
Abstract
The sorption behavior of Cs to Mg-containing calcium silicate hydrate (C–S–H) was examined to consider C–S–H formation under the water-saturated conditions around a radioactive waste repository. The amount of Cs sorption was slightly lower for Mg-containing C–S–H than for C–S–H, because Mg leads to a decrease in sorption sites by facilitating the polymerization of silicate chains. However, the apparent sorption distribution coefficient Kd of Cs on C–S–H with a Ca/Si molar ratio of 0.8 and Mg contents up to 20% was estimated to be 3.4–6.1 mL/g. Furthermore, the amount of Cs sorption was not decreased by the presence of Ba. The apparent Kd obtained in this study was about 10 times higher than that conventionally estimated for host rocks in the plutonic repository, indicating that C–S–H retains the effect of Cs immobilization even when it contains Mg.
Graphical abstract

Similar content being viewed by others
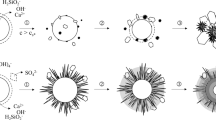

Introduction
The construction of a radioactive waste repository requires a large amount of cementitious materials. Such cementitious materials contain several alkaline species, such as Ca, Na, and K ions; therefore, the pH of the groundwater around the repository rises to approximately pH 11–13 over a long period of time [1, 2]. Under these highly alkaline conditions, silicate minerals in the host rock dissolve and react with silicate ions and Ca ions leached from the cement and form calcium silicate hydrate (C–S–H), a main component of cementitious materials, as a secondary mineral under the water-saturated conditions in flow paths [3,4,5,6].
C–S–H consists of a Ca–O central layer flanked by silicate chains of silica tetrahedra, similar in structure to tobermorite and jennite [7, 8]. The structure and characteristics of C–S–H are strongly dependent on the Ca/Si molar ratio. C–S–H in cement is generally stable at a Ca/Si molar ratio of 1.6, whereas C–S–H formed under water-saturated conditions around a repository is stable at a Ca/Si molar ratio of 0.8 [9].
Some research groups have reported that such C–S–H can sorb significant amounts of radionuclides. Ochs et al. reported that a decrease in pH coincides with a decrease in the Ca/Si ratio of C–S–H, which results in a corresponding increase in surface sites with strong affinity for Cs and, thus an increase in the distribution coefficient, Kd [10]. Missana et al. examined the retention behavior of Cs, Ba, and Ni in C–S–H phases. They showed that the apparent Kd increases with a lower Ca/Si ratio in C–S–H because the surface charge of C–S–H at lower Ca/Si ratios (0.8 and 1.0) is negative, based on ζ-potential measurements [11,12,13]. Seki et al. also reported that C–S–H effectively retards the migration of Cs and Sr from the early stages (day 1) of its formation [14].
However, the incorporation of other ions, such as elements in groundwater or host rock, will change the structure of C–S–H and may also affect the sorption properties. Minato et al. studied the effect of Al incorporation into C–S–H on its structure and the sorption behavior of radionuclides [15]. They reported that the amount of Cs sorption to calcium aluminosilicate hydrate (C–A–S–H) was significantly larger than that to C–S–H due to a similar mechanism as that reported by Richardson [7], i.e., the isomorphic substitution of Al(III) at the Si(IV) position results in a charge deficiency and cations (Na+ and K+) are introduced to compensate for the negative charge. Following such studies, this study has focused on the sorption behavior of Cs to Mg-containing C–S–H, considering that Mg is abundant in groundwater and silicate minerals of the host rock. The influence of Ba, which is generated by the decay of Cs-134 and Cs-137, on the sorption of Cs to Mg-containing C–S–H was also examined. In this study, C–S–H is simulated as a secondary mineral under the water-saturated conditions in flow paths; therefore, the preparation process of C–S–H does not include any drying process.
