
Abstract
Reference-scaled average bioequivalence (RSABE) has been recommended by Food and Drug Administration (FDA), and in its closely related form by European Medicines Agency (EMA), for the determination of bioequivalence (BE) of highly variable (HV) and narrow therapeutic index (NTI) drug products. FDA suggested that RSABE be evaluated by an approximating procedure. Development of an alternative, numerically exact approach was sought. A new algorithm, called Exact, was derived for the assessment of RSABE. It is based upon the observation that the statistical model of RSABE follows a noncentral t distribution. The parameters of the distribution were derived for crossover and parallel-group study designs. Simulated BE studies of HV and NTI drugs compared the power and consumer risk of the proposed Exact method with those recommended by FDA and EMA. The Exact method had generally slightly higher power than the FDA approach. The consumer risks of the Exact and FDA procedures were generally below the nominal error risk with both methods except for the partial replicate design under certain heteroscedastic conditions. The estimator of RSABE was biased; simulations demonstrated the appropriateness of Hedges’ correction. The FDA approach had another, small but meaningful bias. The confidence intervals of RSABE, based on the derived exact, analytical formulas, are uniformly most powerful. Their computation requires in standard cases only a single-line program script. The algorithm assumes that the estimates of the within-subject variances of both formulations are available. With each algorithm, the consumer risk is higher than 5% when the partial replicate design is applied.
Similar content being viewed by others

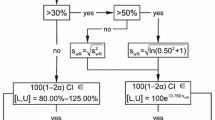
INTRODUCTION
American and European regulatory authorities, the Food and Drug Administration (FDA), and the European Medicines Agency (EMA), respectively, have adopted differing but related procedures for the determination of bioequivalence (BE) of highly variable (HV) drugs and drug products.
FDA recommends that the difference between the logarithmically transformed means of two formulations be standardized by the within-subject standard deviation of the reference product (1). The approach of reference-scaled average BE (RSABE) requires that the reference formulation be measured twice in each subject. For the evaluation of the confidence limits of RSABE, its model needs to be squared, linearized, and probability bounds for each component calculated (2,3). FDA recommended this approach, published a computer program for accomplishing it (4), and implemented it in a draft guidance for progesterone (5).
EMA suggests for HV drugs that the BE limits be proportional to the within-subject standard deviation of the reference formulation and that the classical confidence interval or the two one-sided test (TOST) procedure be applied directly with these limits (6).
Tothfalusi and Endrenyi proposed a third method based on the noncentral t distribution (7). However, the scope of this algorithm was very limited. It could be used only for standard two-period, two-sequence (2 × 2) crossover studies, and the scaling factor for scaled average bioequivalence (SABE) was not the within-subject variation of the reference formulation but the pooled within-subject variation.
This method has not gained acceptance because it could be used only for scaled average bioequivalence (SABE) but not for RSABE. In the present communication, we develop further our initial proposal and show that the method based on the noncentral t distribution can be applied generally for testing RSABE. The method evaluates RSABE exactly, i.e. not approximately. Its calculation and properties will be illustrated for various designs and conditions of BE studies. It will be demonstrated that the proposed exact method can be calculated by a simple procedure.
RSABE was introduced to solve the bioequivalence problem for highly variable drugs and drug products (3,7). Indeed, regulatory authorities accept RSABE to establish bioequivalence only if s WR > 0.294 (CVWR > 30%) (1,5,7).
Recently, however, FDA proposed (8–10) that the bioequivalence of narrow therapeutic index drugs, such as warfarin, should also be assessed with RSABE. The performances of the various methods evaluating RSABE will be evaluated also for NTI drugs.
METHODS
Theory: an Exact Method to Evaluate RSABE
The statistical background of the proposed algorithm will be developed in a stepwise manner. For an easier understanding, we start from the simplest study design to more complex study arrangements.
