
Abstract
The statistical assessments needed to establish anti-drug antibody (ADA) assay cut points (CPs) can be challenging for bioanalytical scientists. Poorly established CPs that are too high could potentially miss treatment emergent ADA or, when set too low, result in detection of responses that may have no clinical relevance. We evaluated 16 validation CP datasets generated with ADA assays at Regeneron’s bioanalytical laboratory and compared results obtained from different CP calculation tools. We systematically evaluated the impact of various factors on CP determination including biological and analytical variability, number of samples for capturing biological variability, outlier removal methods, and the use of parametric vs. non-parametric CP determination. In every study, biological factors were the major component of assay response variability, far outweighing the contribution from analytical variability. Non-parametric CP estimations resulted in screening positivity in drug-naïve samples closer to the targeted rate (5%) and were less impacted by skewness. Outlier removal using the boxplot method with an interquartile range (IQR) factor of 3.0 resulted in screening positivity close to the 5% targeted rate when applied to entire drug-naïve dataset. In silico analysis of CPs calculated using different sample sizes showed that using larger numbers of individuals resulted in CP estimates closer to the CP of the entire population, indicating a larger sample size (~ 150) for CP determination better represents the diversity of the study population. Finally, simpler CP calculations, such as the boxplot method performed in Excel, resulted in CPs similar to those determined using complex methods, such as random-effects ANOVA.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the past several decades, protein therapeutics have been developed to treat a broad array of diseases, including arthritis, eye diseases, and cancer (1,2,3). However, administration of biotherapeutics may elicit an unwanted immune response that can result in the generation of anti-drug antibodies (ADA). These ADA responses have the potential to impact the efficacy of the drug or result in serious adverse safety events (4,5,6).
The assays for detecting antibody responses are typically non-quantitative, titer-based methods that involve potentially three evaluations of a sample: a screening assay to detect antibodies that bind to the drug, a confirmation assay to identify samples with a reduction of assay signal in the presence of excess unlabeled drug, and a titer assay to assess the magnitude of the response.
To determine if an ADA sample is positive, a threshold is established by testing multiple drug-naïve individuals in the assay. These data are examined statistically to set cut points (CPs) with a defined false positivity rate for each tier in the ADA assessment (7, 8). This approach is intended to be conservative to ensure patient safety by minimizing the possibility of false-negative results. However, CPs that are set too low can result in a high percentage of samples identified as positive in the assay that are not due to a true immunogenic response. Consequently, establishing appropriate CPs for screening and confirmatory tiers is a crucial factor in immunogenicity assessment to identify true positive responses and to evaluate the clinical relevance of any ADA detected.
Regulatory agencies have issued recommendations for performing ADA assay validations, and there are numerous industry publications and white papers describing approaches to setting CP values (9,10,11). However, the statistical assessments required to establish CPs can be challenging for the bioanalytical scientists responsible for developing and validating immunogenicity assays. These threshold values also help define the assay sensitivity, drug tolerance, and titers, so other stakeholders such as regulators and members of drug development teams have an interest in understanding how these crucial determinants of ADA positivity are established. In recent years, this has become more important with the increasing evidence that supersensitive ADA assays do not necessarily improve detection of impactful immunogenicity and may in fact weaken the correlation between ADA and clinical response (12,13,14,15,16).
Here, we evaluated 16 validation CP datasets, for 15 human monoclonal antibodies and one receptor-Fc fusion protein, from different disease indications and compared the results from a suite of different CP calculation tools that use different levels of statistical complexity, ranging from simple spreadsheet calculations to linear mixed-effect modeling approaches. We also evaluated the impact of several straightforward yet fundamental factors in CP analysis. These included the impact of biological variability and the optimal number of samples for capturing that variability, outlier removal, and the use of parametric vs. non-parametric CPs. From these analyses, we conclude that simpler CP calculation methods, such as the boxplot method using Excel, result in CPs similar to those obtained from more complex methods. Additionally, biological variation is the key factor driving signal variation in study samples, and these datasets are often non-normal, and therefore including large sample sizes (e.g., 150) better captures the variation of the population. Finally, outlier removal processes that exclude too many samples can underestimate the biological variation and result in inappropriate CPs with false positive error rates (FPERs) substantially greater than the targeted 5% (screening) or 1% (confirmation).
