
Abstract
Basic oxygen furnace slag (BOFS) is a high-volume waste resulting from the production of steel from pig iron. Due to its high free lime content, BOFS is difficult to recycle and/or include into conventional cement systems. Alkali-activation technology offers a pathway to transform industrial wastes such as BOFS into low-carbon cements. Alternative precursors for cement systems are needed as the reliance on commonly used materials like ground granulated blast furnace slag (GGBFS) is becoming unsustainable due to decreasing availability. This study investigates alkali-activated cements incorporating 20 and 30 wt.% of naturally weathered BOFS as a replacement for GGBFS, in both sodium silicate- and sodium carbonate-activated systems. A fraction of BOFS subject to mechanical activation is compared against the untreated BOFS in the 20 wt.% systems. It is observed that in naturally weathered BOFS, a significant portion of the free-lime is found to convert to portlandite, which accelerates alkali-activation kinetics. In sodium silicate-activated systems, the high pH of the activator results in incomplete reaction of the portlandite present in BOFS. The sodium carbonate-activated system shows near complete conversion of portlandite, causing an acceleration in the kinetics of reaction, setting, and hardening. These findings confirm the viability of sodium carbonate activated GGBFS-based systems with only a minor loss in strength properties. BOFS can be utilised as a valuable cement additive for the production of sustainable alkali-activated cements utilising sodium carbonate as a less carbon-intensive activator solution than the more commonly used sodium silicate. Mechanical activation of BOFS offers further optimisation potential for alkali-activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The iron and steel manufacturing sectors play a central role in many developed and emerging economies worldwide. The ongoing growth of both industries is closely tied with persistent CO2 emissions, utilisation of primary raw materials, and significant production of solid waste [1]. Currently, crude steel is produced through two main processes: the primary route involves converting pig iron via the basic oxygen furnace process, and the secondary route utilises scrap and directly reduced iron through the electric arc furnace process. In 2021 global production of crude steel amounted to 1.96 billion tonnes resulting in over 300 million tons of steel slags [2]. Globally about 73% of steel is produced through the basic oxygen furnace route which generates basic oxygen furnace slag (BOFS) as a by-product [3].
The construction industry also contributes significantly to global CO2 emissions and raw resource consumption [4], and there exists a strong drive to improve the sustainable development of both steelmaking and construction sectors. Alkali-activation technology may provide a solution for both [5], enabling the large-scale valorisation of steelmaking wastes [6] through their use in the production of high performance low-carbon binders [7] that contribute to the circular economy [8]. Alkali-activated cements are made from the reaction of solid aluminosilicate precursors and an alkaline solution to form a hardened binder. The most commonly used precursors are ground granulated blast-furnace slag (GGBFS) and fly ash [9]. Both GGBFS and fly ash are now valuable resources with high demand, and are expected to have a reduction in availability due to technological advances in ironmaking and in energy generation, respectively [5].
The main oxides within BOFS are CaO, SiO2, Fe2O3, Al2O3, and MgO, present as mineral phases including calcium silicates, aluminosilicates, aluminoferrites, and the “RO” phase, which is a solid solution of CaO, FeO, MgO, and MnO commonly found in steel slags [10,11,12]. BOFS can vary substantially in mineral composition between sources, and generally exhibits poor hydraulic properties [10, 13]. The high content of free lime (up to 40%) is the main limitation on the use of BOF in construction applications, as free lime is known to cause volumetric expansion of concretes upon its hydration into portlandite which can potentially initiate structural failure [14].
BOFS has been used in asphalt [15, 16], road pavements [17, 18], as an aggregate for traditional concrete [19], and alkali-activated metakaolin [20, 21] with minimal treatment. It also has use as a liming material in agriculture to improve the quality of soil [22]. Despite these various uses, most BOFS is discarded as landfill [1]. The reported utilisation of BOFS as a supplementary cementitious material (SCM), a mineral admixture, or as a precursor in alkali-activation technology is relatively limited. An investigation into the hydration properties of BOFS by Wang et al. [23] reported very slow reaction kinetics, that could be accelerated by increasing the fineness of BOFS particles, increasing the curing temperature, and utilising an alkaline solution. Fine BOFS has been successfully blended as an SCM in Portland cement based materials without a significant reduction in performance [24]. Kourounis et al. [25] found BOFS inclusion in conventional cement systems to improve workability, while generally lowering final compressive strength. Alternative processing techniques such as iron recovery [26], mechanical treatment [27], and air-granulation instead of conventional cooling have demonstrated an improve in BOFS reactivity when used as an SCM [28].
Research into the utilisation of BOFS in alkali-activated cements, blended with GGBFS and fly ash, has been explored in a few cases, necessitating the use of strong activator solutions of NaOH [12] and high curing temperatures [29]. Various strategies have been undertaken to tailor the properties of BOFS. These include techniques such as granulation to modify particle sizes and mineralogy [30], as well as the incorporation of alumina and silica at elevated temperatures to favour amorphous formation [31], aiming to enhance the production of alkali-activated materials.
The performance and final application of alkali-activated materials is significantly impacted by characteristics of the solid precursors, including morphology, particle size, mineral composition, and amorphous fraction. Another critical factor is the nature and concentration of the activator solution [32,33,34,35]. Conventional activators such as sodium/potassium silicates and hydroxides have been identified as significant contributor to the overall CO2 emissions from the alkali-activation process [36]. Sodium carbonate could offer a more accessible, sustainable, and cost-effective alternative to sodium silicate, reducing both costs and total CO2 emitted by two-thirds.