Paired, Reference-Scaled Bioequivalence for Two-Period Crossover Studies
If the period effect is not a concern, then the simplest way to demonstrate bioequivalence is a study with a paired design where each subject gets one of the T(est) and R(eference) drug product first and then, after an appropriate washout period, the same subjects receive the other formulation. The statistical model is
where y iR and y iT are the observed, logarithmically transformed pharmacokinetic parameters in subject i, μ R and μ T are the population means, and the within-subject random error (ε) is assumed to follow a normal distribution with a mean of zero and a variance of σ W 2. Initially, we assume that there is no difference between the within-subject variances of the formulations: σ 2 WT = σ 2 WR = σ 2 W. The difference between μ T and μ R is estimated from the difference of the corresponding means: (Ȳ T − Ȳ R) and σ 2 ȲT − ȲR = 2σ 2 W.
The pivotal index for scaled bioequivalence is
Scaled bioequivalence is shown at least at the 90% level if the relationship
holds where δ is the true, population value of d and θ is a regulatory criterion.
Equation 2 is similar to the criterion for average bioequivalence, but there is a big difference. The sampling distribution of d cannot be described with known analytical distribution functions. Only the sampling distribution of another random variable, d/K is known (11); here K, a constant, is related to the number of subjects, n:
The sampling distribution of d/K is a noncentral t distribution (Tnc) with a noncentrality parameter of d/K and n−1 degrees of freedom.
Since the sampling distribution of d/K is known, we can easily compute the lower and upper 90% confidence limits for d/K:
Supposing that
holds then, after multiplying each part of Eq. 5 by K, we get that Eq. 2 is also true. That is, if Eq. 5 holds with 90% confidence, then Eq. 2 will also be true with 90% confidence. For a somewhat more general proof of this statement, see Steiger (12). Based on this, the classical confidence interval-based approach to establish SABE, by the classical confidence approach, is (11)
-
1-
Estimate Ȳ T − Ȳ R and s W
-
2-
Set K = (2/n)0.5, d = (Ȳ T − Ȳ R)/s W
-
3-
Calculate the noncentrality parameter, d/K
-
4-
Calculate the lower and upper 90% confidence limits:
and
where Tnc is the quantile function of the noncentral t distribution at the 0.05 and 0.95 levels with a noncentrality parameter of d/K and n−1 degrees of freedom. SABE is established if Eq. 5 is true.
This algorithm of the confidence interval approach was initially described by Steiger and Fouladi (13). Equation 5 permits that SABE be claimed if the back-transformed confidence limits (L’ and U’, multiplied by K) are between the regulatory limits.
Two One-sided Tests Procedure
Schuirmann introduced the two one-sided tests (TOST) approach to establish bioequivalence (14). This approach can also be used to establish SABE. The strategy is the same as above: first, try to establish that the transformed d/K variable is between the transformed limits [−θ/K, θ/K] with 90% confidence. If the transformed limits are set correctly, then we can conclude, also with 90% confidence, that δ is between the regulatory limits [−θ, θ]. Tothfalusi and Endrenyi proposed the following TOST algorithm to establish SABE (7):
Define δ, K, and θ as above. But L’ and U’ are different from above:
and
The two one-sided hypothesis test corresponds to L’ > d/K and U’ < d/K. If both hypothesis are rejected, then we establish with 90% confidence that
Multiplying Eq. 7 by K, we get back again Eq. 2 and we can conclude at the 90% level of confidence that SABE has been established.
The TOST approach above was described also by Wellek (15). He showed also that a TOST-based test is uniformly most powerful. When average bioequivalence is tested, then the confidence interval approach and the TOST approach are “operationally” equivalent. But this is not true for SABE; the confidence interval and TOST algorithms can yield different results and they are not equivalent. Because we know that the TOST algorithm is the optimal solution, we shall focus on it and use the confidence interval approach only for illustration.
Scaled Difference Is Estimated with Bias
Hedges (16) investigated the statistical properties of d as an estimator of δ. Hedges proved the following results about d:
-
1-
It is a biased estimate, d underestimates δ.
-
2-
It is possible to get an unbiased estimate of δ if d is divided by a correction factor (cr). The cr correction factor depends only on, and is a complicated function of, the degrees of freedom (df) of s R. Thus, we denote the correction factor as cr(df). Hedges (16) showed that it can be approximated as
Therefore, the procedure for establishing SABE should take into account the bias correction for d. Consequently, SABE can be established with 90% confidence if the relations
and
are satisfied.
