
Abstract
Materials for scintillator radiation detectors need to fulfill a diverse set of requirements such as radiation hardness and highly specific response to incoming radiation, rendering them a target of current materials design efforts. Even though they are amenable to cutting-edge theoretical spectroscopy techniques, surprisingly many fundamental properties of scintillator materials are still unknown or not well explored. In this work, we use first-principles approaches to thoroughly study the optical properties of four scintillator materials: NaI, LaBr3, BaI2, and SrI2. By solving the Bethe–Salpeter equation for the optical polarization function we study the influence of excitonic effects on dielectric and electron-energy loss functions. This work sheds light into fundamental optical properties of these four scintillator materials and lays the ground-work for future work that is geared toward accurate modeling and computational materials design of advanced radiation detectors with unprecedented energy resolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
I. INTRODUCTION
Scintillator radiation detectors have many important applications in the context of high-energy physics, medicine, as well as homeland security.1,2 Examples of inorganic crystal scintillators used for gamma or x-ray detection include halides, oxides, and chalcogenides,2,3 whereas plastic scintillators are often used for neutron/gamma differentiation.4 Developing more accurate and faster radiation detectors requires successful materials design: certain applications, such as radioactive isotope identification, require particularly high-energy resolution,5 i.e., very high specificity of the optical response depending on the energy of incoming radiation. This is currently one of the major design criteria used in the search for new scintillator materials that also need to withstand incoming radiation and, at the same time, provide an easily detectable, optical response.
Sodium iodide (NaI) has been traditionally used in many devices, due ease of fabrication and low cost. To achieve higher energy resolution, new materials are currently investigated, such as Ce-doped lanthanum bromide (LaBr3),6 Eu-doped strontium iodide (SrI2),7,8 and undoped cesium hafnium chloride (Cs2HfCl6).9 Lately, co-doping of scintillators has also proven to be a successful route to improved scintillation properties.10–14
Unfortunately, the complex physics underlying the energy resolution of scintillators presently is still only understood at a qualitative level. Better fundamental, quantitative insight is needed, to enable targeted materials design for scintillators. Furthermore, the length and time scales involved in the entire scintillation mechanism dictate that multiscale modeling has to be used to develop a fully comprehensive picture. Existing models depend on external parameters, such as dielectric functions, stopping power, migration barriers, defect properties, polaron formation, and migration, as well as nonradiative recombination rates.15–21 Another example is the Monte-Carlo model of gamma-ray response that requires accurate knowledge of the electron-energy loss function.22
In principle, all these quantities can be deduced either from experiment or from first-principles modeling. Both the exponential growth of computational resources as well as development of sophisticated and predictive computational approaches renders the second option very attractive. Modern first-principles and theoretical spectroscopy techniques are clearly advanced enough to thoroughly study some of the mechanisms at play. While we and others started to apply these techniques to scintillator materials,12,23–34 in most cases, even basic quantities such as the dielectric function, optical absorption, or electron-energy loss function are still unknown. The influence of quasiparticle effects and excitons, for instance, is very well studied, e.g., for transparent conducting oxides,35–39 but it is largely unknown for scintillator materials. Experiments alone cannot always mitigate this situation: LaBr3 is an example for a material where measuring the optical properties is particularly difficult because high-quality, optically polished crystals are practically unavailable.40
In some cases, thorough experimental and theoretical investigations, targeted at studying the role of excitonic effects, are currently underway: Ucer et al. presented a detailed study on time-resolved pump-probe experiments on pristine and Tl-doped CsI and concluded that self-trapped excitons (STEs) are only present in the former case and that electrons and holes rapidly become trapped on Tl atoms.41 On the other hand, the scintillation mechanism of undoped, recently discovered9 cesium hafnium chloride (Cs2HfCl6) is believed to consist of luminescence from an STE consisting of a hole centered at a Cl-dimer and an electron at a Hf d-orbital.42 More work is needed to quantitatively model these processes and to incorporate this information into multiscale models that can achieve computational materials design of scintillator materials.