Sodium carbonate activated slag cements tend to suffer from delayed hardening, requiring up to 5 days, and low early strength development [37, 38]. The delay in hardening and strength development is strictly related to the lower alkalinity of the system, which is required for the complete dissolution of slag particles. For systems with pH < 12, only the dissolution of calcium from GGBFS is favoured, which is consumed by CO32− to form carbonate salts such as calcite and gaylussite. As the pH slowly increases when the CO32− concentration is reduced, the reaction proceeds analogous to that in NaOH-activated slag, forming primarily a calcium aluminosilicate hydrate gel (C–A–S–H) gel [39]. Additives such as MgO [40], CaO [41], and calcined layered double hydroxides [42] can be utilised to enhance early age properties, or the sodium carbonate can be blended with sodium silicates to improve early strength development [43].
In this study, seven binders were produced using 20 and 30 wt.% of BOFS as a replacement for GGBFS, and activated with either sodium silicate or sodium carbonate solutions. The reaction kinetics during formation of these binders are analysed via isothermal calorimetry, and the setting time and workability assessed. The resulting phase assemblages in the hardened binders are determined via X-ray diffraction (XRD) and Fourier transform infra-red spectroscopy (FTIR), and the compressive strength evaluated, benchmarked against conventional systems. Optimisation of this system using a controlled mechanically activated BOFS fraction is also proposed to increase the strength of sodium carbonate-BOFS/GGBFS cements.
2 Materials and methods
2.1 Materials characterisation and mixing procedure
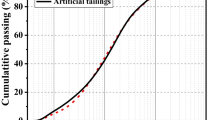
Two types of precursor materials were used: GGBFS supplied by Ecocem (Belgium) and naturally weathered BOFS supplied by ArcelorMittal (Belgium). The BOFS was used as received, with particle sizes ranging up to 250 μm, and a fine faction (BOF*) was obtained by mechanical activation of BOFS using a ball mill to both reduce the particle size and increase the reactivity. This fraction was also sieved to achieve a similar particle size distribution to GGBFS. Particle size and density values are presented in Table 1, and the particle size analysis illustrated in Fig. 1

Particle size distribution of precursors GGBFS, BOFS, and BOF*
The oxide composition is shown in Table 2. The loss of ignition (LOI) value of 8.1 wt.% for BOFS is relatively high due to the conditions of weathering and carbonation to which the sample was exposed.
Scanning electron microscope (SEM) images representing the precursor particles are shown in Fig. 2. The GGBFS precursor consists of small (mostly < 20 µm) angular particles, while the BOFS precursor consists of much larger heterogeneous aggregated particles (many of which are > 100 µm) composed of smaller sub-units of differing morphologies.

Backscattered electron (BSE) images of the raw precursors: a GGBFS, and b BOFS
The XRD patterns of the GGBFS, BOFS, and BOF* are presented in Fig. 3. The major crystalline phases identified in the XRD pattern of BOF slag include portlandite (P, Ca(OH)2, PDF# 00-044-1481) and calcite (C, CaCO3, PDF# 00-005-0586), along with minor phases such as srebrodolskite (S, Ca2Fe2O5, PDF#00-047-1744), wüstite (W, FeO PDF#00-006-0615), hydrous calcium carboaluminoferrite that evidently result from weathering of the srebrodolskite contained in the BOFS (Fc, C8AFcH23, PDF#00-045-0572), and a solid solution of CaO, FeO, MgO, and MnO (known as RO phase PDF#00-053-0926) commonly found in steel slags [10,11,12]. Srebrodolskite is a partially hydraulic phase with fast dissolution and hydration kinetics, considered the most reactive phase in BOFS [44]. The major crystalline phases of the fine fraction of BOF* comprise portlandite and calcite, however with differing phase fractions and minor srebrodolskite and Fc phases.

XRD patterns of GGBFS, BOFS, and BOF*
The XRD pattern of GGBFS indicates an amorphous structure with a broad feature due to diffuse scattering centred at 30° 2θ, and a distinct lack of any crystalline phases.
Sodium hydroxide pellets (98% purity, Sigma Aldrich), sodium carbonate (99.5% purity, Sigma Aldrich), and sodium silicate solution (PQ Corporation) with a molar SiO2/Na2O ratio of 2.07 and solid content of 44.1 wt.%, were used to prepare the activator solutions.
Alkali-activated pastes were prepared by mixing GGBFS with 0, 20, and 30 wt.% of BOFS. Two types of activator solution were prepared. Type “a” was made by initially mixing NaOH pellets with water and adding sodium silicate solution on cooling of the solution, to obtain a molar ratio SiO2/Na2O of 0.56, then combined with the precursors to form a paste with an activator dose of 4 g Na2O per 100 g solid precursor and a water/binder mass ratio (w/b) of 0.38. Type “b” was prepared by dissolving sodium carbonate in water, then mixed with the precursors at a dose of 8 g Na2CO3 per 100 g solid precursor and with w/b of 0.38. Each combination of precursors was activated with both activator types “a” and “b”. An optimised formulation was prepared with 20 wt.% of BOF*, activated with sodium carbonate, type b, and recorded as 20BOFb*. The details of all the formulations are shown in Table 3.
The precursors were dry-blended for 60 s prior to addition of the alkaline activator solution. Pastes were mixed for 4 min at low speed (140 ± 5 rpm), then for 4 min at high speed (285 ± 5 rpm). The pastes were poured into 50 mm cubic moulds and sealed for at least 24 h, then cured in sealed conditions until reaching the specific testing age under laboratory conditions of 20 ± 1 °C.
2.2 Testing programme
2.2.1 Isothermal calorimetry
The reaction kinetics of blended binders were determined via isothermal calorimetry using a TAM Air Conduction Calorimeter (TA Instruments, USA). 20 g of fresh paste were transferred into a glass vial after mixing for ~ 90 s. The vial was sealed and promptly loaded into the calorimeter along with a reference vial filled with water as described by Wadsö [45]. The thermal behaviour was recorded for ~ 250 h. The results are presented as heat release per gram of solid binder, i.e. precursor and solid activators combined.
