
Abstract
The aim of this study was to develop a model to predict individual subject disease trajectories including parameter uncertainty and accounting for missing data in rare neurological diseases, showcased by the ultra-rare disease Autosomal-Recessive Spastic Ataxia Charlevoix Saguenay (ARSACS). We modelled the change in SARA (Scale for Assessment and Rating of Ataxia) score versus Time Since Onset of symptoms using non-linear mixed effect models for a population of 173 patients with ARSACS included in the prospective real-world multicenter Autosomal Recessive Cerebellar Ataxia (ARCA) registry. We used the Multivariate Imputation Chained Equation (MICE) algorithm to impute missing covariates, and a covariate selection procedure with a pooled p-value to account for the multiply imputed data sets. We then investigated the impact of covariates and population parameter uncertainty on the prediction of the individual trajectories up to 5 years after their last visit. A four-parameter logistic function was selected. Men were estimated to have a 25% lower SARA score at disease onset and a moderately higher maximum SARA score, and time to progression (T50) was estimated to be 35% lower in patients with age of onset over 15 years. The population disease progression rate started slowly at 0.1 points per year peaking to a maximum of 0.8 points per year (at 36.8 years since onset of symptoms). The prediction intervals for SARA scores 5 years after the last visit were large (median 7.4 points, Q1-Q3: 6.4–8.5); their size was mostly driven by individual parameter uncertainty and individual disease progression rate at that time.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic cerebellar ataxias are progressive rare neurological diseases (RNDs) affecting the cerebellum, often with multi-systemic damage to other neurological systems, causing debilitating impairment of gait, balance, speech, and fine motor skills. More than 100 ataxia diseases are autosomal-recessive cerebellar ataxias (ARCAs), often starting in early childhood or early adulthood. While each of them is ultra-rare, they constitute prime candidates for molecular treatment trials targeting their specific genetic defects, but robust statistical methodologies allowing to predict progression trajectories and their modification under treatment in very small samples are needed (1).
In this work, we aim to develop models to robustly capture and predict individual disease progression in RND patients. We use Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) as showcase, leveraging data from the Autosomal Recessive Cerebellar Ataxia (ARCA) patient registry (2). This real-world registry includes patients at any stage of their disease, with 0 to 7 longitudinal follow-up visits. Assessment of disease severity is measured through the SARA (Scale for the Assessment and Rating of Ataxia) score, a composite score comprised of eight items evaluated by a clinical assessment developed in 2006 (3). To handle design heterogeneity, we used non-linear mixed effect models (NLMEM) to model the natural progression of the disease measured by the total SARA score, and investigate patient characteristics associated with disease evolution. A feature of real-world data is the large amount of missing covariate information at some or all visits. Several methods have been proposed to deal with missing data (4), single or multiple imputations being the most common approaches (5). Simultaneous imputation and estimation have also been proposed to infer the distribution of missing covariates in NLMEM assuming a known covariate-parameter relationship (6). Here, we combine multiple imputation with a pooled test statistic (7) to iteratively build the covariate model.
We used the final model to predict individual trajectories of disease progression over several years. In this section, we focused on patients in the early stages of disease, who would be the population of most interest in clinical trials. In addition to accounting for individual parameter uncertainty using conditional distributions and evaluating the effect of covariates, we investigated how including population parameter uncertainty influences predictions and prediction intervals.
Methods
Data
For this paper, we analysed data from the ARCA registry (2) of patients with ataxia. Patients were enrolled across more than 30 centers in 15 countries, at any stage of their disease. Disease severity was primarily measured by the Scale for the Assessment and Rating of Ataxia (SARA) score, a composite score comprised of eight items, evaluated by a clinical assessment of gait, stance, speech, sitting, fine motor, and leg movements (3). For each patient, the main variables recorded at each visit were age, SARA score, as well as other clinical scores measuring daily living activities (not analysed here). Covariates were also recorded at inclusion in the study: Age at Onset of symptoms (AOO), as reported by the patient or their caregiver, Body Mass Index (BMI), Inventory of Non Ataxia Signs (INAS (8), secondary disease progression score for non-ataxia symptoms), the genotype of the mutation (missense or loss of function mutation), and sex.
In this work, we considered the largest genetic autosomal-recessive ataxia population in the ARCA registry (extraction date: January 2022) as showcase, namely, Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS), comprising of 173 patients included between 2013 and 2022. The dataset included a total of 349 measurements of SARA scores ranging from 3 to 40 points (median 20.5). The follow-up varied from 0 to 6 years, with one visit for 81 patients, two visits for 44 and more than two visits in 48 patients. Median Time Since Onset of symptoms at inclusion in the registry was 35 years (Q1-Q3: 24–48). Table I summarises the covariates at inclusion. The percentage of missing values in the covariates ranged from 0 (Sex) to around 40% (INAS score).
Figure 1 shows the SARA scores as a function of Time Since Onset of symptoms. On average, the SARA score increases with Time Since Onset of symptoms, with large variations in the individual profiles as some patients exhibited a stable or even decreasing SARA score over time.

Plot of the SARA score as a function of TSO (time since onset of symptoms) in the 173 ARSACS patients. Solid lines represent the repeated assessments for an individual
Modelling SARA Score
Time Since Onset (TSO) was computed as current Age minus Age of Onset (AOO). In the ARSACS population, 22 missing values of AOO were imputed to the median AOO in the data set (2 years of age), consistent with early childhood presentation of ARSACS. In the analysis, AOO was split into three clinically relevant categories: 0–7, 8–14 and 15–40 years (9).