Reference-Scaled Bioequivalence for Parallel Design Studies
In a parallel design bioequivalence study, m subjects get the R formulation and n subjects the T formulation. The statistical model is
where μ R and μ T are, as before, the logarithmic means of the two drug products and both ε i and ε j follow normal distributions with means of zero and variances of σ 2. We initially assume that the population variances of T and R are the same. Still, we have two estimates from the two arms of the parallel design study. Denote these two estimates of the population standard deviations by s T and s R. Ȳ T and Ȳ R estimate the means, μ T and μ R, respectively. RSABE is established if
where θ is again a regulatory constant. We note that there is no regulatory recommendation for setting the value of θ in parallel design studies.
Define now d as
Unlike in Eq. 1 of the paired case, the deviation between the means is scaled now by the (total) standard deviation of the reference formulation (s R). Psychologists call d as Glass’s estimator to measure effect size.
In studies with parallel design, evaluated with reference scaling, d has a noncentral t distribution with m-1 degrees of freedom and the noncentrality parameter of dK−1/cr(m−1). Here, the constant K is now
(16). Regardless of the value of the constant K, we can apply the same TOST approach as described in the section about paired, crossover bioequivalence studies. The only difference is the definition of K, and instead of a naïve estimate d, we should use the bias-corrected form. That is, RSABE for parallel design can be established if the relations
and
are satisfied with 90% confidence. Equations 13a and 13b are true if the logically opposites of Eqs. 13a and 13b are rejected at the 5% level. And if the validity of Eqs. 13a and 13b is established then, after multiplication by K, we can conclude with 90% confidence that the bias-corrected d is in the [−θ, θ] interval.
Crossover Designs
In crossover studies, an unbiased estimate of μ T − μ R can be obtained from the group-by-periods means. For example, consider a four-period, two-sequence design, denoted by TRTR-RTRT. We again assume that there is no difference between the within-subject variances of the two formulations: σ 2 WT = σ 2 WR = σ 2 W. Denote the corresponding group-by-period means in the jth sequence as Ȳ Tji and Ȳ Rji, and the group-by-sequence means as Ȳ Tj and Ȳ Rj.
To simplify the notation, we assume a balanced study with n/2 subjects in both sequences. The difference of μ T − μ R can be estimated by sequences as
After multiplication and rearrangement we get:
The standard error for the formulation difference can be computed by considering only the within-subject error (that is, the subjects are treated as a fixed factor) and by summing the corresponding within-subject error terms on the right side of Eq. 14d (17,18):
We get, after some algebra:
The Ȳ T − Ȳ R difference divided by its standard error follows a noncentral t distribution (11,12). Therefore, after substitution, we get for δ (using notations as before)
Thus, we obtain exactly the same formula as for the paired design except that now K = 1/\( \sqrt{n} \).
Therefore, the RSABE test follows the same pattern as above:
-
1-
Estimate d from (Ȳ T − Ȳ R)/s W
-
2-
The TOST test for SABE is
and
df is now n–2 but generally it is Σn i − s where n j is the number of subjects in sequence j and there are s sequences.
Unequal Within-Subject Variations of the Test and Reference Products: Heteroscedasticity
We assumed until now that the within-subject variances of the Test and Reference formulations were the same: σ 2 WT = σ 2 WR. If this does not hold, then we cannot simply sum the terms on the right of Eq. 15a. The summation must be performed separately for s WT and s WR. Using the four-period, two-sequence example above,
The Ȳ T − Ȳ R difference divided by its standard error is denoted by d’. After simplification of Eq. 18, we get
When the variances of the Test and Reference formulations are truly different then the sampling distribution of δ’ follows only approximately the noncentral t distribution (11,19) and there is no closed analytical formula of the corresponding distribution. But according to the draft bioequivalence requirements (6), the scaling factor must be s WR. Therefore, we proceed as previously and rely on simulations to establish the validity of the formula. Let us denote the estimated σ WT/σ WR ratio by z. Substituting the definition of z into Eq. 19 and rearranging, we get
But what we are seeking is not the distribution of d’ but d/K. Fortunately, it is easy to transform Eq. 20 to the desired form by setting K to
K should be used to evaluate RSABE in a balanced, four-period, two-sequence crossover bioequivalence study (Eqs. 17a and 17b). Contrary to the previous cases, the solution will not be exact in the sense that the sampling distribution of d/K depends on the extent of heteroscedasticity measured by z. But in bioequivalence studies, z–s are only moderately different from 1 and therefore the sampling distribution of d/K can be approximated well by noncentral t. If the approximation is wrong, then the consumer error of the TOST test can be above the nominal 5%. Therefore, the difference between the nominal and actual consumer rates in simulations can be used as a yardstick to measure how good the noncentral approximation is in the case of heteroscedasticity.