Materials and methods
C–S–H phases were synthesized at Ca/Si molar ratios (C/S) of 0.4, 0.8, 1.2, and 1.6. To synthesize the C–S–H phases, specified amounts of CaO (99.9%, Fujifilm Wako Pure Chemical), fumed silica (AEROSIL 300, specific surface area of 300 m2/g, Nippon Aerosil), and ultrapure water were mixed in a Nalgene centrifuge tube (polypropylene copolymer) inside a nitrogen-filled glove box. The total weight of the solid phase was 1.5 g, and the total volume of the liquid phase was 30 mL (20 g/L). For the Mg-containing C–S–H samples, 10 or 20% of CaO was replaced with Mg(NO3)2·6H2O (Guaranteed Reagent, Fujifilm Wako Pure Chemical) at (Ca + Mg)/Si molar ratios of 0.4, 0.8, 1.2, and 1.6. In this case, the pH of the samples was adjusted with NaOH (Wako 1st Grade, Fujifilm Wako Pure Chemical) because the pH was decreased by the addition of Mg(NO3)2·6H2O.
The sorption experiment was conducted under the co-precipitation conditions referred to by Seki et al. [14], i.e., nuclides were simultaneously added to the C–S–H samples during synthesis. CsCl (Wako Special Grade, Fujifilm Wako Pure Chemical) was added as the nuclide to the C–S–H and Mg-containing C–S–H samples during the synthesis to produce a Cs concentration of 1.0 mM. For the case of Ba presence, both CsCl and BaCl2·2H2O (Guaranteed Reagent, Fujifilm Wako Pure Chemical) were each added at a concentration of 0.5 mM to the C–S–H and Mg-containing C–S–H samples with C/S 0.8. The detailed preparation conditions are listed in Table 1.
These samples were shaken on a shaker set to 120 strokes/min at 25 °C, and the curing (sorption) periods were 7 or 42 days. After the given curing times, the slurries were separated into solid and liquid phases by centrifugation at 7500 rpm (6360 G) for 10 min and filtration through a 0.20 µm membrane filter. The solid phase was weighed to determine the water content of each sample. Raman spectra were obtained immediately after separation using a Raman spectrometer (NRS-3300, JASCO). Identification of the Raman spectra peaks was conducted with reference to the peak assignments reported by Garbev et al. [16] and Ortaboy et al. [17]. The pH and concentrations of Ca, Si, Mg, Cs, and Ba in the liquid phase were also measured. The concentrations of Ca, Si, Mg, and Ba were analyzed using inductively coupled plasma atomic emission spectroscopy (SPS7800, SII Nano Technology), and that of Cs was measured using atomic absorption spectroscopy (iCE 3300, Thermo Fisher Scientific).
The sorption ratio and the apparent sorption distribution coefficient (apparent Kd) of Cs to C–S–H and Mg-containing C–S–H after 7 and 42 days are estimated. The sorption ratio is obtained using Eq. 1:
where C0 is the initial amount of the substance (mmol) and C1 is the amount of the substance in the liquid phase at a given time (mmol). In addition, the apparent sorption distribution coefficient is referred to in this study, i.e., the distribution ratio, considering the possibility that Cs has not reached equilibrium. The apparent sorption distribution coefficient, apparent Kd (mL/g), for Cs can be defined as:
where Ms is the amount of the substance in the solid phase (mol), ML is the amount of the substance in the liquid phase, V is the volume of the liquid phase (mL), and m is the mass of the solid phase (g), respectively.
Results and discussion
Characterization of synthesized C–S–H
Figure 1 shows the C/S and Mg contents of the synthesized C–S–H in the absence of Cs and Ba after 7 and 42 days. Note that the C–S–H samples evaluated in this study did not undergo a drying process, and that the C–S–H phases contain water and are gel-like.

Characterization of synthesized C–S–H in the absence of Cs and Ba with respect to the C/S and Mg content; a pH of the liquid phase, b weight of the solid phase, and c, d Raman spectra of the solid phase after 42 days with 0% and 20% Mg
The pH of the liquid phases was higher with C/S increased from 9.4 to 13.0 and was slightly decreased with Mg content, except for C/S 0.4 after 7 days (Fig. 1a). This trend in pH is similar to the amount of Ca added, which suggests that the initial amount of Ca addition affects the final pH of the liquid phase. The pH trend for C/S 0.4 after 7 days was different from the others and increased from 10.1 to 11.0 with the Mg content; this was because the C–S–H structure was unstable during to the early stages of C–S–H formation.
In Fig. 1b, the weight of the solid phases also tended to increase with C/S, which indicates that the amount of water retained in the structure decreases with the interlayer distance of C–S–H at higher C/S, which is consistent with the report by Geng et al. [18]. In addition, the weight of the solid phases decreased with an increase in the Mg content. This may be due to a decrease in interlayer water within the C–S–H samples as the polymerization of the silicate chains in C–S–H progresses with the addition of Mg.