We modelled the SARA score \({y}_{ij}\) recorded at TSO in individual i at time \({t}_{ij}\) (i = 1,…,N, j = 1,…,\({n}_{i}\)) as a continuous variable using non-linear mixed effect models (10), defined through the following equations:
where \({t}_{ij}\) represents time j for individual i, \({y}_{ij}\) the observation of individual i at time \({t}_{ij}\), \(f\left({t}_{ij},{\psi }_{i}\right)\) represents the structural model, depending on the vector of individual parameters for individual i, \({\psi }_{i}\), \(g\left({t}_{ij},{\psi }_{i}\right)\) describes the standard deviation of the residual errors, and \({\epsilon }_{ij}\sim N\)(0,1) is the residual error for the observation \({y}_{ij}\). The individual parameters were assumed to follow a log-normal distribution, with a mean equal to a linear function of fixed effects \(\mu ,\) covariate effects for parameter k \({\beta }_{C,k}\) and \({C}_{i}\), the vector of covariates for individual i. The random effects \({\eta }_{i}\) were assumed to follow a joint multinormal distribution with variance–covariance matrix \(\Omega\). We estimated the population parameters for this model, \(\theta =\left(\mu ,\beta ,\Omega ,\sigma \right)\), using the SAEM algorithm (11).
Base Model Building
Several models (linear, exponential, sigmoidal and Gompertz functions, 3 and 4 parameter logistic equations) were considered to model the progression of the SARA score as a function of TSO. The four-parameter logistic function was parameterised to include clinically relevant parameters as:
where \({S}_{0}\) represents the SARA score at the onset of symptoms \(\left(f\left(0\right)={S}_{0}\right)\), \({S}_{max}\) is the maximum SARA score, \({T}_{50}\) is the time when \(f\left({T}_{50}\right)=\frac{{S}_{0}+{S}_{max}}{2}\), k represents the disease progression rate (year−1). The structural model was selected using the Bayesian Information Criterion (BIC), with a diagonal variance–covariance matrix for random effects (no correlations) and a combined error model.
Additive, proportional and combined residual error models were compared, with a Likelihood Ratio Test (LRT) for nested models and BIC for non-nested models. In a third step, the structure of the covariance matrix was investigated by fitting a model with correlations on all parameters. Correlations were removed if they were less than 0.7. Finally, remaining correlations (if any) were removed using a stepwise procedure (backward-forward approach) using the LRT and removed by block when non-significant. The p-value for all LRT tests was 0.05.
Covariate Model Building Accounting for Missing Covariates
The covariate model building and final interindividual variability (IIV) selection is summarised in Fig. 2. To build the covariate model, we combined multiple imputation (5) with a stepwise algorithm including a pooled log-likelihood ratio test (LRT).

Flowchart of the approach used to build the covariate model, refine the structure of the variance–covariance matrix and obtain the final parameter estimates with their uncertainty
First, the missing values for BMI, INAS and ARSACS genotype were imputed with Multiple Imputation using the MICE package (5). For this, we first selected the structure of each regression model using the subset with complete data for that covariate, with the non-missing covariates (AOO, Age at first visit, Sex, SARA score at first visit) and the individual parameters (Empirical Bayes Estimates, EBE) estimated from a model with no covariates (6) as regressors. Individual parameters were estimated as the mode of the conditional distribution of patient (12). Linear regressions were used for continuous covariates (INAS, BMI) and logistic regressions for categorical covariates (ARSACS genotype). For each regressor, we tested both the regressor itself and its logarithm to choose which to include in the model, based on the Akaike Information Criterion (AIC). The resulting models for each missing covariate were entered in a multivariate regression model for MICE to impute ARSACS genotype, BMI and INAS, by increasing proportion of missing information. Ten data sets were generated using this procedure.
Second, the covariate model was built with a step-wise forward–backward procedure with a pre-selection step based on a p-value for the Pearson correlation coefficient between the EBE of the model without covariates fitted above and the covariates below 0.2. In the forward–backward procedure, we used Meng and Rubin's (7) approach to compute a pooled LRT on all imputed data sets for each covariate-parameter relationship. This allows to compute a single p-value across all imputed data sets for a given covariate-parameter relationship, through a two step procedure detailed in Supplementary Materials 3. This statistic was used throughout the step-wise procedure, with a threshold set at 0.05.
Covariate effects were added iteratively with a first forward approach among the pre-selected covariates. When none of the remaining relationships were significant, a second forward approach was performed to test whether non pre-selected covariate-parameter relationships should be included. Finally, a backward approach was performed on all covariate-parameter relationships included in the model at this stage. In a final step, a backward approach was again applied to the variability components with the same procedure.
Evaluation and Uncertainty
During model building, models were evaluated with standard goodness of fit plots (population fits, individual fits, Visual Predictive Check (VPC), Normalised Prediction Distribution Errors NPDE (13) versus TSO and predictions). The parameter estimates of the final model were reported as the mean parameter estimates across all imputed data sets (7). Standard errors of estimation (SE) were estimated using case bootstrap, which resamples patients uniformly from the original data set with replacement (14). The final model was fit to 200 bootstrapped datasets for each imputed dataset (15) and the resulting distributions of the estimated population parameters pooled to obtain the overall bootstrap distribution. SE were computed as the standard deviation of this pooled distribution over the 10 imputed data sets. Bootstrap SE were also computed for the base model without covariates.