In this light, it is the goal of our research to employ and develop cutting-edge theoretical spectroscopy techniques to establish fundamental knowledge. Here we apply many-body perturbation theory to scintillator materials, to provide predictions for optical properties, such as dielectric functions, for NaI, LaBr3, BaI2, and SrI2. We disentangle the influence of excitonic effects by comparing to the independent-particle approximation, and we provide quantitative insight into optical anisotropy via the complex frequency-dependent dielectric tensor. Furthermore, in the present work, we aim to alleviate the absence of experimental data for electron-energy loss spectra and we discuss trends across the different scintillator materials. At the same time, by providing detailed insight into free excitons in scintillator materials, we lay the ground-work for studying STEs in more complicated scintillator materials in the future.
The remainder of this work is structured as follows: in Sec. II the theoretical framework and computational approach is summarized briefly. Results for optical properties and electron-energy loss functions are presented in Sec. III. Section IV summarizes and concludes our work.
II. THEORETICAL AND COMPUTATIONAL APPROACH
In this work, we use density functional theory43,44 (DFT) to compute Kohn–Sham states and eigenvalues as starting electronic structure for many-body perturbation theory. We use the local-density approximation45 to describe exchange and correlation for SrI2 and the generalized-gradient approximation by Perdew, Burke, and Ernzerhof46 (PBE) is used for the other three materials. The electron–ion interaction was described within the projector-augmented wave method47 and a plane-wave basis48 is used for the wave function expansion. All calculations are carried out within the Vienna Ab-initio Simulation Package49–51 and the Bethe–Salpeter equation (BSE) implementation discussed in Refs. 52 and 53.
A. Atomic geometries
In this work we adopt experimental atomic geometries for NaI and SrI2 as described in Ref. 29 and for LaBr3 as described in Ref. 28. NaI crystallizes in the rocksalt structure in equilibrium, SrI2 belongs to the Pbca space group29 (no. 61 of the International Tables of Crystallography54), and LaBr3 adopts a hexagonal lattice (P63/m, no. 176 in Ref. 54).
For orthorhombic BaI2 (space group Pnma, no. 62 in Ref. 54), we fully relaxed the atomic positions until all Hellman–Feynman force components on any atoms were less than 0.02 eV/Å. This leads to a = 9.01 Å, b = 5.43 Å, and c = 10.87 Å. The Ba atom and both iodine atoms occupy the 4c Wyckoff site with x = 0.244, z = 0.112 (for Ba), x = 0.142, z = 0.425 (for the first I), and x = 0.020, z = 0.836 (for the second I). All unit cells for the materials studied in this work are visualized in Fig. 1.

Unit cells of the materials studied in this work: (a) NaI, (b) LaBr3, (c) BaI2, and (d) SrI2.
B. Single-particle electronic structure
DFT does not provide an accurate description of the electronic structure, since it neglects quasiparticle effects, which manifests itself for instance in the infamous band-gap underestimation.57 In the present work, we need to include quasiparticle effects to accurately describe both band gaps and electronic structure as the foundation of a reliable description of optical properties. Here, we use a scissor operator Δ that shifts all conduction bands to higher energies. The shift Δ is determined using band gaps available in the literature for NaI, LaBr3, and SrI2 (see Table I). For BaI2 we carry out generalized-gradient approximation (GGA) + G0W0 calculations of quasiparticle energies using a Γ-centered 3 × 6 × 3 Monkhorst–Pack58 (MP) k-point grid and 1024 bands (out of which 54 are occupied) to achieve convergence of the screened interaction.59 This yields a band gap of 4.98 eV for BaI2 (see Table I).
In addition, local or semilocal approximations to exchange and correlation suffer from self-interaction errors. This error particularly affects localized orbitals such as d and f states. Hence, we compute the electronic structure of LaBr3 within the rotationally invariant GGA + U approach,60 to correct for the La 4f states that incorrectly appear within the band gap in DFT-PBE calculations.28 Here we use U = 11.0 eV and J = 0.68 eV, acting on these La 4f electrons.61
C. Two-particle excitations: excitons
To achieve an accurate description of optical properties (including excitonic and local-field effects) from first principles, we solve the BSE for the optical polarization function.57 This allows us to include two-particle (electron–hole) excitations in the description of the dielectric function. The underlying DFT Kohn–Sham states are used to compute the optical transition matrix elements in the longitudinal approximation49 and the statically screened Coulomb attraction as well as the unscreened exchange terms that determine the excitonic Hamiltonian.52,53,57 After constructing this Hamiltonian, the dielectric function is computed from it using a time-propagation technique.62 The model dielectric function of Bechstedt et al.63 is used to compute the screened Coulomb interaction W in the excitonic Hamiltonian, using the static electronic dielectric constants obtained on DFT level (see Table I).