2.2.2 Mini-slump
A mini-slump test was conducted on fresh mixtures after 30 min to determine the workability. A slump cone with a height of 57 mm, and internal top and bottom diameters of 19 and 38 mm respectively was used. For each slump test, the slump cone was placed on a flat sheet and gradually filled with the fresh mixture. The cone was then lifted as slowly as possible (< 1 cm/s) [46] and the mean value of the flow diameters measured in two perpendicular directions recorded as the spread.
2.2.3 Setting time
The Vicat technique was used to evaluate initial and final setting times of fresh alkali-activated pastes, with experiments performed in an automatic Matest VICATRONIC apparatus (Impact Test Equipment, UK) equipped with a 1.13 mm needle, following the standard testing procedure EN 196-3 [47]. Fresh pastes were poured into a conical frustum mould with a height of 40 mm, top internal diameter of 60 mm, and bottom internal diameter of 70 mm. The initial setting time was determined as the moment in which the separation between the needle and the base plate reached 6 ± 3 mm. The final setting time was recorded once a maximum penetration depth of 0.5 mm into the specimen was achieved.
2.2.4 X-ray diffraction
Before XRD analysis, hydration was stopped via solvent exchange at each curing time interval. Small hardened paste fragments were immersed in isopropanol for 48 h, then dried at 35 °C, and subsequently crushed and sieved to obtain powder fractions < 67 µm. XRD patterns were obtained using a PANalytical X’Pert3 (PANalytical, USA) diffractometer operating in Bragg–Brentano geometry with a Cu Kα radiation source at 45 kV and 40 mA, fitted with a PIXcel-Medipix3 detector. Samples were analysed over the 2θ range of 5–70°, using a step size of 0.02°. Phase identification was carried out using ICDD PDF + 4 (ICDD, USA) and HighScore plus (Malvern Panalytical, UK).
2.2.5 Fourier-transform Infrared spectroscopy
FTIR analysis was performed using a PerkinElmer Frontier FTIR spectrometer (PerkinElmer, USA) coupled with a triglycine sulfate detector. 2 mg of precursor material were mixed and ground together with 200 mg of KBr. The mixed powder was transferred to a 13 mm pellet die and pressed with 10 tonnes of force to a form a transparent pellet which was subsequently tested in the spectrometer. FTIR spectra were collected between 4000 and 400 cm−1, with a resolution of 2 cm−1.
2.2.6 Compressive strength
The compressive strength of alkali-activated mixtures was determined after 7 and 28 days on 50 mm cubes according to the relevant sections of ASTM C109 [48] considering the difference in the nature of the materials. Fresh pastes were poured into moulds and sealed. After 24 h (or as soon as possible thereafter) the cube specimens were demoulded, resealed, and stored in a curing chamber with relative humidity of 45 ± 1% at 20 ± 1 °C until testing age. The strength values for the mixtures at each testing age are reported as the mean value of three tests.
3 Results and discussion
3.1 Reaction kinetics
Figure 4a and b show the heat evolution rates of BOFS-GGBFS blends activated with sodium silicate and with sodium carbonate, respectively. Five stages can be identified in the heat evolution profile of AAMs: pre-induction, induction, acceleration, deceleration, and diffusion period. The pre-induction period during the initial hour of reaction is associated with the dissolution and wetting of particles. This pre-induction signal is not clearly distinguishable within the data in Fig. 4a due to its rapid early onset and the delay in recording the early data points using isothermal calorimetry with external mixing. The induction period follows, characterised by very low heat release, in which dissolution of the precursors proceeds and the initial formation reaction products through precipitation polycondensation occurs. The induction period terminates after a considerable amount of heat is released, signalling the initial acceleration stage. The 0BOFa sample exhibits an acceleration period after ~ 17 h. This behaviour is consistent with previous studies of similar systems [43]. Inclusion of BOFS resulted in a progressive shortening of the induction stage, which became ~ 12.5 h for 20BOFa, and ~ 8 h for 30BOFa. The acceleration peak in these two systems is also reduced in intensity compared to the pure GGBFS mixture. The acceleration-deceleration process is complete within 48 h for all three samples.

Rate of heat evolution (a) and total heat release (b) of BOFS-GGBFS mixtures activated with sodium silicate; rate of heat evolution (c) and total heat release (d) of BOFS-GGBFS mixtures activated with sodium carbonate, measured at 20 °C
Figure 4c illustrates the heat evolution for the mixtures activated with sodium carbonate. 0BOFb shows an initial pre-induction peak during the first hour, followed by a second signal centred around 3 h which can be associated with initial formation of carbonate species [40]. This is followed by a long induction period, interrupted at ~ 35 h by the appearance of low intensity convoluted signals associated with the conversion of formed carbonates, e.g. gaylussite, to stable calcite [43]. The acceleration period occurs around ~ 80 h, forming a wide peak between 80 and 125 h corresponding to the delayed condensation and precipitation of C–A–S–H gels. Extended induction periods in sodium carbonate activated slag binders have been observed in several previous studies [37, 38, 49]. The profiles of 20BOFb and 30BOFb in Fig. 4c similarly include minor peaks after 2–3 h, associated with initial carbonate formation. The subsequent induction period is reduced in duration, ~ 12 h for 20BOFb and ~ 5 h for 30BOFb, compared to 0BOFb. The high intensity and convoluted shape of the initial peak in the 20BOFb* profile differs from that seen in 20BOFb due to the higher reactivity obtained from mechanical processing of BOFS precursor [27].