Methods and Materials
Bridging Immunogenicity Assays
All ADA assays were developed and validated as per the regulatory guidance (9, 10). The presence of ADA in samples was determined using a non-quantitative, titer-based, bridging immunoassay using a 3-tiered approach. The bridging assay procedure employed biotinylated drug and ruthenium-labeled drug as the bridge components. For most assays, samples were subjected to an acid dissociation step (to dissociate potential antibody:drug complexes) prior to incubation with the labeled drugs to improve detection of ADA in the presence of drug. ADA complexed with labeled drugs were then captured on streptavidin-coated plates and detected by an electrochemiluminescent (ECL) signal (Counts) measured on a Meso Scale Discovery (MSD) plate reader.
Study Samples and Cut Point Experiment Design
Commercially obtained drug-naïve healthy and/or disease-state samples and baseline samples from clinical studies with diseased populations were used for validation CP determination. Informed consent from patients participating in clinical studies had been obtained prior to sample collection. Cut point exercises were performed by at least two analysts over multiple days with a balanced design. Table I specifies the number of samples run per study and number of observations obtained per sample for a particular study. Signal to noise (\(S/N\)) normalization was performed for all samples prior to outlier remove and assessment of CPs described in the methods below.
Linear Mixed-Effects ANOVA Method
The CP data were analyzed using a linear mixed-effects analysis of variance (ANOVA) model programmed in SAS® Version 9.4 (SAS Institute, Inc., Cary, NC, USA) as generally described in Devanarayan et al. (17), Shankar G et al. (8), and Mire-Sluis AR et al. (17) (7, 8, 17). Linear mixed-effects ANOVA models including fixed effects due to sample group and/or analyst and random effects due to sample (nested within group), run (nested within analyst), plate number, and/or residuals were used to remove analytical and biological outliers based on the conditional residual and best linear unbiased prediction (BLUP) values, respectively, as well as identify sources of variation in the sample results. Biological variation was defined as sample-to-sample variation, whereas analytical variation was defined as variation due to other factors, such as analyst or plate number. For studies where each sample was only run once, biological variation was captured under the residual effect. The parametric CP values were calculated using the Tukey’s biweight mean and SD by multiplying the SD by the 95th or 99th quantile of the t-distribution and adding the product to the mean, for the screening (log-transformed \(S/N\) values) and confirmation (%Inhibition values) CPs, respectively. Non-parametric screening and confirmation CPs were calculated as the 95th or 99th empirical percentile, respectively. Data normality was evaluated after removal of outliers by using the Shapiro-Wilk test and skewness.
Excel Boxplot Method
An Excel template was created to determine CPs using the boxplot method of outlier removal with a user-determined interquartile range (IQR) factor. Parametric screening CP factors were calculated as described in Devanarayan (2017), i.e., Mean + 1.645 × SD of log-transformed \(S/N\) values, followed by an inverse log transformation to target a 5% FPER (7). Similarly, parametric confirmation CPs were calculated as the Mean + 2.33 × SD of %Inhibition values to target a 1% FPER. Non-parametric screening CP factors were determined by calculation of the 95th percentile for the log-transformed \(S/N\) values, followed by an inverse log transformation. For the confirmation CP value, the non-parametric CPs were determined based on the 99th percentile of %Inhibition values. Non-parametric CPs were calculated using the percentile (inclusive) function. Data normality was determined after outlier removal by calculating the skewness of the log(\(S/N\)) and %Inhibition values using Excel’s built-in SKEW function. If skewness is greater than 1.0, the data were considered non-normal and the non-parametric CP was selected. Otherwise, the data were considered approximately normal and the parametric CP was recommended.
JMP Boxplot Method
A JMP add-in was developed using JMP® Version 16.1.0 (SAS Institute, Inc., Cary, NC, USA) to determine CP values using the boxplot method for outlier exclusion. The add-in imports the CP data from an Excel workbook, performs a log10 transformation of \(S/N\) values, plots and excludes outlier values, and computes screening and confirmation CP values as described in the Excel boxplot method. The add-in generates a report which displays the CP values, sample positivity (with and without outliers excluded), and outlier plots for a range of IQR factors.