Prediction of Individual Trajectories Accounting for Uncertainty
To build individual predictions, 100 individual parameter vectors were drawn from each patient's conditional distribution in each imputed data set (1000 samples in total). Predicted SARA scores were computed for each vector up to 5 years after the last visit and pooled. Finally, for each patient and each time, we computed the 5th, 50th and 95th percentile of simulation to get a median prediction and a Prediction Interval (PI) for the trajectories.
To account for population parameter uncertainty, we repeated this procedure for each imputed data set, by first sampling 200 population parameter vectors before computing the conditional distributions and sampling 100 individual sets of parameters. The resulting 200,000 samples constituted the conditional distribution under uncertainty and were used to build individual predictions as above.
Individual predictions for the model with covariates were obtained with and without population parameter uncertainty, and with population parameter uncertainty for the model without covariates. In each case, we computed the predicted SARA score at 5 years after each patient's last visit along the width of their PI and the ratios of the predicted SARA score the ratio of the width of PI at 5 years, with and without covariates, with and without population parameter uncertainty. Individual predictions are reported in the following for the 70 patients with a maximum SARA score less than 20 points (table in supplementary materials 1).
Implementation
Analyses were performed in R version 4.2.0 (16). Parameter estimation was performed with the saemix package version 3.1 for R (17) (with 800 and 400 iterations for the first and second phases respectively, 10 chains, and 10,000 samples for the estimation of the likelihood through Importance Sampling). Multiple Imputation was performed using the MICE R package (5).
Results
Model Selection
Describing the evolution of SARA score with TSO, through a linear model yielded an intercept of 1.7 (RSE = 32%) (SARA score for TSO = 0) and a progression rate of 0.5 (RSE = 4%) points per year, but this model predicted negative SARA scores at onset in some patients or very high SARA scores as the patients grew older, which was incompatible with scores bounded between 0 and 40. An exponential model performed worse than the linear model in terms of BIC (\(\Delta\) BIC = + 4). The four-parameter logistic equation defined in (3) resulted in a better performance (\(\Delta\) BIC = -59 compared to the linear model). The NPDE of the models displayed in Fig. 3 also showed that this model performed better than the others as it adapted to the bounded nature of the score. This structural model was therefore selected for the following. An additive error model was chosen, and we did not find significant correlations between the parameters.

NPDE (Normalised Prediction Distribution Errors) versus TSO of the models without covariates (top) and with covariates (bottom). The observed 5%, 50% and 95% percentiles are represented as solid black lines, the red band represents the simulated (1000 replicates) median npd with its 90% prediction interval, the blue line represents the simulated 5% and 95% percentiles (1000 replicates) with their 90% prediction intervals
The individual parameter estimates from the base model without covariates were used to test for potential covariate-parameter relationships. There was no difference between parameters between the first two categories of AOO which were then regrouped as early onset (reference class: \(AOO<15.\) The initial screening process generated 8 covariate effects (sex, BMI, AOO > 15 on \({S}_{0};\) sex and AOO > 15 on \({S}_{max};\) INAS and BMI on k; AOO > 15 on \({T}_{50}),\) three covariate-parameter effects were selected during the first forward step, none during the second and none were removed during the backward procedure. The final model included 3 relationships: \({S}_{0}\) was 25% lower in men, \({S}_{max}\) was 8% higher in men compared to women (class of reference in the modelling), and \({T}_{50}\) was 35% lower in late onset patients compared to early onset patients. The parameter estimates and their residual standard errors (RSE) for the model with/without covariates can be found in Table II. All parameters of the model are well estimated with RSE under 50%. For the final model, the eta-shrinkage was of 91% for \({S}_{0}\), 73% for α and 52% for \({T}_{50}\). The residual error was slightly less than 2 points of SARA score, reflecting the intrasubject variability seen in Fig. 1.
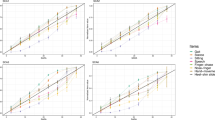
The NPDE of the models with and without covariates reported in Fig. 3 show that both models fit the data well, with no trend in the median profile. In Fig. 4, we simulated 100 parameter vectors from the population distribution of the parameters for each patient, and computed the median and the 5th-95th percentiles of the predicted SARA score for the four covariate categories. The model describes well disease progression over time in the different categories, although there are few patients with late onset.

Prediction bands of the evolution of the SARA scores versus TSO, stratified by covariate group (with women (top), males (bottom), AOO < 15 (left), AOO > 15 (right)) for the 173 ARSACS patients. Each band was obtained using 100 simulations of the random effects from the population distribution obtained with the final model to predict the evolution of SARA scores for each subject in the group over the range 0 (onset of symptoms) to 70 years after onset. The limits of the band correspond to the 90% simulation interval at each time point. The observed SARA scores are overlayed for each group, with lines joining the repeated measurements for subjects with more than one visit
Prediction of Individual Trajectories Accounting for Uncertainty
Figure 5 shows the individual disease progression predicted for the base and covariate models, along with the prediction intervals, for one randomly selected patient in each covariate group, accounting for population parameter uncertainty. Although residual uncertainty wasn't included in these predictions, they still show considerable uncertainty 5 years after the last visit.