Such calculations of converged results for optical properties across a large energy range are numerically very challenging, since different competing requirements have to be fulfilled: the sampling of the Brillouin zone needs to be fine enough to converge the onset of the optical absorption spectrum, but at the same time a large number of conduction bands is required to compute optical properties for high-energy optical transitions. Due to the large computational cost of the BSE approach, we employ the BSE cutoff energies (maximum noninteracting electron–hole pair energy taken into account) and MP k-point grids as listed in Ref. 64. Convergence is improved by displacing each grid by a small random vector to lift degeneracies that occur for unshifted MP meshes.
For computing the electron-energy loss function, we also need to converge the real part of the dielectric function (i.e., related to the imaginary part via a Kramers–Kronig relation) at low photon energies. This is achieved by including optical transitions with transition energies between the BSE cutoff and 200 eV on the level of DFT (as described in Refs. 35 and 65).
III. OPTICAL PROPERTIES
A. Dielectric functions
Using the computational framework described in Sec. II, we computed the optical properties (dielectric functions) of ideal crystals of NaI (see Fig. 2), LaBr3 (see Fig. 3), BaI2 (see Fig. 4), and SrI2 (see Fig. 5). Due to the absence of experimentally determined (for instance via ellipsometry) dielectric functions, these results represent highly accurate predictions. In these figures, we compare the results within the independent-quasiparticle approximation, i.e., where quasiparticle effects on the Kohn–Sham eigenvalues are taken into account using the DFT + Δ scheme, to the solution of the BSE, that, in addition, includes excitonic and local-field effects. This allows us to draw conclusions about the influence of excitonic effects in the following.

Imaginary part of the dielectric function ε(ω) computed using the DFT + Δ approximation (top) as well as the BSE approach (bottom) for NaI.

Imaginary part of the dielectric function ε(ω) computed using the DFT + Δ approximation (top) as well as the BSE approach (bottom) for LaBr3. The optical anisotropy is shown for both.

Imaginary part of the dielectric function ε(ω) computed using the DFT + Δ approximation (top) as well as the BSE approach (bottom) for BaI2. The optical anisotropy is shown for both.

Imaginary part of the dielectric function ε(ω) computed using the DFT + Δ approximation (top) as well as the BSE approach (bottom) for SrI2. The optical anisotropy is shown for both.
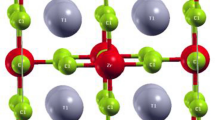
Excitonic effects are particularly dramatic for NaI (see Fig. 2), where the onset of optical absorption is dominated by a bound-excitonic state that occurs as a pronounced peak in the spectrum. It can be seen that this is an additional peak at about 5.5 eV in the lower panel of Fig. 2, that is absent in the upper panel. We visualize part of the two-particle electron–hole wave function of this bound-exciton state in Fig. 6 by fixing the position of the hole at an iodine atom and plotting the resulting electron distribution. The spherical shape confirms its similarity to a 1 s state within a hydrogen-like Wannier–Mott-exciton picture. In Fig. 6, it can be clearly seen that this electron density is centered around the iodine atom at which the hole is fixed. We previously studied this bound-excitonic state in more detail and determined the exciton-binding energy to be 216 meV and the Bohr radius to be 9 Å.29

The two-particle electron–hole wave function is visualized for the lowest bound-exciton state in NaI. The position of the hole is fixed on a I atom in the center of the spherical structure that represents the electronic part.
Furthermore, comparing the upper and lower panels in Fig. 2 shows that peaks that appear in the DFT + Δ spectrum at higher energies (labeled A, B, C in the figure) are shifted to lower photon energies and show slightly redistributed spectral weight when excitonic effects are taken into account. This red shift as well as the spectral redistribution has been traced back to excitonic effects for other materials (e.g., GaN, MgO, or ZnO) that show a similar structure of the uppermost valence and the lowest conduction band.35,66 In our calculations, we found that the peaks A, B, and C are attributed to transitions from the I 5p electrons into the conduction bands. The largest contribution to A is transitions into empty Na s states and to B and C transitions into empty I d states. Empty I s states contribute significantly only to A, while B and C also include important contributions from transitions into empty Na and I p states.