The add-in calculates skewness and performs the Shapiro-Wilk test to determine data normality for the log(\(S/N\)) and %Inhibition distributions to select the parametric or non-parametric CP. If skewness is greater than 1.0 or if the null hypothesis of the Shapiro-Wilk test is rejected (\(p\) value < 0.05), the data were considered non-normal and the non-parametric CP was selected. Otherwise, the data were considered approximately normal and the parametric CP was selected. These values were calculated for the log(\(S/N\)) values for the screening assay and %Inhibition values for the confirmation assay.
Results
A review was conducted of data from CP experiments for 16 clinical ADA assays validated at Regeneron’s bioanalytical laboratory in accordance with current regulatory guidance and/or recommendations (Table I) (9,10,11). All methods were bridging electrochemiluminescent (ECL) assays on the (MSD) platform. Fifteen of the 16 molecules examined were mAb therapeutics of differing isotypes (IgG1 or hinge stabilized IgG4), and one was a receptor-Fc biologic. The therapeutic proteins were specific for a variety of targets including soluble or membrane-bound, endogenous or exogenous molecules.
The datasets were generated using drug-naïve samples from either disease-state or healthy individuals and either baseline clinical study samples or samples obtained from commercial vendors. For all datasets, assay signal for each sample was normalized to the negative control (NC) response (signal-noise, \(S/N\)) on each plate by dividing the Mean Counts for each individual serum sample by the NC Mean Counts from the corresponding plate.
The survey of datasets was performed to evaluate the contribution of different sources of variability and compare the data distributions to better inform decisions about CP determination. Subsequently, these datasets were used to compare CPs calculated with SAS® software using linear mixed-effects ANOVA models and an in-house CP determined using a “boxplot” approach with two widely available software applications: Excel or JMP. The different calculation methods had minimal impact on the screening and confirmation CPs (Table II).
Biological Variability Within Validation Study Sample Sets
Variation in sample responses can be attributed to a combination of analytical and biological factors. To investigate the sources of screening assay response variability (i.e., log(\(S/N\))) in the CP experiments for the 16 validation studies, a linear mixed ANOVA model was used (Table I). An average of almost 90% of the variation was explained by the sample (biological) random effect. For one dataset, 97% of the variability in assay response was due to biological factors, and for all (evaluated) datasets, at least 78% of the variability was due to individual samples (for Studies E and F, each sample was run only once; therefore, any biological variation would be captured in the residual effect). This indicates that sample-to-sample variation, or biological variability, explains a large majority of the overall log(\(S/N\)) differences observed and therefore is the key component in the CP determination. For the 16 datasets, the contribution from analyst, assay run, and residual factors (which capture the measurement-to-measurement or analytical variability) explains on average approximately 10% of the variation observed. A similar trend was observed in the confirmation assay, where biological variability was the largest contributor of %Inhibition variation for 12 out of 14 studies (Supplemental Table I).
Distribution of the Datasets: Non-Normality and Right Skewness
For 15 of the 16 sample populations, the normality assumption was not confirmed (\(p < 0.05\)) by the Shapiro–Wilk test (Table I). This is consistent with published data indicating CP datasets are typically non-normally distributed (18). Another measure of relative symmetry of a distribution is skewness. All 16 datasets had a positive or right skew. Eight of the 16 datasets had a coefficient greater than or equal to 1.0, which can be considered highly skewed, while only 3 had a coefficient less than 0.5, considered to have low skewness (19). Both these metrics of distribution indicate that a large majority of the screening datasets were non-normal and/or asymmetric. Similar results were observed for the distribution of the confirmation datasets, indicating these datasets were also typically non-normal and/or asymmetric (Supplemental Table I).
Impact of IQR Factor and Skewness on Outlier Determination, Cut Point, and Sample Positivity
The interquartile range (IQR) is the spread of the middle 50% of the data (25th–75th percentile or first to third quartile). A common approach to outlier determination is to set a “fence” using an IQR multiple (factor) to identify “outside values” (20). However, the approach suggested by Tukey (1977) of applying a 1.5-fold factor to classify “outside values” assumes an approximately symmetrical distribution, which is not the case for skewed CP datasets.