Examples of individual predictions with population parameter uncertainty (median + 90% PI without residual error) for the models with (red) and without (green) covariates for four patients with various covariate combination. Uncertainty covers population and individual parameter uncertainty but the predictions themselves do not include residual error
The difference for the predictions with and without covariates is only apparent if AOO is greater than 15 years of age. Indeed, in late onset patients, the population \({T}_{50}\) was estimated to be 24.8 years instead of 38.1 years, close to the TSO at last visit recorded for these patients, and because progression is fastest around \({T}_{50},\) taking into account covariates had more impact on the predicted progression in this data set.
To better understand the effect of including covariates and/or including population parameter uncertainty on the prediction of the trajectories, we compared the predicted SARA score at 5 years and its prediction interval for all patients in the different configurations (Fig. 6). We restricted the comparison to subjects having a maximum SARA score below 20 points (70 patients), and we summarised the differences by looking at violin plots of the predicted SARA score and its width at 5 years with covariates and population uncertainty (in red): the majority of patients were predicted to increase (median + Q1-Q3) by + 2.7 points (1.9–3.6) without covariates (with uncertainty), and by + 2.9 points (1.9–3.9) with covariates (with uncertainty) 5 years after their last visit, but the uncertainty around that prediction, measured by the width of the PI, was 7.4 points (6.4–8.5) without covariates (with uncertainty) and 7.4 points (6.1–8.3) with covariates (with uncertainty).

Violin plot of the predicted SARA score and the width of 90% PI width at 5 years with uncertainty (top), and its ratio with/without uncertainty, with and without covariates (with uncertainty) (bottom), in the 70 patients with a maximum SARA score below 20
The violin plots of the corresponding ratios comparing the model with or without covariates in yellow show that including covariates had little impact on the predicted SARA score at 5 years (median -0.2%) and on its width of PI (median: -5%). The violin plots of the corresponding ratios comparing the model with or without parameter uncertainty in purple show that including population parameter uncertainty had no impact on the predicted SARA score at 5 years (median: + 0%), and little impact on the width of its prediction interval (median: + 4% with half of the subjects varying by less than + 10%).
For more detail, in Fig. 7 we show the distribution of the two ratios, stratified by covariate category. Including covariates impacted the predicted SARA score at 5 years for the groups with AOO > 15 years, increasing the median by + 17% in women and + 22% in men, and decreased the median width of the prediction interval for the group women + AOO < 15 years (median: -9%) whereas the group man + AOO < 15 has a median ratio of 1. It could be due to the fact that, in the model with covariates, women have a higher \({S}_{0}\) (8.1) and a lower \({S}_{max}\) (34.3) than in the model with no covariates (resp 6.9 and 35.8), resulting in a slower progression, and therefore narrower prediction intervals at 5 years. Taking into account population parameter uncertainty had no impact on the predicted SARA score at 5 years, and slightly increased the width of the prediction interval for men with AOO < 15 years (median: + 6%), likely due to the fact that the parameters \({\beta }_{male,{S}_{0}}\) and \({\beta }_{male,{S}_{max}}\) have higher RSE (50% and 38% respectively) than the rest of the parameters.

Violin plot of the ratio of the predicted SARA score and the width of the 95% PI at 5 years with/without uncertainty, with and without covariates (with uncertainty) stratified by the two covariates in the model, for the 70 ARSACS patients with a maximum SARA score below 20 points, including 39 women (2 with AOO > 15) and 31 men (2 with AOO > 15)
Discussion
In this work, we modelled the evolution of a clinician-reported outcome (SARA score) in a rare neurological disease patient group with an ultra-rare disease (ARSACS) using NLMEM. We used an innovative approach to build the covariate model in the presence of missing values for several covariates. We investigated the impact of including covariates in the model and how taking into account population parameter uncertainty affected the predicted SARA score at 5 years and its prediction intervals.
The evolution of SARA scores were best described as a non-linear function of TSO, as in (18). In the literature, sigmoid models have been used to model scores for other neurodegenerative diseases. For example, MDS-UPDRS score in Parkinson's disease were modelled with bounded functions such as the Gompertz equation (19). Three parameter logistic functions have been used to model cognitive scores CDR-SB (20) or CAMCOG (21) in Alzheimer's disease. For ataxia in particular, Jacobi et al. used mixed effect models to analyse the progression of several scores (including the SARA score) in 677 patients with Spinocereballar Ataxia (SCA) as a function of time from ataxia onset (12), and they showed that when modelling the whole disease progression, a non-linear model fitted best. Alternative models consider the change with time since first visit: Maas et al. (22) performed a multivariable linear regression on 223 patients with SCA3 to investigate predictors of disease progression, and reported that the disease progression rate was almost three times higher for patients with TSO > 10 years, with a large IIV. Using a linear model in the ARSACS dataset led to higher IIV on the rate of progression. We also tested a linear model to SARA score evolution as a function of time since first visit, which had a much higher BIC, suggesting that modelling the SARA score as a function of TSO was more informative.