Figure 1c, d shows the Raman spectra of the solid phases with 0% and 20% Mg in the absence of Cs and Ba after 42 days. Similar Raman spectra were obtained with the addition of 10% and 20% Mg and the curing time of 7 and 42 days; therefore, the results for 0% and 20% are shown in Fig. 1. All of the spectra shown in Fig. 1c, d were normalized with respect to the peak of the Si–O–Si symmetric bending of Q2 at 671 cm−1 (Q2-SB), which appeared in all of the samples. The ν3(SiO4) asymmetric stretching of Q2 Si–O–(Ca) or Si–O–(H) at 1015 cm−1 (Q2-ASS) and the ν3(SiO4) symmetric stretching of Q2 at 1010 cm−1 (Q2-SS) were also observed in all samples. Besides, the ν1(SiO4) symmetric stretching of Q1 at 870–900 cm−1 (Q1-SS) appeared only at C/S 1.2 and 1.6, which indicates that the degree of polymerization of silicate chains in C–S–H decreased under higher C/S conditions. The lattice vibrations of Ca–O polyhedra at 300–350 cm−1 (Ca–O) were also observed in the C/S 1.2 and 1.6 samples. This indicates that an excess of Ca for C–S–H formation precipitated as portlandite (Ca(OH)2) in C–S–H with C/S 1.2 and 1.6 samples under high pH conditions. Furthermore, the peak for the symmetric stretching of Q3 at 1040 cm−1 (Q3-SS) appeared in all 20% Mg-containing C–S–H samples for all C/S, which indicates that the polymerization of silicate chains is further enhanced by the addition of Mg. This is consistent with the results for the weight of the solid phases shown in Fig. 1b, where Mg enhances the polymerization of silicate chains and decreases the structural water in C–S–H. In addition, Q2 of magnesium silicate hydrate (M–S–H) at 870 cm−1 was confirmed on Mg 20% of C/S 1.6, indicating that Mg-containing C–S–H in this study may be a complex of C–S–H and small amount of M–S–H.
Sorption behavior of Cs to C–S–H
The sorption ratio and the apparent Kd of Cs to C–S–H and Mg-containing C–S–H after 7 and 42 days are shown in Fig. 2. The effect of the curing period on the sorption ratio and apparent Kd varied with C/S and no clear trend was evident. However, the sorption amount for all conditions, except C/S 0.4, was not significantly different with the curing period, which indicates that Cs sorption to C–S–H or Mg-containing C–S–H had almost reached an apparent steady state within 7 days. On the other hand, for C/S 0.4, the sorption amount for 42 days with 10% and 20% Mg content was lower than that for 7 days. This may be due to the degree of polymerization that progressed with time and a decrease in the silanol groups (–SiO−) of C–S–H, which contribute to the sorption of Cs.

Effects of curing period, C/S, and Mg content on the sorption behavior; a sorption ratio and b apparent Kd of Cs for C–S–H
The amount of Cs sorption decreased with an increase in C/S for both 7 and 42 days of curing time. This is because the surface charge of lower C/S is negative and therefore Cs is electrostatically attracted, i.e., Ca ions sorb onto the silanol groups of silicate chains and take the form -SiOCa+ at high C/S, as reported by Lothenbach and Nonat [19], which inhibits the sorption of cationic Cs at a positive surface potential of C–S–H. The amount of Ca remaining in the liquid phase, i.e., not incorporated into the solid phase, was slightly higher in the Cs addition sample than that in the blank sample (Table S2). In addition, the sorption ratio and apparent Kd of the samples with C/S 1.2 and 1.6 were similar, which may be due to the excess Ca during C–S–H formation at higher C/S forming portlandite, and the C/S of the final C–S–H is slightly lower than 1.6. Lothenbach and Nonat also showed that the formation of portlandite increases at C/S 1.5 and above.