The spectra of the other three materials show either much weaker additional peaks/spectral features attributed to bound-excitonic states (see BaI2 and SrI2 in Figs. 4 and 5) or none is visible at all (see LaBr3 in Fig. 3). Partly, this can be attributed to the larger dielectric screening in these materials (see Table I): NaI has the lowest electronic dielectric constant of all four cases investigated here. Consequently the electron–hole interaction is strongest in this material, which is confirmed by the strong excitonic effects observed in the dielectric function. In addition, also the band structures of the materials differ: especially in the case of LaBr3, we attribute the lack of a bound-exciton peak at the absorption onset to the more complicated band structure (see Ref. 28) that does not have a pronounced valence-band maximum and conduction-band minimum but rather dispersionless bands. The spectral red shift of higher-energy peaks due to excitonic effects is clearly visible for all materials.
In Figs. 3–5, it can be seen that the spectra of LaBr3, BaI2, and SrI2 feature a fairly broad (more than 2 eV of full width at half maximum) single peak structure starting right above the respective absorption onsets. This clearly distinguishes the spectra of these three materials from the one of NaI (see Fig. 2). Using our data, we are able to trace this feature back to transitions into empty d states that do not occur in NaI but in the other three materials. For LaBr3 this feature arises mostly due to transitions from Br 4p electrons into empty La d states and for BaI2 it is mostly transitions from I 5p into empty Ba d states. In the case of SrI2, it is transitions from I 5p electrons into empty Sr d states that cause the appearance of this spectral feature.
The four different scintillator materials also differ regarding their optical anisotropy: cubic NaI is optically isotropic due to its lattice symmetry. However, also the spectra of the noncubic materials BaI2 and SrI2 show almost no dependence on the light polarization (see Figs. 4 and 5). This agrees well with the dielectric functions computed by Singh using the random phase approximation.67 In the case of LaBr3, the overall anisotropy is still not very strong; however, the energy position and the spectral shape of the absorption edge depend on the polarization of the light (see Fig. 3). This is due to the energy splitting of the uppermost Br 4p derived valence bands by about 88 meV.
B. Electron-energy loss function
To understand the energy loss processes of electrons in the scintillator materials, we use the complex dielectric function to compute the electron-energy loss function for zero momentum transfer according to
For all four materials studied in this work, the results are shown in Fig. 7. In this figure, we compare results computed within the independent-particle approximation, i.e., neglecting excitonic effects, to electron-loss functions derived from the BSE data (i.e., taking excitonic effects into account).

Figure 7 clearly illustrates the impact of excitonic effects on −Im ε−1 across the entire energy range studied here. For all four materials, we observe a remarkable red shift when the electron–hole interaction is included, along with significant spectral redistribution. In particular for NaI, which shows the strongest excitonic effects due the weak screening of the electron–hole interaction, the difference between both computational results are large: a peak that occurs at around 16 eV in independent-particle approximation occurs at ≈13.5 eV due to excitonic effects and also its height is significantly reduced. In the case of NaI, we also compare to an experimental result68 and find very good agreement with our theoretical data that takes excitonic effects into account. Even though the y axis scaling of the experiment is arbitrary, it is immediately clear that both absolute peak positions and relative peak heights agree very well with our computational result. The data for all four materials illustrate that both quasiparticle and excitonic effects need to be treated accurately, to achieve reliable predictions of electron-energy loss function that can be used, for instance, in multiscale modeling approaches.22
IV. CONCLUSIONS
In summary, we have used cutting-edge theoretical spectroscopy techniques to investigate the frequency-dependent, complex dielectric functions, and electron-energy loss functions for zero momentum transfer for four different scintillator materials. Using these results, we illustrate the influence of excitonic effects, which we find to be very strong in NaI, due to the weak screening of the electron–hole interaction. We also point out the formation of bound-excitonic states that are visible near the absorption onset. These first-principles results allow us to explain the origin of three sub peaks in the dielectric function of NaI, and to trace back a broad, peak-like feature in the spectra of LaBr3, BaI2, and SrI2 to the presence of d electrons in these materials. At the same time, the optical anisotropy is small for these four scintillator materials.