Total SARA score was modelled as a continuous dependent variable, but it is actually a sum of sub-items measuring different neurological functions. An alternative could be to consider the discrete nature of the sub-scores, using Item Response Theory (IRT) (23) for example. IRT modelling was successfully applied to other composite scores for neurodegenerative diseases (23,24,25). The model involves a latent variable, which informs on the disease severity of a patient and can translate to the probability of scoring a certain grade on any sub scale. For example, a patient having a high score on the gait test could have the same total score as another patient with a lower score on the gait test, but a higher one on the fine motor movement tests, when both subscales could progress at different stages of disease severity. In the context of clinical trials, Buatois et al. (26) showed that, when modelling the MDS-UPDRS scale for patients with Parkinson's Disease, an IRT analysis yielded higher power with no assumption on the drug effect profile. Hamdan et al. (27) developed a cross-sectional IRT model to describe the disease severity in the patients from the ARCA registry. The resulting total score adding sub-items has a sigmoid shape similar to the logistic function we used, so that the total score analysis can be considered as a continuous approximation to the IRT model. Hamdan et al. (27) developed a cross-sectional IRT model to describe the disease severity in the patients from the ARCA registry. They then developed a longitudinal IRT model while investigating Markovian features (work in progress).
In our analysis, we used an innovative approach to build the covariate model in the presence of missing covariates, combining multiple imputation and a pooled test statistic. Methods have been investigated for model selection in the presence of multiple imputation. Covariates can be selected independently on each data set, with a voting at the end (28), or a pooled metric can be applied to select covariates on all imputed data sets simultaneously (7). The pooled LRT p-value we used is asymptotically equivalent the pooled p-value initially derived from Wald's test (7), and was shown to be preferable for testing random effects (29). Another advantage is that using the LRT does not require normality in the complete data model, although it may be more conservative in smaller samples (29). These two approaches were compared with others through simulation studies involving linear and logistic regression models by Wood et al. (28). They notably presented a stacking method, where all imputed data sets are merged into one data set, and a split imputation approach, where covariate selection is performed independently on each imputed data set, with a voting step at the end of the procedure. The stacking approach gave consistent estimates compared to the pooled approach we used, but a penalisation term needs to be included in the likelihood to conserve valid standard error estimates. The split imputation approach was shown to yield underestimated standard errors, and to increase the selection rate of nuisance covariates as it assumes all imputed covariates are real data. Overall, Wood et al. conclude that the pooled p-value method we used was the only one preserving type 1 error in their setting.
The pooled p-value metric used to select covariate effect uses the likelihood of the model across imputed data sets. However, since Importance Sampling was used to evaluate the likelihood of the model, some covariate-parameter relationships could have been penalised due to the stochasticity of the method. To reduce that impact, for the model fitting, at each iteration of the model building, for a given covariate-parameter candidate, the same random seed was assigned to all imputed data sets, to ensure that if only non-imputed covariates are in the model, the likelihood would be the same across all imputed data sets. Another feature is that the method penalises covariate-parameter relationships when covariates have high degree of missing information. The penalisation is twofold, first through the term r (formulas in Supplementary Materials 3), which is proportional to the average difference between the LRT evaluated at the MLE (\(\widehat{d}\)) and the LRT evaluated at the mean MLE \(\left(\overline{d}\right).\) Larger differences between the imputed data sets will translate into larger differences between the MLEs across all imputed data sets, increasing r. A second penalty occurs via \(\nu .\) When there is no difference between \(\widehat{d}\) and \(\overline{d},\) \(\nu =+\infty\), and the F-distribution will converge to a chi square distribution with k degrees of freedom (for k = 1 here, we get the standard threshold of 3.84 for the LRT at 0.05). When r increases, \(\nu\) decreases and the threshold for statistical significance will increase, increasing the p-value and making the test stricter. During covariate selection, the effect of INAS on k was likely not selected because of these two penalties (\(\overline{d}=8.9\), r = 1.25 therefore the statistic becomes D = 3.95 and with \(\nu =17,\) the threshold for statistical significance at 0.05 is 4.45). In the preselection stage, the correlation between the EBEs and the covariates were used. Out of the 24 parameter-covariate relationships, 8 were pre-selected (with the correlation), and the three final covariate effects were among the pre-selected. Although the eta-shrinkage of the EBEs of the parameters was quite high, it may not have had a big impact, since the chosen p-value threshold was large (0.2) and since, even if a covariate effect was not selected during the first stage, it is tested during the second forward step. The covariate genotype of the mutation type of ARSACS (loss of function) was not found significant in our model. It could be due to the fact that, in the complete case, 82% of patients have a loss of function mutation. For the selected covariates, our analysis showed an effect of sex and onset. In (22), for SCA3, disease progression was found to be significantly faster for men. In our model, although being a man decreases \({S}_{0}\) and increases \({S}_{max},\) with the structural model equation equation, it could be interpreted as a faster progression in men at all disease stages (of + 18% with the estimated parameters). In the end, none of the covariates with missing values were selected, so the uncertainty was computed using bootstrap on the original data set. However, a straightforward extension in the presence of imputed covariates would be to pool the bootstrap distributions over the imputed data sets to obtain the uncertainty distribution.