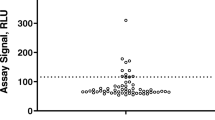
To evaluate the impact of the IQR factor on the screening positivity, percentage of outliers removed, and the screening CP factor, screening data from 16 datasets were analyzed using the JMP boxplot method with IQR factors ranging from 1.5 to 4.0 (Fig. 1). As the IQR factor increased, the percentage of naive or baseline samples that would screen as positive decreased, in most cases reaching a plateau at an IQR factor of 3.0 (Fig. 1a). The range of outlier-inclusive screening positivity ranged from 6.0 to 14.0% using an IQR factor of 1.5, while it ranged from 4.7 to 8.8% using an IQR factor of 3.0.

Effect of IQR factor used for outlier removal on a observations screening positive (outlier-inclusive), b percentage of screening outliers removed, and c screening cut point factor (SCP). All studies used the non-parametric cut point except for Studies I and J, which used the parametric cut point
The plateau in screening positivity at IQR factor ≥ 3.0 was related to changes in the number of outliers removed from the datasets. At 1.5 IQR factor, two datasets (Studies F and K) had more than 8% of the data points removed as outliers, and nine datasets (Studies A, B, D, F, G, K, M, O, and P) had more than 4% of data points removed (Fig. 1b). However, in each case, an IQR factor of 3.0 resulted in less than half the number of outliers being identified compared to an IQR factor of 1.5. Importantly, the total percentage of samples screening positive is approximately the percentage of outliers removed plus the target 5% false positive rate specified in regulatory guidance and white papers. Consequently, those datasets with a large percentage of outliers removed (Fig. 1b) also had an outlier-inclusive screening positivity substantially greater than the recommended 5% rate.
Figure 2 shows similar data for the impact of IQR factor on the confirmation positivity rate, outlier removal, and CP. Here, the impact of a high percentage of outlier removal on the outlier-inclusive positivity is even more impactful for the confirmation CP, with a target FPER of 1% (Fig. 2). An IQR factor of 1.5 resulted in 5 of the 16 studies having an outlier-inclusive positivity greater than 5%, including one dataset with more than 10% of samples confirmed positive. Similar to the screening assay, an IQR factor of 3.0 substantially reduced the number of outliers identified.

Effect of IQR factor used for outlier removal on a observations confirming positives (outlier-inclusive), b percentage of confirmation outliers removed, and c confirmation cut point factor (CCP). All studies used the non-parametric cut point except for Studies I and J, which used the parametric cut point
The distributions for the screening and confirmation range in skewness from relatively low to highly right-skewed (Table I and Supplemental Table I). The positivity rate when using parametric CP values demonstrated an increase that corresponded with increasing skewness (for both screening and confirmation assays; Fig. 3). However, when using non-parametric CP estimates, the screening positivity rate was close to 5% regardless of the level of skewness. Similar results were obtained for the confirmation, with a positivity rate close to 1% for non-parametric CP estimates. Studies I and J were both relatively symmetric and allowed for the use of the parametric CP estimate. However, in both these cases, the non-parametric CPs (SCP 1.40 and 1.41, CCP 52.0% and 41.0%, respectively, for I and J) were similar to the parametric CPs (SCP 1.30 and 1.41, CCP 47.0% and 40.2%, respectively). This suggests that although parametric CP estimations are suitable for distributions with low skewness, non-parametric estimates may be applicable for distributions with a wide range of skewness.