The cumulative heat is shown in Fig. 4b for all mixtures activated with sodium silicate, and Fig. 4d for those with sodium carbonate. The 0BOFa mixture exhibits the greatest overall heat release after 250 h, with increasing additions of BOF reducing the total heat evolution by 10% and 15% for 20BOFa and 30BOFa respectively. Interestingly, the converse trend is seen using sodium carbonate as the activator. 20BOFa and 20BOFb show similar profiles after the initial 100 h, and release similar amounts of heat (188.8 J/g and 188.9 J/g after 250 h). The combination of highly reactive GGBFS and the limited reactivity of portlandite in the high alkalinity systems, compared with the complete reaction of portlandite and reduced reactivity of GGBFS in the sodium carbonate environment, result in a similar cumulative contribution to the reaction kinetics and evolved heat. The cumulative heat of 20BOFb* reaches ~ 211 J/g, very close to the 217 J/g of 0BOFa, however the slope of the curve appears reduced at this point. After 250 h, 30BOFb has released more heat than 30BOFa, which may mean that the reactivity of portlandite gives a greater contribution to the exothermic profile.
3.2 Mini-slump and setting times
Table 4 shows the mini-slump results of the fresh mixtures. Inclusions of BOFS are seen to increase the workability of the systems activated with sodium silicate via the inclusion of larger particles and a consequent reduction in surface area. The lower reactivity of BOFS, with respect to GGBFS, and the limited solubility of portlandite in high alkalinity systems may also act to increase workability. The same trend is seen in mixtures activated with sodium carbonate, where the lower flow diameter of these samples than the silicate-activated mixes is consistent with the added silicates having a plasticising effect by dispersing the precursor particles [50].
Inclusion of BOFS results in an increasing trend in both initial and final setting times (Table 4) for systems activated with sodium silicate, underpinned by the lower reactivity of BOFS compared to GGBFS. Mix 0BOFb has a setting time of over 3 days, as indicated in the heat evolution profile (Fig. 4b), corroborating that the strength bearing phases do not form during the initial days of reaction [49, 51]. The presence of BOFS is thus noted to be responsible for achieving setting times within a reasonable timeframe. Increasing the BOFS content from 20 to 30 wt.% results in a slight increase in setting time, suggesting that in a sodium carbonate activated system containing some BOFS, the formation of hydration products of GGBFS is not inhibited by the formation of carbonate salts. The shorter setting times of 20BOFb* can be attributed to increased reactivity achieved through mechanical activation of BOFS, which reduces the particle size, allowing portlandite contained within BOFS to react more promptly.
3.3 X-ray diffraction
Figure 5 illustrates the XRD patterns of samples activated with sodium silicate and sodium carbonate after 2 and 28 days of curing. In the 2-day 0BOFa pattern seen in Fig. 5a, the crystalline phases identified are calcium aluminosilicate hydrates (C–A–S–H), calcite (C, CaCO3 PDF #01-086-0174) due to atmospheric carbonation [52], and a hydrotalcite-group phase (HT, Mg4Al2(CO3)(OH)12∙3H2O PDF #01-089-0460). The 28-day 0BOFa pattern shows an increase in C–A–S–H peak intensity and a reduction in the full width at half maximum (FWHM), indicating increased short range ordering and a higher degree of polymerisation, as expected with increasing curing time. C–A–S–H, calcite, and the hydrotalcite-group phase are identified in the 2-day patterns of 20BOFa and 30BOFa, along with AFm-like phases including calcium hemi- (Hc, C4Ac0.5H12, PDF #00-041-0221) and mono-carboaluminate (Mc, C4AcH11 PDF#00-041-0219). The presence of carbonates helps the stabilisation of these AFm phases; the formation of Hc and Mc phases is promoted by available CO32− present in the BOFS as calcite, and may also be influenced by the presence of an analogous AFm-like phase, Fc, in the naturally weathered BOFS (Fig. 3). Remnant precursor peaks identified as portlandite (P, Ca(OH)2 PDF#00-044-1481) show that there is incomplete reaction of the BOFS in high alkalinity environments, with potentially lower reaction kinetics due to its coarse particle size.

XRD patterns of BOFS and GGBFS activated with sodium silicate after a 2 days, and b 28 days of curing, and activated with sodium carbonate after c 2 days, and d 28 days
In contrast, the two main crystalline phases identified in the 2-day 0BOFb pattern shown in Fig. 5c are gaylussite (Ga, Na2Ca(CO3)2∙5H2O, PDF #00-021-0343) and calcite, which are reported in sodium carbonate activated systems [39, 40, 43]. There is minimal formation of C–A–S–H gel and HT present after 2 days. C–A–S–H and HT phases do appear after curing for 28 days, with minor Hc/Mc phases accompanied by a reduction in gaylussite phase fraction. This follows the mechanism proposed by Bernal et al. [39], whereby (i) CO32− released by the activator reacts with the Ca2+ from the dissolution of GGBFS to initially form gaylussite and calcite; (ii) as the reaction proceeds, gaylussite releases Na+ and converts to calcite; (iii) the reduction of CO32− concentration promotes precipitation of C–A–S–H instead of CaCO3 whilst increasing OH− concentration gives a highly alkaline pore solution, (iv) the reaction proceeds similarly to that of NaOH activated slag after 4 days. Identification of secondary carbonate phases in the 2-day 0BOFb XRD pattern supports the interpretation of the 0BOFb calorimetric peaks seen in Fig. 4a at 3 h and 35 h as the formation of these carbonate phases, with the subsequent peak after ~ 4 days due to the polycondensation and precipitation of C–A–S–H and HT phases [39, 43].