In our model, \(S_0\) represents the score at onset of symptoms, and was estimated to be different from 0, while \({S}_{max}\) was estimated to be around 35. We could not identify individual variability for \({S}_{max},\) implying that all patients will reach a SARA score around those values. In our model, the parameters which most influence the disease progression are k and \({T}_{50},\) which control respectively the sigmoidicity of the curve and the time to fast increase of SARA score. For example, with the parameters estimated for the model with covariates, for women, such values would correspond to an increase of SARA score ranging from 0.05 to 0.75 points/year for early onsets and 0.28–0.82 points per year for late onset patients (TSO = 5 years to\({T}_{50})\), with marginally higher values for men. An analysis of disease progression over the time of follow-up was performed by Traschütz et al. on all major genotypes from the ARCA registry (30). They modelled the evolution of the SARA score with the time since inclusion in the study using linear mixed effect models. They notably found that the slope of progression was 0.12 points/year in the ARSACS group, not statistically different from 0. This can be explained by our model where, depending on the TSO, the slope of disease progression can range from 0 to 0.98 points/year. Gagnon et al. (31) investigated the progression of the SARA score in 19 patients with ARSACS after a two year follow up, reporting a mean yearly progression of 1.3 points/year. In this study, the mean age was 38.3 years, ranging from 25 to 59 years of age, so in our model, most of these patients would be around \({T}_{50},\) at a period of fast progression, assuming an early onset. The interpretation of the effect of the selected covariates on disease progression is unclear. For example, we found that having an AOO greater than 15 years of age resulted as having a T50 13 years lower, meaning that those patients would have a rapid progression at approximately the same age as those with an AOO of 2 years. It is possible therefore that recall bias affected patients self-reporting as late onset if they discounted early symptoms of the disease.
We applied our final model to predict the individual trajectories of patients along with their prediction interval over 5 years after their last visit. Predicting patient trajectories on the individual level could be used for simulations of clinical trials, to estimate the power of a design or to predict which patients are the most informative. We limited the results presented to the patients with a SARA score below 20 points, as clinical trials with disease-modifying drugs currently focus on early or at most moderate disease stages in neurodegenerative diseases where neurodegeneration is not yet too (and likely irreversibly) advanced (32). We investigated the impact of including covariates on the individual predictions. We found that there was little impact on the individual predictions, mainly because those covariates, while being statistically significant, explained only a small portion of the IIV of the parameters of the model. The inclusion of population parameter uncertainty also had little impact on the width of prediction intervals, which we can attribute to the low uncertainty (RSE) on population parameters. This approach will however be extended to other populations in the registry, and since they have fewer patients, we expect the RSE of the population parameters to be higher.
In this work, we developed a workflow using real world data (patient registries) for a rare neurodegenerative disease, handling missing patient information as well as high heterogeneity. Such a workflow could be extended to other Autosomal Recessive Cerebellar Ataxias or other rare diseases.
In the ARSACS dataset, the increase in SARA score and the width of their PI during those 5 years were higher the closer T50 was to the TSO of the observation period (correlation coefficient: -0.65, p-value < 10–8). This suggests that patients recruited at a TSO between 30 and 50, at an intermediate stage of disease, could be the most informative when designing a clinical trial. Our model also predicts a median increase of SARA score of 3 points in 5 years, but with a median width of prediction interval of 7 points. This large uncertainty somewhat undermines the practical usefulness to predict individual progression. This could be explained by our sparse design (only about half of the patient have more than one visit), so many patient trajectories are predicted by the population parameter values, and with individual parameter SE driven mainly by the random effect on \({T}_{50}.\) On the other hand, the covariate category or the number of visits were not strongly associated with the precision of the PI in our analyses, but the dataset had mostly sparse sampling. Repeated longitudinal measurements should however help to get a more accurate prediction of their individual parameters.
In our work, we developed a method to predict individual trajectories at 5 years including uncertainty. We plan to apply this approach to an independent data set to evaluate the predictive ability of our model. In the meantime, we performed an internal evaluation analysis using simulations. Supplementary material 4 reports this evaluation, showing that individual prediction method is unbiased.
In this work, describing the evolution of the SARA score using modelling approaches on a population of ARSACS patients informed us on the heterogeneity of disease progression, which mainly depends on the time since onset. Modelling can then help clinicians determine the likely evolution for a given patient. It could also provide insight on which patients would be the most informative in the context of a clinical trial, helping with patient stratification. A perspective of this work is to investigate model-based designs as an alternative to end of treatment analyses to enhance the power of small size clinical trials.
Conclusion
In this work, we modelled disease progression in patients with a rare neurological disease, using ARSACS as showcase, and investigated the impact of the inclusion of covariates and population parameter uncertainty on individual trajectories of progression and their associated prediction intervals. We combined multiple imputation with a pooled LRT statistic method to select a covariate model with missing covariate values. Although the implementation can be somewhat complex and computationally cumbersome, it ensured missing covariates were properly accounted for. We found that covariates had a moderate impact in explaining disease progression, and that including population parameter uncertainty had little impact on the individual prediction intervals. Our current population analysis was limited by the size of the studied population and could be extended to a larger one, in order to look for other predictors of disease progression. The small impact of the inclusion of population parameter uncertainty could be explained by the low uncertainty in population parameter estimates, and the proposed workflow could be more relevant in other ataxia diagnoses with fewer patients.
Data Availability
Code for the fits, covariate model selection and individual predictions are available in a repository (10.5281/zenodo.10958731) as well as a simulated data set to make the code executable as standalone and two main scripts in addition with a readme.
Change history
24 May 2024
A Correction to this paper has been published: https://doi.org/10.1208/s12248-024-00932-8
References
Synofzik M, van Roon-Mom WMC, Marckmann G, van Duyvenvoorde HA, Graessner H, Schüle R, et al. Preparing n-of-1 antisense oligonucleotide treatments for rare neurological diseases in europe: genetic, regulatory, and ethical perspectives. Nucleic Acid Ther. 2022;32(2):83–94.