Relationship between validation sample population skewness and naïve sample positivity for the a screening anti-drug antibody assay and b confirmation anti-drug antibody assay for 16 clinical validation studies. SCP: screening cut point value, CCP: confirmation cut point value
Clinical Impact on Number of Baseline Samples Used to Estimate Population-Specific Cut Point Factors
Observing that biological factors explain the majority of variation in assay response, we wanted to evaluate the impact of using larger numbers of individuals in an attempt to capture a true representation of the diversity of the study population. To do this, we investigated the impact of sample size using in-study baseline assay responses from two large phase 3 studies (\(N>3000\)) for two different molecules. Similar to the 16 CP validation datasets, the distribution of log(\(S/N\)) of the baseline samples for both studies exhibited high skewness (5.17 and 2.21, excluding samples which confirmed as drug-specific). For each study, random baseline selections of different sizes (50, 100, 150, and 300 subjects) were sampled 100 times with replacement after each subset (subjects were eligible to be selected once per sampling but were returned to the dataset between samplings). For example, log(\(S/N\)) values from 50 subjects were randomly selected from the clinical assay dataset and these values were used to generate an estimated cut point; this procedure was repeated 100 times per sample size for the entire dataset (see Supplemental Fig. 1). The JMP boxplot method was used to exclude outliers (IQR × 3.0) and calculate the non-parametric CP factor for each of the 100 samplings. A boxplot representation of the distributions of CPs for the 100 samplings at each sample size is shown in Fig. 4. In addition, the population CP value calculated for the entire baseline dataset is indicated by the dashed horizontal reference lines in Fig. 4, both with and without outlier removal. As expected, for both studies, higher sample sizes were associated with a tighter range of potential calculated CPs, centered around the 95th percentile of all baselines excluding outliers. For example, increasing the sample size from 50 to 150 reduced the standard deviation of calculated CPs 45% for Clinical Study 1 (0.40 vs. 0.22) and 56% for Clinical Study 2 (0.43 vs. 0.19). In addition, the median calculated CP became closer to the overall population 95th percentile (excluding outliers) as sample sizes increased. However, increasing the sample size from 150 to 300 reduced the standard deviation by 32% for both Clinical Study 1 (0.22 vs. 0.15) and Clinical Study 2 (0.19 vs. 0.13), suggesting diminishing returns as sample size increases beyond 150 individuals.

Larger sample sizes randomly selected from baseline datasets associated with tighter distribution of possible CPFs. Two phase three studies (Clinical Studies 1 and 2) for different molecules were selected where the number of baseline sample screening assay results was large (\(N>3000\)). Subsets of baseline samples for each study were randomly selected, with replacement between subsets, 100 times with sample sizes of 50, 100, 150, and 300. Outlier exclusion was performed for each random sample using the boxplot method with an IQR factor of 3.0. The distribution of non-parametric cut point factors (empirical 95th percentile) is shown as box-and-whiskers, where the whiskers denote the 5th and 95th percentile of observed CPFs (outliers not shown). The 95th percentile of the entire baseline set for each study is shown as dashed reference lines, before and after outlier removal with IQR factor of 3.0
To further assess the clinical impact of SCPs set with different sample sizes, we investigated the effect of cut points generated with 50 or 150 individuals on in-study baseline samples. The range of different SCPs shown in Fig. 4 (from sample sizes of 50 and 150) was applied to the \(S/N\) results for the predose clinical samples for each study, excluding samples identified as having preexisting ADA using the validated assays, and the resulting FPER in the screening assay was calculated for each SCP. Therefore, a set of 100 FPERs was determined for the corresponding set of 100 SCPs for each sample size of 50 and 150.
A screening FPER in the range of 2% to 11% is generally considered acceptable (7). For Clinical Study 1, SCP set using a sample size of 50 subjects resulted in 36 out of 100 FPERs falling outside of the 2–11% benchmark. In contrast, using a larger sample size of 150 resulted in only 12 out of 100 FPERs falling outside of the accepted range (Table III). A similar trend was observed for Clinical Study 2. When the sample size for the dataset was 150, only 3 out of 100 FPERs were outside the 2–11% range, versus 10 out of 100 FPERs when SCPs were set with sample sizes of 50.
To understand the clinical impact of outlier removal, we evaluated the effect of varying IQR factor on baseline sample positivity using these same studies. To do this, we estimated non-parametric SCPs using the entire baseline datasets after excluding outliers using an IQR factor of either 1.5 or 3.0 (Table IV). These SCPs were applied back to the baseline \(S/N\) dataset to estimate an overall FPER. As shown in Table IV, the use of IQR factor of 3.0 results in FPER closer to the target 5%, while using an IQR factor of 1.5 results in around 10% FPER (9.5% or 10.1% for Clinical Study 1 and 2, respectively) in the screening assay for both studies.
Discussion
Cut point determination is a critical parameter for immunogenicity assays. The concept of establishing a threshold with a defined false-positive rate based on statistical analysis of background assay responses is theoretically straightforward. However, decisions about the population type and size used for CP experiments as well as the statistical approaches employed can substantially impact the resulting threshold values. Selecting the most appropriate strategies in several key steps is critical to establishing suitable CPs. Furthermore, demystifying the CP setting process is crucial to enable bioanalytical scientists to address questions from health authorities about assay CPs and potential impact on downstream factors such as high placebo positivity rates or poor correlation with clinical outcomes. To that end, we systematically investigated 16 validation CP datasets from Regeneron’s bioanalytical laboratory, across a range of molecules and indications, to elucidate key factors impacting CP determination.