The amount of Cs sorption decreased with an increase in the Mg content for all conditions except after 7 days with C/S 0.4, whereas high sorption distribution coefficients of apparent Kd = 3.4–6.1 mL/g were still confirmed in C–S–H with C/S 0.8 and Mg content up to 20%. The decrease in the sorption capacity by Mg addition is due to the incorporation of Mg into C–S–H in the tetrahedral or octahedral form, which leads to the promotion of silicate chain polymerization in the C–S–H structure. Cs is sorbed to C–S–H primarily through the silanol groups (–SiO−) in the silicate chains of C–S–H; therefore, the accelerated polymerization of these silicate chains by the presence of Mg results in a decrease in the available sorption sites.
Figure 3 shows the sorption behavior of Cs and Ba on C–S–H with C/S 0.8 when both are present at 0.5 mM. Higher Ba sorption was observed for both 7 and 42 days, which may be due to the stronger interaction of C–S–H with Ba, a divalent cation, while Cs is a monovalent cation, which is consistent with the results for Cs and Sr sorption reported by Seki et al. [14]. A slight decrease in Cs sorption was observed for 7 days in the presence of Ba; however, no significant difference was confirmed for 42 days with or without Ba (Figs. 2 and 3). The slight decrease in Cs sorption for 7 days may be due to the unstable structure of C–S–H due to the insufficient curing time of C–S–H, as described in 3.1. In addition, the Nuclear Waste Management Organization of Japan has reported that the sorption distribution coefficient of Cs for host rocks in the plutonic repository is 0.04 mL/g up to 1000 years after repository closure and 0.4 mL/g after more than 1000 years [20]. All apparent Kd values for Cs on C–S–H or Mg-containing C–S–H in this study were higher than those of the host rocks in the plutonic repository. It can be assumed that the sorption capacity of C–S–H for Cs is maintained, even Mg-containing C–S–H and in the presence of Ba, and is expected to retard the migration of Cs through C–S–H.

Effect of the presence of Ba on the sorption of Cs on C–S–H and Mg-containing C–S–H at C/S 0.8; a sorption ratio and b apparent Kd of Cs (red) and Ba (black)
Conclusion
The sorption behavior of Cs to Mg-containing C–S–H was evaluated experimentally with consideration for the incorporation of Mg, which changes the C–S–H structure and affects the sorption behavior of Cs to C–S–H around a radioactive waste repository. C–S–H preparation and sorption experiments were conducted without drying processes to simulate the C–S–H form as a secondary mineral under the water-saturated conditions around a repository. Raman spectra of the solid phases indicated that the incorporation of Mg into the C–S–H structure reduces the Cs sorption sites by facilitating the polymerization of silicate chains. The sorption ratio and the apparent sorption distribution coefficient, apparent Kd, for Cs decreased slightly with an increase in the Mg content of C–S–H. However, a high sorption ratio and apparent Kd were confirmed at Mg contents up to 20%, Kd = 5.5–10.2 mL/g for C/S 0.8. Moreover, the amount of Cs sorption was not decreased by the presence of Ba in Cs. The apparent Kd for all the C–S–H samples examined in this study exceeded the sorption distribution coefficient of 0.04–0.4 mL/g for host rocks in the plutonic repository, which indicates that C–S–H contributes to the immobilization of Cs without decreasing its sorption performance, even when C–S–H contains Mg and also Ba. C–S–H thus maintains the sorption capacity of Cs, even when it contains up to 20% Mg and in the presence of Ba; therefore, C–S–H is expected to retard the migration of Cs by sorption around radioactive waste disposal sites.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
A. Atkinson, N.M. Everitt, R.M. Guppy, Time Depndence of pH in a Cementitious Repository, MRS Online Proceedings Library (OPL) 127 (1988).