Traschütz A, Reich S, Adarmes AD, Anheim M, Ashrafi MR, Baets J, et al. The ARCA registry: a collaborative global platform for advancing trial readiness in autosomal recessive cerebellar ataxias. Front Neurol. 2021;12:677551.
Schmitz-Hübsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology. 2006;66(11):1717–20.
Gastonguay MR, French JL, Heitjan DF, Rogers JA, Ahn JE, Ravva P. Missing data in model-based pharmacometric applications: points to consider. J Clin Pharmacol. 2010;50(S9):63S-74S.
van Buuren S, Groothuis-Oudshoorn K. mice: Multivariate imputation by chained equations in R. J Stat Softw. 2011;45:1–67.
Johansson ÅM, Karlsson MO. Comparison of methods for handling missing covariate data. AAPS J. 2013;15(4):1232–41.
Meng XL, Rubin DB. Performing likelihood ratio tests with multiply-imputed data sets. Biometrika. 1992;79(1):103–11.
Jacobi H, Rakowicz M, Rola R, Fancellu R, Mariotti C, Charles P, et al. Inventory of Non-Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum. 2013;12(3):418–28.
Rummey C, Corben LA, Delatycki M, Wilmot G, Subramony SH, Corti M, et al. Natural History of Friedreich Ataxia: Heterogeneity of Neurologic Progression and Consequences for Clinical Trial Design. Neurology. 2022;99(14):e1499–510.
Lavielle M. Mixed effects models for the population approach: Models, tasks, methods and tools. CRC Press; 2014. (Chapman & Hall/CRC Biostatistics Series).
Delyon B, Lavielle M, Moulines E. Convergence of a stochastic approximation version of the EM algorithm. Ann Stat. 1999;27(1):94–128.
Lavielle M, Ribba B. Enhanced Method for Diagnosing Pharmacometric Models: Random Sampling from Conditional Distributions. Pharm Res. 2016;33(12):2979–88.
Nguyen T, Mouksassi M, Holford N, Al-Huniti N, Freedman I, Hooker A, et al. Model Evaluation of Continuous Data Pharmacometric Models: Metrics and Graphics. CPT Pharmacometrics Syst Pharmacol. 2017;6(2):87–109.
Thai HT, Mentré F, Holford NHG, Veyrat-Follet C, Comets E. A comparison of bootstrap approaches for estimating uncertainty of parameters in linear mixed-effects models. Pharm Stat. 2013;12(3):129–40.
Jonsson EN, Nyberg J. A quantitative approach to the choice of number of samples for percentile estimation in bootstrap and visual predictive check analyses. CPT: Pharmacometrics Syst Pharmacol. 2022;11(6):673–86.
R Core Team. R: A language and environment for statistical computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2022. Available from: https://www.R-project.org. Accessed 2024 Apr 23.
Comets E, Lavenu A, Lavielle M. Parameter Estimation in Nonlinear Mixed Effect Models Using saemix, an R Implementation of the SAEM Algorithm. J Stat Softw. 2017;80:1–41.
Jacobi H, Schaprian T, Schmitz-Hübsch T, Schmid M, Klockgether T, the EUROSCA and RISCA Study Groups. Disease progression of spinocerebellar ataxia types 1, 2, 3 and 6 before and after ataxia onset. Ann Clin Transl Neurol. 2023;10(10):1833–43.
Holford N, Nutt JG. Disease progression, drug action and Parkinson’s disease: Why time cannot be ignored. Eur J Clin Pharmacol. 2008;64(2):207–16.
Muralidharan KK, Kowalski KG, Tong X, Haeberlein SB, Rajagovindan R, Nestorov I. Characterization of exposure-Clinical Dementia Rating-Sum of Boxes relationship in subjects with early Alzheimer’s disease from the aducanumab Phase 3 trials. J Pharmacokinet Pharmacodyn. 2023;50(1):45–62.
Oulhaj A, Refsum H, Beaumont H, Williams J, King E, Jacoby R, et al. Homocysteine as a predictor of cognitive decline in Alzheimer’s disease. Int J Geriatr Psychiatry. 2010;25(1):82–90.
Maas RPPWM, Teerenstra S, Lima M, Pires P, Pereirade Almeida L, van Gaalen J, et al. Differential Temporal Dynamics of Axial and Appendicular Ataxia in SCA3. Mov Disord. 2022;37(9):1850–60.
Zou H, Aggarwal V, Stebbins GT, Müller MLTM, Cedarbaum JM, Pedata A, et al. Application of longitudinal item response theory models to modeling Parkinson’s disease progression. CPT Pharmacometrics Syst Pharmacol. 2022;11(10):1382–92.
Ueckert S, Plan EL, Ito K, Karlsson MO, Corrigan B, Hooker AC, et al. Improved utilization of ADAS-cog assessment data through item response theory based pharmacometric modeling. Pharm Res. 2014;31(8):2152–65.
Novakovic AM, Krekels EHJ, Munafo A, Ueckert S, Karlsson MO. Application of Item Response Theory to Modeling of Expanded Disability Status Scale in Multiple Sclerosis. AAPS J. 2017;19(1):172–9.