One driver of unsuitable CPs is selection of an inappropriate sample population and size to establish the background signal in the assay. (An exception is for rare diseases, where sample size is limited.) Testing individuals from the disease indication of interest and representing demographics of the target population are highly recommended. The current recommendation is to analyze around 50 treatment-naïve subjects from the target indication, with each subject analyzed by two analysts over three days. This balanced design results in six observations per subject and 300 observations overall, which allows the experimenter to describe variation due to analyst, run, plate order, and sample groups (8, 10, 11). However, determination of the “true” CP from statistical analysis of screening results from all baseline samples in two phase 3 datasets indicates that a sample size of 50 is less likely than larger sample sizes to generate appropriate values. Data extracted from our internal LIMS database suggest 150 samples generate CPs closer to the “true” CP of the population, within a clinical trial, with approximately half as much variation in CP estimates as when using 50 samples (Fig. 4). In addition, as we and others have shown, in well controlled assays the major source of variability in assay signal from drug naïve individuals is biological (21). Other sources of variability (e.g., analyst, day, and plate) that are captured by repeated observations of a sample constitute only a minor fraction of the total (Table I). Therefore, CPs established using larger samples sizes, such as 150, are more likely to generate appropriate CPs by better capturing the biological diversity of the study population. This is shown in Table III where a smaller proportion of SCPs set using 150 subjects fall outside of the 2–11% FPER benchmark compared to CPs set using 50 subjects. The generation of SCPs that are appropriate across studies within the same indication will likely lead to more meaningful associations in the immunogenicity assessment.
Another consideration in ensuring appropriate CPs is the normality and skewness assessment of the distribution of assay responses. Skewness and normality testing can provide a quantitative method for objective determination of assay response distribution, as described herein. A skewness of ± 1.0 is commonly used as a threshold for whether a sample distribution is considered normal or non-normal. A normality test such as the Shapiro-Wilk test can also determine normality of data where the null hypothesis of data normality is rejected with a \(p\) value < 0.05. Fifteen out of 16 studies shown here were considered non-normal by one of these criteria suggesting the use of the non-parametric method for CP calculation (Table I). However, even when the data distribution was normal, the parametric and non-parametric CPs were very similar (Fig. 3) indicating that the non-parametric CP may be suitable for most distributions (assuming a sufficient number of observations—at least 300 recommended in 2019 FDA guidance) (10).
Finally, a key driver of unsuitably low CPs is outlier removal. The standard outlier removal method is to set “fences” determined by adding (or subtracting) a multiple of the interquartile range to the median and excluding values outside these limits (20). However, this approach assumes a symmetrical distribution. Two pieces of evidence indicate screening CP datasets are rarely symmetrical. First, in the screening assay, very few outliers are identified on the low side of the signal distribution. Second, the distribution of most CP datasets before outlier removal or transformation is not normal (Table I) (18). Therefore, although a 1.5 multiple of the IQR removes only 0.7% of individuals in a symmetrical dataset, in non-normal datasets, this percentage increases significantly, potentially more than 10% (7, 22). This may lead to excluding the actual 95th percentile (SCP) or 99th percentile (CCP) of the sample distribution that subsequent calculations estimate. This high percentage elevates the observed screening positivity rate of the baseline samples in the patient populations, which is approximately the percentage of outliers (in some cases > 10%) plus the 5% FP rate statistically built into the calculations (positivity rate). Excessive outlier removal is even more impactful when setting the confirmation CP. In the confirmation assay, the desired FP rate is 1%. Removal of 10% of the population or more as outliers before applying the built-in 1% rate can lead to dramatically elevated rates of reported ADA positivity.