The 2-day 20BOFb, 30BOFb, and 20BOFb* XRD patterns in Fig. 5c show the appearance of several reaction products, identified as C–A–S–H, HT, Hc, Mc, calcite, and minor traces of gaylussite. The portlandite phase fraction is diminished compared to that seen in Fig. 5a and b, confirming the favoured reactivity of portlandite in systems of lower alkalinity. Portlandite provides an increase in pH via the release of OH−, which then promotes the dissolution of GGBFS and formation of C–A–S–H gel from dissolved Si and Al species. The formation of AFm phases, Mc and Hc, is beneficial in promoting gaylussite dissolution by driving demand for carbonate, also resulting in precipitation of both amorphous and crystalline CaCO3 polymorphs [41, 42]. The phase evolution from 2 to 28-day in XRD patterns of 20BOFb, 20BOFb*, and 30BOFb seen from Fig. 5c, d shows an increase in peak intensity of C–A–S–H and AFm, especially for Mc, which is expected to be more stable that Hc overtime [53]. The peak intensity of portlandite shows no significant decrease, which may be due to the cessation of further reaction.
3.4 Fourier-transform infrared spectroscopy
FTIR spectra of samples activated with sodium silicate and with sodium carbonate are shown in Fig. 6a and b respectively. Similar bands are found for all samples, indicating that reaction products with comparable nature have formed. The H-OH bending band is seen at 1660 cm−1, associated with partially bonded water after hydration [54]. Bands located in the range 1489–1419 cm−1 and at ~ 870 cm−1 indicate the presence of carbonates, specifically the C-O asymmetrical stretching and asymmetrical bending vibrations. The existence of these bands is consistent with the present of incompletely-crystalline calcium carbonate in different polymorphs, and gaylussite, with specific peaks located at 1450, 874, 855, 713, and 668 cm−1 [49, 55,56,57]. Carbonated hydrotalcite and AFm-like phases (Mc, and Hc) may also contribute to these bands [57]. The band shift observed for 0BOFb at 1489–1419 cm−1 in Fig. 6b with increasing curing time supports the mechanism by which a high initial concentration of gaylussite is formed, which over time is converted to calcite and other polymorphs of CaCO3 [42].

FTIR spectra of BOFS and GGBFS activated with a sodium silicate after 2 days and 28 days, and b activated with sodium carbonate after 2 days and 28 days of curing
The band associated with formation of silicate binding phases appears at 970–950 cm−1, assigned to the asymmetric stretching vibration mode of the Si–O–T (T = Si or Al in tetrahedral coordination) bonds. This band location is in agreement with the C–(A)–S–H structure formed by the activation of slag in alkaline media [58, 59], appearing narrower and sharpened for all samples with increasing curing time as a result of increased structural order of the gel phase as the reaction evolves [60]. The 2-day cured 0BOFb has a broad and rounded peak due to the delay in C–A–S–H gel formation which is identified in the XRD analysis above. A shift of this band towards lower wavenumbers is also recorded in all samples as the crosslinking and the degree of polymerisation of the structure increase due to the incorporation of Al, from 2 to 28 days [49, 61]. The features at ~ 700 cm−1 and 500–400 cm−1 are bands associated with symmetric stretching vibration of Si–O bands, and deformation of SiO4 tetrahedra, respectively. These last bands also become narrower as the reaction proceeds.
3.5 Compressive strength
The compressive strength results are shown in Fig. 7. 0BOFa reaches 44 MPa and 56 MPa after 7 and 28 days respectively. After 7 days 0BOFb exhibits a compressive strength of 33 MPa, whereby the strength bearing phases (C–A–S–H gels) have formed after the initial 4 days of reaction. All other mixtures have strength values between 20 and 35 MPa after 7 days. The respective compressive strength gains for mixtures including BOF between 7 and 28 days are greater than for 0BOFa. 20BOFa and 20BOFb obtain similar strength values, between 25 and 32 MPa after 7 days and 39–44 MPa after 28 days. The strength values of 20BOFb* are comparable to 0BOFb,45 and 46 MPa after 28 days respectively, indicating that the mechanical activation process positively influences the reactivity of BOFS. Further replacement of GGBFS by BOFS (30 wt.%) results in a slightly inferior compressive strength (up to 30–35 MPa). All the studied alkali-activated cements made with BOFS inclusions could potentially qualify for structural applications. It is demonstrated that the use of waste BOFS in GGBFS blends can achieve desirable fresh and mechanical properties when utilising sodium carbonate as a low cost activator.

Compressive strength of specimens based on BOFS and GGBFS and activated with sodium silicate and sodium carbonate, after 7 and 28 days of curing. Error bars represent the standard deviation among three replicate specimens
4 Conclusions
This work has investigated the use of naturally weathered BOFS as a partial replacement for GGBFS in alkali-activated cements produced using two different activator solutions: sodium silicate and sodium carbonate.
The reaction kinetics governing the formation of C–A–S–H are accelerated with naturally weathered BOFS inclusion by exploiting the fast reactivity of portlandite. Sodium carbonate activated-GGBFS has a 4-day induction period, reduced to ~ 10 h with 20 wt.% BOFS. The two-component system including 20 wt.% BOFS with GGBFS, and different activator solutions, resulted in comparable heat evolution.
Workability is affected by the activator type and by BOFS inclusion. The presence of sodium silicate results in higher workability due to a plasticising effect, and BOFS inclusions also increase workability due to the coarser particles and lower reactivity with respect to GGBFS. Setting times also increase with BOFS inclusion when using silicate activators, but BOFS addition accelerates the setting of sodium carbonate activated GGBFS which otherwise does not set until 3 days.