Buatois S, Retout S, Frey N, Ueckert S. Item Response Theory as an Efficient Tool to Describe a Heterogeneous Clinical Rating Scale in De Novo Idiopathic Parkinson’s Disease Patients. Pharm Res. 2017;34(10):2109.
Hamdan A, Hooker AC, Chen X, Traschütz A, Schüle R, Synofzik M, et al. Item Response Theory Analysis of the Scale for the Assessment and Rating of Ataxia in Autosomal Recessive Cerebellar Ataxias [Internet]. Abstr 10626; 2023; PAGE 31. Available from: www.page-meeting.org/?abstract=10626.
Wood AM, White IR, Royston P. How should variable selection be performed with multiply imputed data? Stat Med. 2008;27(17):3227–46.
van Buuren S. Flexible Imputation of Missing Data, Second Edition. 2nd ed. Boca Raton: Chapman and Hall/CRC; 2018. p. 444.
Traschütz A, Adarmes-Gómez AD, Anheim M, Baets J, Brais B, Gagnon C, et al. Responsiveness of the Scale for the Assessment and Rating of Ataxia and Natural History in 884 Recessive and Early Onset Ataxia Patients. Ann Neurol [Internet]. [cited 2023 Aug 28];n/a(n/a). Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/ana.26712.
Gagnon C, Lessard I, Lavoie C, Côté I, St-Gelais R, Mathieu J, et al. An exploratory natural history of ataxia of Charlevoix-Saguenay. Neurology. 2018;91(14):e1307–11.
Benatar M, Wuu J, McHutchison C, Postuma RB, Boeve BF, Petersen R, et al. Preventing amyotrophic lateral sclerosis: insights from pre-symptomatic neurodegenerative diseases. Brain. 2022;145(1):27–44.
Acknowledgements
This work was funded by the European Joint Programme on Rare Diseases (EJP RD) Joint Transnational Call 2019 for the EJP RD WP20 Innovation Statistics consortium “EVIDENCE-RND” focusing on “Innovative Statistical Methodologies to Improve Rare Diseases Clinical Trials in Limited Populations” under the EJP RD Grant Agreement (n°825575) (to M.K, R.S. and M.S.) Moreover, work in this project was supported by the Clinician Scientist programme "PRECISE.net" funded by the Else Kröner-Fresenius-Stiftung (to M.S., R.S. and A.T). This work was supported by members of the Evidence-RND consortium, which includes Alzahra Hamdan, Andrew C. Hooker, Xiaomei Chen, Mats O. Karlsson, Nicole Maria Heussen, Ralf-Dieter Hilgers, Thomas Klockgether, France Mentré, Yevgen Ryeznik, Oleksandr Sverdlov.
Funding
This work was funded by the European Joint Programme on Rare Diseases (EJP RD) Joint Transnational Call 2019 for the EJP RD WP20 Innovation Statistics consortium “EVIDENCE-RND” focusing on “Innovative Statistical Methodologies to Improve Rare Diseases Clinical Trials in Limited Populations” under the EJP RD Grant Agreement (n°825575) (to M.K, R.S. and M.S.) Moreover, work in this project was supported by the Clinician Scientist programme "PRECISE.net" funded by the Else Kröner-Fresenius-Stiftung (to M.S., R.S. and A.T).
Author information
Authors and Affiliations
Consortia
Contributions
1- Conceived and designed the analysis;
2- Contributed data;
3- Performed the analysis;
4- Wrote the manuscript
5- Comments and revision
NH: 1,3,4,5
FM: 1,4,5
AT: 1,2,5
CG: 2,5
RS: 1,2,5
ARCA Study Group: 2
EVIDENCE-RND consortium: 1,5
MS: 1,2,4,5
EC: 1,4,5
Corresponding author
Ethics declarations
Relevant Conflicts Of Interest/Financial Disclosures
Dr. Klockgether is receiving research support from the Bundesministerium für Bildung und Forschung (BMBF), the National Institutes of Health (NIH) and Servier. Within the last 24 months, he has received consulting fees from Biogen, UCB and Vico Therapeutics, all unrelated to the present manuscript.
Dr. Synofzik has received consultancy honoraria from Ionis, UCB, Prevail, Orphazyme, Servier, Reata, GenOrph, AviadoBio, Biohaven, Zevra, and Lilly, all unrelated to the present manuscript.
Drs Hooker and Karlsson has received consultancy fees from, and own stock in Pharmetheus, all unrelated to this manuscript.
Dr Comets has received consultancy fees from Sanofi, unrelated to this manuscript.
The other authors have no potential conflicts of interest to report.
Ethical Approval
For this study data from the ARCA Registry (arca-registry.org) was used. No new personal data was collected and/or processed. Probands recruited to the ARCA Registry study consented to secondary use of their data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
For ARCA Study Group and EVIDENCE-RND authors, see Supplementary Materials 2.
Matthis Synofzik and Emmanuelle Comets are shared last authorship.
The original online version of this article was revised due to a retrospective Open Access order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hendrickx, N., Mentré, F., Traschütz, A. et al. Prediction of Individual Disease Progression Including Parameter Uncertainty in Rare Neurodegenerative Diseases: The Example of Autosomal-Recessive Spastic Ataxia Charlevoix Saguenay (ARSACS). AAPS J 26, 57 (2024). https://doi.org/10.1208/s12248-024-00925-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-024-00925-7