Ishii-Watabe et al. (2018) suggest that CP outlier exclusion using an IQR factor in the range of 1.5 to 3.0 is suitable, as long as the statistical approach to CP factor generation can be scientifically justified (11). Kubiak et al. (2018) demonstrate using simulated data that outlier removal with an IQR factor of 1.5 strips biological variability from the dataset, resulting in CPs close to the minimum CPs which may be irrelevant for use with study samples (21). Our data suggest an IQR factor of 3.0 is appropriate for most datasets, as it more frequently results in an outlier-inclusive positivity closer to the target FPER (Figs. 1 and 2; Table IV).
Inappropriately high or low CP values are an ongoing challenge in immunogenicity testing (21, 23, 24). Values that are too high may result in failure to detect treatment emergent ADA. Conversely, CPs that are too low can result in elevated false positive rates in baseline or placebo groups that may lead to questions about the selectivity of the assay (as well as unnecessary bioanalysis in confirmation, titer, and neutralizing antibody assays). In addition, low immunogenicity CPs may result in detection of positive responses in the assay that dilute the true positive responses, leading to a weaker correlation of the impact of ADA on clinical outcomes such as safety and efficacy (12,13,14,15,16).
Conclusion
Based on our analysis of 16 validation CP datasets covering a range of healthy and disease-state drug-naïve populations and different drugs/targets, we observed that the majority of sample variability in CP determination is due to the natural biological diversity in the patient population. Consequently, using a larger number of samples representative of the study population is more likely to generate suitable CP values. In addition, excessive outlier removal by definition reduces the biological diversity in the population and therefore using a low IQR factor (e.g., 1.5) for outlier removal is often not appropriate. Our data suggest the boxplot method for outlier removal, as can be performed in Excel, achieves similar CPs as more complex methods, such as linear mixed models. Using these straightforward approaches to CP setting can ensure clinically meaningful treatment-emergent responses are identified while generating sensitive ADA assays to ensure patient safety.
References
Heier JS, Brown DM, Chong V, Korobelnik JF, Kaiser PK, Nguyen QD, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119(12):2537–48. https://doi.org/10.1016/j.ophtha.2012.09.006.
van de Putte LB, Atkins C, Malaise M, Sany J, Russell AS, van Riel PL, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63(5):508–16. https://doi.org/10.1136/ard.2003.013052.
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33. https://doi.org/10.1056/NEJMoa1606774.
Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–8. https://doi.org/10.1001/jama.2011.406.
Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian J-J, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–75. https://doi.org/10.1056/NEJMoa011931.
Rosenberg AS. Immunogenicity of biological therapeutics: a hierarchy of concerns. Dev Biol (Basel). 2003;112:15–21.
Devanarayan V, Smith WC, Brunelle RL, Seger ME, Krug K, Bowsher RR. Recommendations for systematic statistical computation of immunogenicity cut points. AAPS J. 2017;19(5):1487–98. https://doi.org/10.1208/s12248-017-0107-3.
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81. https://doi.org/10.1016/j.jpba.2008.09.020.
European Medicines Agency. Guideline on immunogenicity assessment of therapeutic proteins. Committee for Medicinal Products for Human Use; 2017. Available from: https://www.ema.europa.eu/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf. Accessed 30 Mar 2023.
US Food and Drug Administration. Immunogenicity testing of therapeutic protein products—developing and validating assays for anti-drug antibody detection. Guidance for Industry. U.S. Department of Health and Human Services Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Center for Biologics Evaluation and Research (CBER); 2019. Available from: https://www.fda.gov/ucm/groups/fdagov-public/@fdagovdrugs-gen/documents/document/ucm629728.pdf. Accessed 30 Mar 2023.
Ishii-Watabe A, Shibata H, Nishimura K, Hosogi J, Aoyama M, Nishimiya K, et al. Immunogenicity of therapeutic protein products: current considerations for anti-drug antibody assay in Japan. Bioanalysis. 2018;10(2):95–105. https://doi.org/10.4155/bio-2017-0186.
Hassanein M, Partridge MA, Shao W, Torri A. Assessment of clinically relevant immunogenicity for mAbs; are we over reporting ADA? Bioanalysis. 2020;12(18):1325–36. https://doi.org/10.4155/bio-2020-0174.
Partridge MA, Karayusuf EK, Shyu G, Georgaros C, Torri A, Sumner G. Drug removal strategies in competitive ligand binding neutralizing antibody (NAb) assays: highly drug-tolerant methods and interpreting immunogenicity data. AAPS J. 2020;22(5):112. https://doi.org/10.1208/s12248-020-00497-2.