We compute electron-energy loss functions for all materials and also find a strong influence of excitonic effects. This result clearly illustrates the importance of accurately describing electronic many-body effects when using first-principles techniques to predict quantities for multiscale modeling approaches. Furthermore, these data constitute the foundation for future studies of STEs, for instance in doped materials or in undoped cesium hafnium chloride, all of which are currently attracting attention in the scintillator community.
Overall, it is our goal to use and develop efficient and accurate computational techniques that provide deep insight into the fundamental properties of materials for scintillator-radiation detectors. One of the biggest challenges in this field is to achieve computational accuracy and efficiency: in many cases the materials of interest are complicated with large unit cells and localized d or f electrons. At the same time, predictions need to be accurate enough to allow the results to be used in larger length scale models. The techniques discussed here and demonstrated for four materials have excellent application potential and promise exciting progress to address some of the challenges of the scintillator community.
References
P.A. Rodnyi: Physical Processes in Inorganic Scintillators (CRC Press, Boca Raton, 1997).
G.F. Knoll: Radiation Detection and Measurement, 4th ed. (Wiley, New York, 2010).
P.A. Rodnyi: Physical Processes in Inorganic Scintillators (CRC Press, Boca Raton, 1997).
N. Zaitseva, A. Glenn, L. Carman, R. Hatarik, S. Hamel, M. Faust, B. Schabes, N. Cherepy, and S. Payne: Pulse shape discrimination in impure and mixed single-crystal organic scintillators. IEEE Trans. Nucl. Sci. 58(6), 3411–3420 (2011).
K.E. Nelson, T.B. Gosnell, and D.A. Knapp: The effect of energy resolution on the extraction of information content from gamma-ray spectra. Nucl. Instrum. Methods Phys. Res., Sect. A 659(1), 207–214 (2011).
E.V.D. van Loef, P. Dorenbos, C.W.E. van Eijk, K. Krämer, and H.U. Güdel: High-energy-resolution scintillator: Ce3+ activated LaBr3. Appl. Phys. Lett. 79, 1573 (2001).
N.J. Cherepy, G. Hull, A.D. Drobshoff, S.A. Payne, E. van Loef, C.M. Wilson, K.S. Shah, U.N. Roy, A. Burger, L.A. Boatner, W-S. Choong, and W.W. Moses: Strontium and barium iodide high light yield scintillators. Appl. Phys. Lett. 92(8), 083508 (2008).
C.M. Wilson, E.V. van Loef, J. Glodo, N. Cherepy, G. Hull, S. Payne, W-S. Choong, W. Moses, and K.S. Shah: Strontium iodide scintillators for high energy resolution gamma ray spectroscopy. Proc. SPIE 7079, 707917 (2008).
A. Burger, E. Rowe, M. Groza, K.M. Figueroa, N.J. Cherepy, P.R. Beck, S. Hunter, and S.A. Payne: Cesium hafnium chloride: A high light yield, non-hygroscopic cubic crystal scintillator for gamma spectroscopy. Appl. Phys. Lett. 107(14), 143505 (2015).
M.S. Alekhin, D.A. Biner, K.W. Krämer, and P. Dorenbos: Improvement of LaBr3:5% Ce scintillation properties by Li+, Na+, Mg2+, Ca2+, Sr2+, and Ba2+ co-doping. J. Appl. Phys. 113(22), 224904 (2013).
M.S. Alekhin, J.T.M. de Haas, I.V. Khodyuk, K.W. Krämer, P.R. Menge, V. Ouspenski, and P. Dorenbos: Improvement of gamma-ray energy resolution of LaBr3:Ce3+ scintillation detectors by Sr2+ and Ca2+ co-doping. Appl. Phys. Lett. 102, 161915–161915 (2013).
D. Åberg, B. Sadigh, A. Schleife, and P. Erhart: Origin of resolution enhancement by co-doping of scintillators: Insight from electronic structure calculations. Appl. Phys. Lett. 104(21), 211908 (2014).
