enterobacter cloacae in golf course turf for the biological-control of Dollar Spot. Plant Dis 1991; 75:510–514. http://dx.doi.org/10.1094/PD-75-0510
Wilson CL, Franklin JD, Pusey PL. Biological control of Rhizopus rot of peach with Enterobacter cloacae. Phytopathology 1987; 77: 303–305. http://dx.doi.org/10.1094/Phyto-77-303
Wisniewski M, Wilson C, Hershberger W. Characterization of inhibition of Rhizopus stolonifer germination and growth by Enterobacter cloacae. Can J Bot 1989; 67:2317–2323. http://dx.doi.org/10.1139/b89-296
Tsuda K, Kosaka Y, Tsuge S, Kubo Y, Horino O. Evaluation of the endophyte Enterobacter cloacae SM10 isolated from spinach roots for biological control against Fusarium wilt of spinach. J Gen Plant Pathol 2001; 67:78–84. http://dx.doi.org/10.1007/PL00012993
Pratella GC, Mari M, Guizzardi M, Folchi A. Preliminary studies on the efficiency of endophytes in the biological control of the postharvest pathogens Monilinia laxa and Rhizopus stolonifer in stone fruit. Postharvest Biol Technol 1993; 3:361–368. http://dx.doi.org/10.1016/0925-5214(93)90016-V
Hoffmann H, Roggenkamp A. Population genetics of the nomenspecies Enterobacter cloacae. Appl Environ Microbiol 2003; 69:5306–5318. PubMed http://dx.doi.org/10.1128/AEM.69.9.5306-5318.2003
Paauw A, Caspers MP, Schuren FH, Leversteinvan Hall MA, Deletoile A, Montijn RC, Verhoef J, Fluit AC. Genomic diversity within the Enterobacter cloacae complex. PLoS ONE 2008; 3:e3018. PubMed http://dx.doi.org/10.1371/journal.pone.0003018
Hormaeche E, Edwards PR. A proposed genus Enterobacter. Int J Syst Evol Microbiol 1960; 10:71–74.
National Center for Biotechnology Information. Genome resource. http://www.ncbi.nlm.nih.gov/genome
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteriaphyl. nov. In: Garrity GM, Brenner DJ, Kreig NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second Edition ed. Volume Volume 2, Part B. New York: Springer; 2005. p. 1.
Validation of publication of new names and new combinations previously effectively published outside the IJSEM. List no. 106. Int J Syst Evol Microbiol 2005; 55:2235–2238. http://dx.doi.org/10.1099/ijs.0.64108-0
Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class nov. In: Garrity GM, Brenner DJ, Kreig NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second Edition ed. Volume Volume 2. New York: Springer; 2005. p 1.
Williams KP, Kelly DP. Proposal for a new class within the phylum Proteobacteria, Acidithiobacillia classis nov., with the type order Acidithiobacillales, and emended description of the class Gammaproteobacteria. Int J Syst Evol Microbiol 2013; 63:2901–2906. PubMed http://dx.doi.org/10.1099/ijs.0.049270-0
Garrity GM, Holt JG. Taxonomic Outline of the Archaea and Bacteria. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 155–166.
Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Evol Microbiol 1980; 30:225–420.
Rahn O. New principles for the classification of bacteria. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg 1937; 96:273–286.
Commission J. Conservation of the family name Enterobacteriaceae, of the name of the type genus, and designation of the type species OPINION NO. 15. Int J Syst Evol Microbiol 1958; 8:73–74.
Sakazaki R. Genus VII. Enterobacter Hormaeche and Edwards 1960, 72; Nom. cons. Opin. 28, Jud. Comm. 1963, 38. In: R. E. B, Gibbons NE, editors. Bergey’s Manual of Determinative Bacteriology. Eighth Edition ed. Baltimore: The Williams and Wilkins Co.; 1974. p 324–325.
Board E. OPINION 28 Rejection of the bacterial generic name Cloaca Castellani and Chalmers and acceptance of Enterobacter Hormaeche and Edwards as a bacterial generic name with type species Enterobacter cloacae (Jordan) Hormaeche and Edwards. Int J Syst Evol Microbiol 1963; 13:38.