Song S, Yang L, Trepicchio WL, Wyant T. Understanding the supersensitive anti-drug antibody assay: unexpected high anti-drug antibody incidence and its clinical relevance. J Immunol Res. 2016;2016:3072586. https://doi.org/10.1155/2016/3072586.
Van Stappen T, Vande Casteele N, Van Assche G, Ferrante M, Vermeire S, Gils A. Clinical relevance of detecting anti-infliximab antibodies with a drug-tolerant assay: post hoc analysis of the TAXIT trial. Gut. 2018;67(5):818–26. https://doi.org/10.1136/gutjnl-2016-313071.
van Schouwenburg PA, Krieckaert CL, Rispens T, Aarden L, Wolbink GJ, Wouters D. Long-term measurement of anti-adalimumab using pH-shift-anti-idiotype antigen binding test shows predictive value and transient antibody formation. Ann Rheum Dis. 2013;72(10):1680–6. https://doi.org/10.1136/annrheumdis-2012-202407.
Mire-Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289(1–2):1–16. https://doi.org/10.1016/j.jim.2004.06.002.
Zhang J, Li W, Roskos LK, Yang H. Immunogenicity assay cut point determination using nonparametric tolerance limit. J Immunol Methods. 2017;442:29–34. https://doi.org/10.1016/j.jim.2017.01.001.
Bulmer MG. Principles of statistics. New York, N.Y.: Dover Publications, Inc.; 1979.
Tukey JW. Exploratory data analysis. Reading, Mass.: Addison-Wesley Pub. Co.; 1977.
Kubiak RJ, Zhang J, Ren P, Yang H, Roskos LK. Excessive outlier removal may result in cut points that are not suitable for immunogenicity assessments. J Immunol Methods. 2018;463:105–11. https://doi.org/10.1016/j.jim.2018.10.001.
Gijbels I, Hubert M. Robust and nonparametric statistical methods. Comprehensive Chemometrics. 2009:189–211. https://doi.org/10.1016/b978-044452701-1.00093-4.
Loo L, Harris S, Milton M, Meena, Lembke W, Berisha F, et al. 2021 White paper on recent issues in bioanalysis: TAb/NAb, viral vector CDx, shedding assays; CRISPR/Cas9 & CAR-T immunogenicity; PCR & vaccine assay performance; ADA assay comparability & cut point appropriateness (part 3 - recommendations on gene therapy, cell therapy, vaccine assays; immunogenicity of biotherapeutics and novel modalities; integrated summary of immunogenicity harmonization). Bioanalysis. 2022;14(11):737–93. https://doi.org/10.4155/bio-2022-0081.
Stevenson L, Richards S, Pillutla R, Torri A, Kamerud J, Mehta D, et al. 2018 White paper on recent issues in bioanalysis: focus on flow cytometry, gene therapy, cut points and key clarifications on BAV (part 3 - lba/cell-based assays: immunogenicity, biomarkers and PK assays). Bioanalysis. 2018;10(24):1973–2001. https://doi.org/10.4155/bio-2018-0287.
Funding
This research was funded by Regeneron Pharmaceuticals, Inc.
Author information
Authors and Affiliations
Contributions
J.G., S.M., E.S., and Q.Y. developed the cut point calculation tools. J.G., S.M., J.T., Q.Y., E.N., and K.F. analyzed the cut point datasets. K.K. and K.F. performed sample analysis for clinical studies. A.T., J.C., G.S., and M.A.P. oversaw the validation of the ADA assays. J.R. and A.T. oversaw the sample analysis. J.G., S.M., J.T., E.S., Q.Y., E.N., J.C., and M.A.P. wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
All authors are employees of Regeneron Pharmaceuticals, Inc., and may hold stock and/or stock options in the company.
Disclaimer
The views expressed in this article are those of the authors and do not reflect official policy of Regeneron Pharmaceuticals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garlits, J., McAfee, S., Taylor, JA. et al. Statistical Approaches for Establishing Appropriate Immunogenicity Assay Cut Points: Impact of Sample Distribution, Sample Size, and Outlier Removal. AAPS J 25, 37 (2023). https://doi.org/10.1208/s12248-023-00806-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-023-00806-5