XRD analysis showed that in sodium silicate-activated systems, which have higher relative alkalinity, not all of the portlandite from the naturally weathered BOFS reacted. In sodium carbonate-activated systems the reaction of portlandite is nearly complete, providing an immediate increase in the pH and allowing GGBFS to react and form C–A–S–H gels in the initial stages of reaction. The typical delay observed in the sodium carbonate-activated slag system via the formation of carbonate salts is therefore avoided. This is confirmed also through the analysis of FTIR data, which shows that the only sodium carbonate-based system where C–A–S–H did not significantly form at early age was the sample containing only GGBFS.
Specimens made with 20 wt.% BOFS inclusion obtain compressive strength values of up to 39–45 MPa after 28 days. The beneficial effect of mechanical activation of BOFS is evident in sodium carbonate activated systems, where final strength reaches 45 MPa, comparable to the pure GGBFS sample activated with sodium carbonate.
This work facilitates the use of less GGBFS in combination with sodium carbonate without compromising setting times. After natural weathering, BOFS can be used, as a valuable additive in the acceleration of the reaction of GGBFS. Mechanical processing can further increase the reactivity of BOFS, and ultimately increase the strength of a sodium carbonate-activated BOFS-containing cement to be comparable with a GGBFS-sodium silicate cement. Additions of BOFS offer a new avenue for reduction of the activator dosage in these cements, and the usage of sodium carbonate as an activator for GGBFS, providing a useful valorisation pathway for materials with otherwise very restricted potential applications.
References
Naidu TS, Sheridan CM, van Dyk LD (2020) Basic oxygen furnace slag: review of current and potential uses. Miner Eng. https://doi.org/10.1016/j.mineng.2020.106234
World Steel Association (2022) World crude steel production from 2012 to 2021, Statista. https://www.statista.com/statistics/267264/world-crude-steel-production/?locale=en (Accessed 3 Apr 2024).
World Steel Association (2023) Steel and raw materials. https://worldsteel.org/wp-content/uploads/Fact-sheet-raw-materials-2023.pdf (Accessed 3 Apr 2024).
Habert G, Miller SA, John VM, Provis JL, Favier A, Horvath A, Scrivener KL (2020) Environmental impacts and decarbonization strategies in the cement and concrete industries. Nat Rev Earth Environ 1:559–573. https://doi.org/10.1038/s43017-020-0093-3
Provis JL, Bernal SA (2014) Geopolymers and related alkali-activated materials. Annu Rev Mater Res 44:299–327. https://doi.org/10.1146/annurev-matsci-070813-113515
Fernández-Jiménez A, Cristelo N, Miranda T, Palomo Á (2017) Sustainable alkali activated materials: precursor and activator derived from industrial wastes. J Clean Prod 162:1200–1209. https://doi.org/10.1016/j.jclepro.2017.06.151
Duxson P, Provis JL, Lukey GC, van Deventer JSJ (2007) The role of inorganic polymer technology in the development of ‘green concrete. Cem Concr Res 37:1590–1597. https://doi.org/10.1016/j.cemconres.2007.08.018
Marsh ATM, Velenturf APM, Bernal SA (2022) Circular economy strategies for concrete: implementation and integration. J Clean Prod. https://doi.org/10.1016/j.jclepro.2022.132486
Duxson P, Provis J (2008) Designing precursors for geopolymer cements. J Am Ceram Soc 91:3864–3869. https://doi.org/10.1111/j.1551-2916.2008.02787.x
Zhang N, Wu L, Liu X, Zhang Y (2019) Structural characteristics and cementitious behavior of basic oxygen furnace slag mud and electric arc furnace slag. Constr Build Mater 219:11–18. https://doi.org/10.1016/j.conbuildmat.2019.05.156
Kong Y, Wang P, Liu S (2018) Microwave pre-curing of Portland cement-steel slag powder composite for its hydration properties. Constr Build Mater 189:1093–1104. https://doi.org/10.1016/j.conbuildmat.2018.09.088
Kabay N, Miyan N, Özkan H (2021) Basic oxygen furnace and ground granulated blast furnace slag based alkali-activated pastes: characterization and optimization. J Clean Prod. https://doi.org/10.1016/j.jclepro.2021.129483
Yildirim IZ, Prezzi M (2011) Chemical, mineralogical, and morphological properties of steel slag. Adv Civ Eng. https://doi.org/10.1155/2011/463638
Deng M, Hong D, Lan X, Tang M (1995) Mechanism of expansion in hardened cement pastes with hard-burnt free lime. Cem Concr Res 25:440–448. https://doi.org/10.1016/0008-8846(95)00030-5
Xue Y, Wu S, Hou H, Zha J (2006) Experimental investigation of basic oxygen furnace slag used as aggregate in asphalt mixture. J Hazard Mater 138:261–268. https://doi.org/10.1016/j.jhazmat.2006.02.073
Pathak S, Choudhary R, Kumar A, Damena DT (2019) Feasibility assessment of the use of basic oxygen furnace slag in open graded asphalt courses. Int J Pavement Res Technol 12:664–673. https://doi.org/10.1007/s42947-019-0079-2