M. Tyagi, H.E. Rothfuss, S.B. Donnald, M. Koschan, and C.L. Melcher: Effect of Ca2+ co-doping on the scintillation kinetics of Ce doped Gd3Ga3Al2O12. IEEE Trans. Nucl. Sci. 61(1), 297–300 (2014).
I.V. Khodyuk, S.A. Messina, T.J. Hayden, E.D. Bourret, and G.A. Bizarri: Optimization of scintillation performance via a combinatorial multi-element co-doping strategy: Application to NaI:Tl. J. Appl. Phys. 118(8), 084901 (2015).
A.V. Vasil’ev: From luminescence non-linearity to scintillation non-proportionality. IEEE Trans. Nucl. Sci. 55, 1054 (2008).
S.A. Payne, N.J. Cherepy, G. Hull, J.D. Valentine, W.W. Moses, and W-S. Choong: Nonproportionality of scintillator detectors: Theory and experiment. IEEE Trans. Nucl. Sci. 56(4), 2506–2512 (2009).
S. Kerisit, K.M. Rosso, B.D. Cannon, F. Gao, and Y. Xie: Computer simulation of the light yield nonlinearity of inorganic scintillators. J. Appl. Phys. 105(11), 114915 (2009).
G. Bizarri, W.W. Moses, J. Singh, A.N. Vasil’ev, and R.T. Williams: The role of different linear and non-linear channels of relaxation in scintillator non-proportionality. J. Lumin. 129(12), 1790–1793 (2009).
R.T. Williams, J.Q. Grim, Q. Li, K.B. Ucer, and W.W. Moses: Excitation density, diffusion-drift, and proportionality in scintillators. Phys. Status Solidi A 248(2), 426–438 (2011).
Z. Wang, R.T. Williams, J.Q. Grim, F. Gao, and S. Kerisit: Kinetic monte carlo simulations of excitation density dependent scintillation in CsI and CsI(Tl). Phys. Status Solidi B 250(8), 1532–1540 (2013).
S.A. Payne, S. Hunter, L. Ahle, N.J. Cherepy, and E. Swanberg: Nonproportionality of scintillator detectors. III. Temperature dependence studies. IEEE Trans. Nucl. Sci. 61(5), 2771–2777 (2014).
F. Gao, Y.L. Xie, Z.G. Wang, S. Kerisit, D.X. Wu, L.W. Campbell, R.M. Van Ginhoven, and M. Prange: Monte Carlo simulation of gamma-ray response of BaF2 and CaF2. J. Appl. Phys. 114(17), 173512 (2013).
M. Klintenberg, S.E. Derenzo, and M.J. Weber: Potential scintillators identified by electronic structure calculations. Nucl. Instrum. Methods Phys. Res., Sect. A 486, 298 (2002).
C. Ortiz, O. Eriksson, and M. Klintenberg: Data mining and accelerated electronic structure theory as a tool in the search for new functional materials. Comput. Mater. Sci. 44(4), 1042–1049 (2009).
W. Setyawan, R.M. Gaume, S. Lam, R.S. Feigelson, and S. Curtarolo: High-throughput combinatorial database of electronic band structures for inorganic scintillator materials. ACS Comb. Sci. 13(4), 382–390 (2011). PMID: 21644557.
A. Canning, A. Chaudhry, R. Boutchko, and N. Grønbech-Jensen: First-principles study of luminescence in Ce-doped inorganic scintillators. Phys. Rev. B: Condens. Matter Mater. Phys. 83, 125115 (2011).
Q. Li, J.Q. Grim, K.B. Ucer, A. Burger, G.A. Bizarri, W.W. Moses, and R.T. Williams: Host structure dependence of light yield and proportionality in scintillators in terms of hot and thermalized carrier transport. Phys. Status Solidi RRL 6(8), 346–348 (2012).
D. Åberg, B. Sadigh, and P. Erhart: Electronic structure of LaBr3 from quasiparticle self-consistent GW calculations. Phys. Rev. B: Condens. Matter Mater. Phys. 85, 125134 (2012).
P. Erhart, A. Schleife, B. Sadigh, and D. Åberg: Quasiparticle spectra, absorption spectra, and excitonic properties of NaI and SrI2 from many-body perturbation theory. Phys. Rev. B: Condens. Matter Mater. Phys. 89, 075132 (2014).