Brady C, Cleenwerck I, Venter S, Coutinho T, De Vos P. Taxonomic evaluation of the genus Enterobacter based on multilocus sequence analysis (MLSA): Proposal to reclassify E. nimipressuralis and E. amnigenus into Lelliottia gen. nov. as Lelliottia nimipressuralis comb. nov. and Lelliottia amnigena comb. nov., respectively, E. gergoviae and E. pyrinus into Pluralibacter gen. nov. as Pluralibacter gergoviae comb. nov. and Pluralibacter pyrinus comb. nov., respectively, E. cowani, E. radicincitans, E. oryzae and E. arachidis into Kosakonia gen. nov. as Kosakonia cowani comb. nov., Kosakonia radicincitans comb. nov., Kosakonia oryzae comb. nov. and Kosakonia arachidis comb. nov., respectively, and E. turicensis, E. helveticus and E. pulveris into Cronobacter as Cronobacter zurichensis nom. nov., Cronobacter helveticus comb. nov. and Cronobacter pulveris comb. nov., respectively, and emended description of the genera Enterobacter and Cronobacter. Syst Appl Microbiol 2013; 36:309–319. PubMed http://dx.doi.org/10.1016/j.syapm.2013.03.005
Riggs PJ, Chelius MK, Iniguez AL, Kaeppler SM, Triplett EW. Enhanced maize productivity by inoculation with diazotrophic bacteria. Aust J Plant Physiol 2001; 28:829–836; 10.1071/Pp01045.
Riggs PJ, Moritz RL, Chelius MK, Dong Y, Iniguez AL, Kaeppler SM, Casler MD, Triplett EW. Isolation and characterization of diazotrophic endophytes from grasses and their effects on plant growth. In: Finan T, O’Brian M, Layzell D, Vessey K, Newton W, editors. Nitrogen Fixation: Global perspectives. New York, NY: CAB International; 2002. p 263–267.
Drew JC, Triplett EW. Whole genome sequencing in the undergraduate classroom: Outcomes and lessons from a pilot course. Journal of Microbiology & Biology Education 2008; 9:3–11. PubMed http://dx.doi.org/10.1128/jmbe.v9.89
Holt JG, Kreig NR, Sneath PHA, Staley JT, Williams ST. Bergey’s Manual of Determinative Bacteriology. Baltimore: Williams & Wilkins; 1994.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001; 17:754–755. PubMed http://dx.doi.org/10.1093/bioinformatics/17.8.754
Minin V, Abdo Z, Joyce P, Sullivan J. Performance-b ased selection of likelihood models for phylogeny estimation. Syst Biol 2003; 52:674–683. PubMed http://dx.doi.org/10.1080/10635150390235494
Schroeder BK, Waters TD, du Toit LJ. Evaluation of onion cultivars for resistance to Enterobacter cloacae in storage. Plant Dis 2010; 94:236–243. http://dx.doi.org/10.1094/PDIS-94-2-0236
Liu WY, Chung KM, Wong CF, Jiang JW, Hui RK, Leung FC. Complete genome sequence of the endophytic Enterobacter cloacae subsp. cloacae strain ENHKU01. J Bacteriol 2012; 194:5965. PubMed http://dx.doi.org/10.1128/JB.01394-12
Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 2013; 10:563–569. PubMed http://dx.doi.org/10.1038/nmeth.2474
Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity G, Kodira CD, Kyrpides N, Madupu R, Markowitz V, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS 2008; 12:137–141. PubMed http://dx.doi.org/10.1089/omi.2008.0017
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989.
Sequin. www.ncbi.nlm.nih.gov/Sequin
Acknowledgements
This project was supported by the Department of Plant Pathology, College of Agricultural, Human and Natural Resource Sciences, PPNS #0625, Agricultural Research Center, Project No. WNP00652 Washington State University, Pullman, WA 99164-6430, USA and the Washington State University ADVANCE program (NSF no. 0810927). We also acknowledge support from the National Science Foundation through grants from the Division of Undergraduate Education (DUE 0920151, DUE 1161177). We also acknowledge the efforts of the following undergraduate and graduate students at the University of Florida who initiated this project with 454 data: Jessica Anderson, Kate Bailey, Emily Barbieri, Ashley Bartczakm Steve Basak, Changhao Bi, Larea Boone, Alyson Brinker, Shauna Brown, Abrar Chaudry, Chris DeFraia, Lauren Drouin, Jacob Esquenazi, Crysten Haas, Dustin Hill, Anabel Hugh, Maigan Hulme, David James, Berenice Jaramillo, Rainy Johnson, Katherine Kamataris, Andrew Karlesky, Amelia Kaywell, Edward Lin, Megan Matassini, Adrienne Maxwell, Megan McCarthy, Edward Miller, Sarah Mollo, Brelan Moritz, Courtney Myhr, Matoya Robinson, Matthew Rogers, Hilary Seifert, Ryan Shienbaum, John Thomas, Angela Trujillo, Ashley Watford, and Stacy Watts. The authors would also like to thank Drs. Mark Mazzola and Mohammad Arif for critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Humann, J.L., Wildung, M., Pouchnik, D. et al. Complete genome of the switchgrass endophyte Enterobacter clocace P101. Stand in Genomic Sci 9, 726–734 (2014). https://doi.org/10.4056/sigs.4808608
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.4808608