Kambole C, Paige-Green P, Kupolati WK, Ndambuki JM, Adeboje AO (2017) Basic oxygen furnace slag for road pavements: a review of material characteristics and performance for effective utilisation in southern Africa. Constr Build Mater 148:618–631. https://doi.org/10.1016/j.conbuildmat.2017.05.036
Mahieux P-Y, Aubert J-E, Escadeillas G (2009) Utilization of weathered basic oxygen furnace slag in the production of hydraulic road binders. Constr Build Mater 23:742–747. https://doi.org/10.1016/j.conbuildmat.2008.02.015
Ding Y-C, Cheng T-W, Liu P-C, Lee W-H (2017) Study on the treatment of BOF slag to replace fine aggregate in concrete. Constr Build Mater 146:644–651. https://doi.org/10.1016/j.conbuildmat.2017.04.164
Sun K, Peng X, Chu SH, Wang S, Zeng L, Ji G (2021) Utilization of BOF steel slag aggregate in metakaolin-based geopolymer. Constr Build Mater. https://doi.org/10.1016/j.conbuildmat.2021.124024
Kareken G, Shon C-S, Tukaziban A, Kozhageldi N, Mardenov M, Zhang D, Kim JR (2023) Geopolymer as a key material to utilize basic oxygen furnace slag (BOFS) as an aggregate. Mater Today Proc. https://doi.org/10.1016/j.matpr.2023.10.093
Branca TA, Pistocchi C, Colla V, Ragaglini G, Amato A, Tozzini C, Mudersbach D, Morillon A, Rex M, Romaniello L (2014) Investigation of (BOF) converter slag use for agriculture in europe. Metall Res Technol 111:155–167. https://doi.org/10.1051/metal/2014022
Wang Q, Yan P (2010) Hydration properties of basic oxygen furnace steel slag. Constr Build Mater 24:1134–1140. https://doi.org/10.1016/j.conbuildmat.2009.12.028
Zhang T, Yu Q, Wei J, Li J, Zhang P (2011) Preparation of high performance blended cements and reclamation of iron concentrate from basic oxygen furnace steel slag. Resour Conserv Recycl 56:48–55. https://doi.org/10.1016/j.resconrec.2011.09.003
Kourounis S, Tsivilis S, Tsakiridis PE, Papadimitriou GD, Tsibouki Z (2007) Properties and hydration of blended cements with steelmaking slag. Cem Concr Res 37:815–822. https://doi.org/10.1016/j.cemconres.2007.03.008
Guo H, Yin S, Yu Q, Yang X, Huang H, Yang Y, Gao F (2018) Iron recovery and active residue production from basic oxygen furnace (BOF) slag for supplementary cementitious materials. Resour Conserv Recycl 129:209–218. https://doi.org/10.1016/j.resconrec.2017.10.027
Singh SK, Jyoti, Vashistha P (2021) Development of newer composite cement through mechano-chemical activation of steel slag. Constr Build Mater 268: 121147. https://doi.org/10.1016/j.conbuildmat.2020.121147
Jawad Ahmed M, Franco Santos W, Brouwers HJH (2023) Air granulated basic oxygen furnace (BOF) slag application as a binder: effect on strength, volumetric stability, hydration study, and environmental risk. Constr Build Mater 367:130342. https://doi.org/10.1016/j.conbuildmat.2023.130342
Sithole T, Okonta F, Freeman N (2020) Mechanical properties and structure of fly ash modified basic oxygen furnace slag based geopolymer masonry blocks. J Solid Waste Technol Manag 46:372–383. https://doi.org/10.5276/JSWTM/2020.372
Lopez Gonzalez PL, Novais RM, Labrincha J, Blanpain B, Pontikes Y (2023) The impact of granulation on the mineralogy of a modified-BOF slag and the effect on kinetics and compressive strength after alkali activation. Cem Concr Compos 140:105038. https://doi.org/10.1016/j.cemconcomp.2023.105038
Lopez Gonzalez PL, Novais RM, Labrincha JA, Blanpain B, Pontikes Y (2019) Modifications of basic-oxygen-furnace slag microstructure and their effect on the rheology and the strength of alkali-activated binders. Cem Concr Compos 97:143–153. https://doi.org/10.1016/j.cemconcomp.2018.12.013
Ben Haha M, Lothenbach B, Le Saout G, Winnefeld F (2011) Influence of slag chemistry on the hydration of alkali-activated blast-furnace slag—part I: effect of MgO. Cem Concr Res 41:955–963. https://doi.org/10.1016/j.cemconres.2011.05.002
San Nicolas R, Cyr M, Escadeillas G (2013) Characteristics and applications of flash metakaolins. Appl Clay Sci 83–84:253–262. https://doi.org/10.1016/j.clay.2013.08.036
Humad AM, Habermehl-Cwirzen K, Cwirzen A (2019) Effects of fineness and chemical composition of blast furnace slag on properties of alkali-activated binder. Materials (Basel). https://doi.org/10.3390/ma12203447
Ibrahim M, Maslehuddin M (2021) An overview of factors influencing the properties of alkali-activated binders. J Clean Prod. https://doi.org/10.1016/j.jclepro.2020.124972
Ouellet-Plamondon C, Habert G (2015) Life cycle assessment (LCA) of alkali-activated cements and concretes. In: Pacheco-Torgal F, Labrincha JA, Leonelli C, Palomo A, Chindaprasirt P (Eds.) Handb. Alkali-Activated Cem. Mortars Concr., Woodhead Publishing, pp. 663–686. https://doi.org/10.1533/9781782422884.5.663.