A. Chaudhry, R. Boutchko, S. Chourou, G. Zhang, N. Grønbech-Jensen, and A. Canning: First-principles study of luminescence in Eu2+-doped inorganic scintillators. Phys. Rev. B: Condens. Matter Mater. Phys. 89, 155105 (2014).
A. McAllister, D. Åberg, A. Schleife, and E. Kioupakis: Auger recombination in sodium-iodide scintillators from first principles. Appl. Phys. Lett. 106(14), 141901 (2015).
P. Erhart, B. Sadigh, A. Schleife, and D. Åberg: First-principles study of codoping in lanthanum bromide. Phys. Rev. B: Condens. Matter Mater. Phys. 91, 165206 (2015).
H. Shi and M-H. Du: Discrete electronic bands in semiconductors and insulators: Potential high-light-yield scintillators. Phys. Rev. Appl. 3, 054005 (2015).
D. Wu, M.P. Prange, F. Gao, and S. Kerisit: First-principles search for efficient activators for LaI3. J. Lumin. 176, 227–234 (2016).
A. Schleife, C. Rödl, F. Fuchs, J. Furthmüller, and F. Bechstedt: Optical and energy-loss spectra of MgO, ZnO, and CdO from ab initio many-body calculations. Phys. Rev. B: Condens. Matter Mater. Phys. 80, 035112 (2009).
A. Riefer, F. Fuchs, C. Rödl, A. Schleife, F. Bechstedt, and R. Goldhahn: Interplay of excitonic effects and van Hove singularities in optical spectra: CaO and AlN polymorphs. Phys. Rev. B: Condens. Matter Mater. Phys. 84, 075218 (2011).
A. Schleife and F. Bechstedt: Ab initio description of quasiparticle band structures and optical near-edge absorption of transparent conducting oxides. J. Mater. Res. 27 (Focus Issue: Oxide Semiconductors), 2180–2189 (2012).
F. Fuchs and F. Bechstedt: Indium-oxide polymorphs from first principles: Quasiparticle electronic states. Phys. Rev. B: Condens. Matter Mater. Phys. 77, 155107 (2008).
J.B. Varley and A. Schleife: Bethe–Salpeter calculation of optical-absorption spectra of In2O3 and Ga2O3. Semicond. Sci. Technol. 30(2), 024010 (2015).
H.T. van Dam, S. Seifert, W. Drozdowski, P. Dorenbos, and D.R. Schaart: Optical absorption length, scattering length, and refractive index of LaBr3:Ce3+. IEEE Trans. Nucl. Sci. 59(3), 656–664 (2012).
K.B. Ucer, G. Bizarri, A. Burger, A. Gektin, L. Trefilova, and R.T. Williams: Electron thermalization and trapping rates in pure and doped alkali and alkaline-earth iodide crystals studied by picosecond optical absorption. Phys. Rev. B: Condens. Matter Mater. Phys. 89, 165112 (2014).
B. Kang and K. Biswas: Carrier self-trapping and luminescence in intrinsically activated scintillator: Cesium hafnium chloride (Cs2HfCl6). J. Phys. Chem. C 120(22), 12187–12195 (2016).
P. Hohenberg and W. Kohn: Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
W. Kohn and L.J. Sham: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
D.M. Ceperley and B.J. Alder: Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 45, 566–569 (1980).
J.P. Perdew, K. Burke, and M. Ernzerhof: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
P.E. Blöchl: Projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 50, 17953–17979 (1994).
Cutoff energies of the plane-wave basis were chosen to be 228 eV (NaI), 219 eV (LaBr3), 300 eV (BaI2), and 228 eV (SrI2).
M. Gajdoš, K. Hummer, G. Kresse, J. Furthmüller, and F. Bechstedt: Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B: Condens. Matter Mater. Phys. 73, 045112 (2006).
G. Kresse and D. Joubert: From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 59, 1758–1775 (1999).
G. Kresse and J. Furthmüller: Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B: Condens. Matter Mater. Phys. 54, 11169–11186 (1996).
C. Rödl, F. Fuchs, J. Furthmüller, and F. Bechstedt: Ab initio theory of excitons and optical properties for spin-polarized systems: Application to antiferromagnetic MnO. Phys. Rev. B: Condens. Matter Mater. Phys. 77, 184408 (2008).