U.R. Berner, Evolution of pore water chemistry during degradation of cement in a radioactive waste repository environment. Waste Manage. 12, 201–219 (1992)
M.C. Braney, A. Haworth, N.L. Jefferies, A.C. Smith, A study of the effects of an alkaline plume from a cementitious repository on geological-materials. J. Contam. Hydrol. 13, 379–402 (1993)
E.B. Moyce, C. Rochelle, K. Morris, A.E. Milodowski, X. Chen, S. Thornton, J.S. Small, S. Shaw, Rock alteration in alkaline cement waters over 15 years and its relevance to the geological disposal of nuclear waste. Appl. Geochem. 50, 91–105 (2014)
C.A. Rochelle, A.E. Milodowski, K. Bateman, E.B.A. Moyce, A. Kilpatrick, A long-term experimental study of the reactivity of basement rock with highly alkaline cement waters: reactions over the first 15 months. Mineral. Mag. 80(6), 1089–1113 (2018)
J. Wilson, K. Bateman, Y. Tachi, The impact of cement on argillaceous rocks in radioactive waste disposal systems: a review focusing on key processes and remaining issues. Appl. Geochem. 130, 104979 (2021)
I.G. Richardson, Tobermorite/jennite- and tobermorite/calcium hydroxide-based models for the structure of C-S–H: applicability to hardened pastes of tricalcium silicate, β-dicalcium silicate Portland cement, and blends of Portland cement with blast-furnace slag, metakaolin, or silica fume. Cem. Concr. Res. 34(9), 1733–1777 (2004)
I.G. Richardson, The calcium silicate hydrates. Cem. Concr. Res. 38(2), 137–158 (2008)
G.M.N. Baston, A.P. Clacher, T.G. Heath, F.M.I. Hunter, V. Smith, S.W. Swanton, Calcium silicate hydrate (C–S–H) gel dissolution and pH buffering in a cementitious near field. Mineral. Mag. 76(8), 3045–3053 (2012)
M. Ochs, I. Pointeau, E. Giffaut, Caesium sorption by hydrated cement as a function of degradation state: experiments and modelling. Waste Manage. 26(7), 725–732 (2006)
T. Missana, M. Garcia-Gutierrez, U. Alonso, O. Almendros-Ginesta, Nickel retention by calcium silicate hydrate phases: Evaluation of the role of the Ca/Si ratio on adsorption and precipitation processes. Appl. Geochem. 137, 105197 (2022)
T. Missana, M. Garcia-Gutierrez, M. Mingarro, U. Alonso, Analysis of barium retention mechanisms on calcium silicate hydrate phases. Cem. Concr. Res. 93, 8–16 (2017)
T. Missana, M. García-Gutiérrez, M. Mingarro, U. Alonso, Comparison between cesium and sodium retention on calcium silicate hydrate (C–S–H) phases. Appl. Geochem. 98, 36–44 (2018)
T. Seki, R. Tamura, T. Chida, Y. Niibori, Sorption behavior of cesium and strontium during the formation process of calcium silicate hydrate as a secondary mineral under the condition saturated with groundwater. MRS Adv. 8, 224–230 (2023)
D. Minato, S. Watanabe, S. Harasawa, K. Yamada, Adsorption behavior of Cs for calcium-aluminosilicate-hydrate. Cem. Sci. Concr. Technol. 69(1), 53–60 (2015)
K. Garbev, P. Stemmermann, L. Black, C. Breen, J. Yarwood, B. Gasharova, Structural features of C-S–H(I) and its carbonation in air-A raman spectroscopic study Part I: Fresh Phases. J. Am. Ceram. Soc. 90(3), 900–907 (2007)
S. Ortaboy, J. Li, G. Geng, R.J. Myers, P.J.M. Monteiro, R. Maboudian, C. Carraro, Effects of CO2 and temperature on the structure and chemistry of C–(A–)S–H investigated by Raman spectroscopy. RSC Adv. 7, 48925–48933 (2017)
G. Geng, R.J. Myers, M.J.A. Qomi, P.J.M. Monteiro, Densification of the interlayer spacing governs the nanomechanical properties of calcium-silicate-hydrate. Sci. Rep. 7, 10986 (2017)
B. Lothenbach, A. Nonat, Calcium silicate hydrates: solid and liquid phase composition. Cem. Concr. Res. 78, 57–70 (2015)
Nuclear Waste Management Organization of Japan (NUMO), The NUMO Pre-siting SDM-Based Safety Case, in: NUMO-TR-21–01 (Ed.) 2021.
Acknowledgments
This work was supported by KAKENHI Grants-in-Aid (Nos. 21H04664 and 22K14627) from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Data collection and analysis were performed by Ryota Oasa. The first draft of the manuscript was written by Tsugumi Seki, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seki, T., Oasa, R., Chida, T. et al. Sorption behavior of cesium ions to Mg-containing calcium silicate hydrate in a co-precipitation process. MRS Advances (2024). https://doi.org/10.1557/s43580-023-00757-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1557/s43580-023-00757-1