Yuan B, Yu QL, Brouwers HJH (2017) Time-dependent characterization of Na2CO3 activated slag. Cem Concr Compos 84:188–197. https://doi.org/10.1016/j.cemconcomp.2017.09.005
Adesina A (2020) Influence of various additives on the early age compressive strength of sodium carbonate activated slag composites: an overview. J Mech Behav Mater 29:106–113. https://doi.org/10.1515/jmbm-2020-0011
Bernal SA, Provis JL, Myers RJ, San Nicolas R, van Deventer JSJ (2014) Role of carbonates in the chemical evolution of sodium carbonate-activated slag binders. Mater Struct 48:517–529. https://doi.org/10.1617/s11527-014-0412-6
Bernal SA (2016) Advances in near-neutral salts activation of blast furnace slags. RILEM Tech Lett 1:39–44
Wang J, Lyu X, Wang L, Cao X, Liu Q, Zang H (2018) Influence of the combination of calcium oxide and sodium carbonate on the hydration reactivity of alkali-activated slag binders. J Clean Prod 171:622–629. https://doi.org/10.1016/j.jclepro.2017.10.077
Ke X, Bernal SA, Provis JL (2016) Controlling the reaction kinetics of sodium carbonate-activated slag cements using calcined layered double hydroxides. Cem Concr Res 81:24–37. https://doi.org/10.1016/j.cemconres.2015.11.012
Bernal SA, San Nicolas R, van Deventer JSJ, Provis JL (2016) Alkali-activated slag cements produced with a blended sodium carbonate/sodium silicate activator. Adv Cem Res 28:262–273. https://doi.org/10.1680/jadcr.15.00013
Kaja AM, Melzer S, Brouwers HJH, Yu Q (2021) On the optimization of BOF slag hydration kinetics. Cem Concr Compos 124:104262. https://doi.org/10.1016/j.cemconcomp.2021.104262
Wadsö L (2010) Operational issues in isothermal calorimetry. Cem Concr Res 40:1129–1137. https://doi.org/10.1016/j.cemconres.2010.03.017
Tan Z, Bernal SA, Provis JL (2017) Reproducible mini-slump test procedure for measuring the yield stress of cementitious pastes. Mater Struct 50:1–12. https://doi.org/10.1617/s11527-017-1103-x
British Standards Institution (2016) BS EN 196-3:2016 Methods of testing cement - Part 3: Determination of setting times and soundnes
ASTM C109/C109M-20 (2020) Standard test method for compressive strength of hydraulic cement mortars
Fernández-Jiménez A, Puertas F (2003) Effect of activator mix on the hydration and strength behaviour of alkali-activated slag cements. Adv Cem Res 15:126–136. https://doi.org/10.1680/adcr.13.3.115.39288
Kashani A, Provis JL, Qiao GG, van Deventer JSJ (2014) The interrelationship between surface chemistry and rheology in alkali activated slag paste. Constr Build Mater 65:583–591. https://doi.org/10.1016/j.conbuildmat.2014.04.127
Fernandez-Jimenez A, Puertas F (2001) Setting of alkali-activated slag cement. Influence of activator nature. Adv Cem Res 13:115–121. https://doi.org/10.1680/adcr.13.3.115.39288
Bernal SA, Provis JL, Walkley B, San Nicolas R, Gehman JD, Brice DG, Kilcullen AR, Duxson P, Van Deventer JSJ (2013) Gel nanostructure in alkali-activated binders based on slag and fly ash, and effects of accelerated carbonation. Cem Concr Res 53:127–144. https://doi.org/10.1016/j.cemconres.2013.06.007
Georget F, Lothenbach B, Wilson W, Zunino F, Scrivener KL (2022) Stability of hemicarbonate under cement paste-like conditions. Cem Concr Res 153:106692. https://doi.org/10.1016/j.cemconres.2021.106692
Ismail I, Bernal SA, Provis JL, Hamdan S, van Deventer JSJ (2013) Microstructural changes in alkali activated fly ash/slag geopolymers with sulfate exposure. Mater Struct 46:361–373. https://doi.org/10.1617/s11527-012-9906-2
Puertas F, Palacios M, Vázquez T (2006) Carbonation process of alkali-activated slag mortars. J Mater Sci 41:3071–3082. https://doi.org/10.1007/s10853-005-1821-2
Li N, Farzadnia N, Shi C (2017) Microstructural changes in alkali-activated slag mortars induced by accelerated carbonation. Cem Concr Res 100:214–226. https://doi.org/10.1016/j.cemconres.2017.07.008
Jones GC, Jackson B (1993) Infrared transmission spectra of carbonate minerals. Springer, Dordrecht
García-Lodeiro I, Fernández-Jiménez A, Blanco MT, Palomo A (2008) FTIR study of the sol–gel synthesis of cementitious gels: C-S–H and N–A–S–H. J Sol-Gel Sci Technol 45:63–72. https://doi.org/10.1007/s10971-007-1643-6
Yu P, Kirkpatrick RJ, Poe B, McMillan PF, Cong X (1999) Structure of calcium silicate hydrate (C-S-H): near-, mid-, and far-infrared spectroscopy. J Am Ceram Soc 82:742–748. https://doi.org/10.1111/j.1151-2916.1999.tb01826.x
Walkley B, Nicolas RS, Sani M-A, Rees GJ, Hanna JV, van Deventer JSJ, Provis JL (2016) Phase evolution of C-(N)-A-S-H/N-A-S-H gel blends investigated via alkali-activation of synthetic calcium aluminosilicate precursors. Cem Concr Res 89:120–135
Ismail I, Bernal SA, Provis JL, San Nicolas R, Hamdan S, van Deventer JSJ (2014) Modification of phase evolution in alkali-activated blast furnace slag by the incorporation of fly ash. Cem Concr Compos 45:125–135. https://doi.org/10.1016/j.cemconcomp.2013.09.006
Acknowledgements
This research study was carried out in the framework of the “By-products for sustainable concrete in the urban environment” (URBCON) project, funded by the Interreg North-West Europe Programme (NWE725) under the EU Cohesion Policy and financed by the European Regional Development Fund (ERDF). The authors also express their gratitude towards Ecocem Benelux B.V. and ArcelorMittal (Belgium) for providing the GGBFS and BOFS used in this study, and PQ Silicates (UK) for generously donating the sodium silicate.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest associated with this research. Co-author JLP holds the role of Past Editor-in-Chief for the journal Materials & Structures, but was not involved in any way in the review or editorial processes for this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stefanini, L., Walkley, B. & Provis, J.L. Basic oxygen furnace (BOF) slag as an additive in sodium carbonate-activated slag cements. Mater Struct 57, 153 (2024). https://doi.org/10.1617/s11527-024-02425-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-024-02425-8