F. Fuchs, C. Rödl, A. Schleife, and F. Bechstedt: Efficient O(N2) approach to solve the Bethe–Salpeter equation for excitonic bound states. Phys. Rev. B: Condens. Matter Mater. Phys. 78, 085103 (2008).
T. Hahn: International Tables for Crystallography Volume A: Space-group Symmetry (Springer, Dordrecht, 2005).
G. Lucovsky, R.M. Martin, and E. Burstein: Localized effective charges in diatomic crystals. Phys. Rev. B: Condens. Matter Mater. Phys. 4, 1367–1374 (1971).
B. Liu, M. Gu, Z. Qi, X. Liu, S. Huang, and C. Ni: First-principles study of lattice dynamics and thermodynamic properties of LaCl3 and LaBr3. Phys. Rev. B: Condens. Matter Mater. Phys. 76, 064307 (2007).
G. Onida, L. Reining, and A. Rubio: Electronic excitations: Density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys. 74, 601–659 (2002).
H.J. Monkhorst and J.D. Pack: Special points for Brillouin-zone integrations. Phys. Rev. B: Condens. Matter Mater. Phys. 13, 5188–5192 (1976).
M. Shishkin and G. Kresse: Implementation and performance of the frequency-dependent GW method within the PAW framework. Phys. Rev. B: Condens. Matter Mater. Phys. 74, 035101 (2006).
A.I. Liechtenstein, V.I. Anisimov, and J. Zaanen: Density-functional theory and strong interactions: Orbital ordering in Mott–Hubbard insulators. Phys. Rev. B: Condens. Matter Mater. Phys. 52, R5467–R5470 (1995).
M.T. Czyżyk and G.A. Sawatzky: Local-density functional and on-site correlations: The electronic structure of La2CuO4 and LaCuO3. Phys. Rev. B: Condens. Matter Mater. Phys. 49, 14211–14228 (1994).
W.G. Schmidt, S. Glutsch, P.H. Hahn, and F. Bechstedt: Efficient O(N2) method to solve the Bethe–Salpeter equation. Phys. Rev. B: Condens. Matter Mater. Phys. 67, 085307 (2003).
F. Bechstedt, R. Del Sole, G. Cappellini, and L. Reining: An efficient method for calculating quasiparticle energies in semiconductors. Solid State Commun. 84, 765–770 (1992).
16 × 16 × 16 and 17 eV (NaI), 4 × 4 × 8 and 15 eV (LaBr3), 8 × 4 × 4 and 14 eV (BaI2), 4 × 6 × 6 and 6.7 eV (SrI2).
A. Schleife: Electronic and optical properties of MgO, ZnO, and CdO (Südwestdeutscher Verlag für Hochschulschriften, Saarbrücken, 2011).
L.X. Benedict and E.L. Shirley: Ab initio calculation of ε2(ω) including the electron–hole interaction: Application to GaN and CaF2. Phys. Rev. B: Condens. Matter Mater. Phys. 59, 5441–5451 (1999).
D.J. Singh: Near optical isotropy in noncubic SrI2: Density functional calculations. Appl. Phys. Lett. 92(20), 201908 (2008).
M. Creuzburg: Energieverlustspektren der alkalihalogenide und der metalle Cu, Ag und Au und vergleich mit optischen messungen. Z. Phys. 196(5), 433–463 (1966).
ACKNOWLEDGMENTS
We acknowledge fruitful discussions with B. Sadigh and R. T. Williams. Part of this work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 with support from the National Nuclear Security Administration Office of Nonproliferation Research and Development (NA-22). This research is part of the Blue Waters sustained-petascale computing project, which is supported by the National Science Foundation (awards OCI-0725070 and ACI-1238993) and the state of Illinois. Blue Waters is a joint effort of the University of Illinois at Urbana–Champaign and its National Center for Supercomputing Applications.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This is an Open Access article, distributed under the terms of the Creative Commons Attribution licence (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted re-use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article

Cite this article
Schleife, A., Zhang, X., Li, Q. et al. Excitons in scintillator materials: Optical properties and electron-energy loss spectra of NaI, LaBr3, BaI2, and SrI2. Journal of Materials Research 32, 56–63 (2017). https://doi.org/10.1557/jmr.2016.395
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2016.395