Other Designs
RSABE can be tested with the general formula of Eqs. 17a and 17b where the value of K depends on three factors: the design, the extent of heteroscedasticity (z), and n j the number of subjects in sequence j The general formula for K is
where C Tij and C Rij are elements of the contrast matrixes. C T and C R , j is the running index for sequences, and i is the running index for periods. The formula is just a generalization of the examples above, and to illustrate how to use it, we give three examples.
In parallel design studies, C T1 = [1, 0] and C R2 = [0, −1], the other row vectors are zero. Assuming that there are m and n subjects in the T and R arms of the parallel study, respectively, we get
For an RRT-RTR-TRR design, among the elements of the first row of C T , (C T1) is [0, 0, 1/3] while C Rj is [1/6, −1/6, 0]. The other rows are just permutations of the first row.
Assuming n/3 subjects in each sequence, we get
For a two-sequence, three-period TRT-RTR design, the contrast vector for sequence 1 is C T11 = [½, 0, ½], and for sequence 2, the vector is [0, ½, 0]. Squaring the elements of the vectors and assuming that there are n/2 subjects in each sequence, we get
In this case, the degree of freedom of the noncentral t distribution is n/2−1, because s WR is estimated only from the data of the RTR sequence.
For a general method to calculate elements of C T and C R , see Ratkowsky (20). The R code to evaluate RSABE with the Exact method for different designs can be downloaded as electronic supplementary material of this paper from the Journal’s homepage.
The FDA Method
The currently recommended method by the FDA to evaluate RSABE is based on the work of Hyslop et al. (2) for a model of individual BE. Implementation for RSABE was described in (3,9).
The SABE regulatory model of Eq. 1 can be applied also to RSABE except that s W should be replaced by s WR, the within-subject standard deviation of the reference product. Let us express then Eq. 1 in its squared form:
and then linearized
Substituting the estimates of μ T, μ R, and σ WR, the two components of Eq. 27 are
and
With the distributions of the two terms, their confidence limits can be calculated:
Here, SE is the standard error of the difference between the means. t and χ 2 are inverse cumulative distribution functions evaluated at the probability level of α = 0.95. The degrees of freedom to calculate C m and C s may not be the same but in a general form equal df = Σn i − s where n j is the number of subjects contributing to estimate in sequence j and there are s sequences. For example in a TRT-RTR design, df for C m is n T + n R–2 while df for C s is only n R−1.
The confidence interval of the of sum random variables, from the individual confidence intervals, is obtained by the method of Howe (21). The squared lengths of the individual confidence intervals are
The confidence interval of the sum is
BE is demonstrated by the RSABE approach if the 95% upper confidence bound of C.I. is negative or zero (2–4).
Bias of the FDA Approach
The upper confidence bound of C.I. will now be evaluated under a limiting condition. Assume that the means of the two drug products are equal: Ȳ T − Ȳ R = 0. The two components are then, from Eqs. 28a and 28b,
and
and also, from Eq. 29a
Assume now further that the BE limit is just barely touched. Then, interpreting and accordingly modifying Eq. 1
or, with the earlier assumption of \( {\overline{\mathrm{Y}}}_{\mathrm{T}}\hbox{--} {\overline{\mathrm{Y}}}_{\mathrm{R}} \)ȲT-ȲR = 0:
Therefore, the confidence limits are
The squared lengths of the confidence interval are
The components of the confidence interval are
The upper confidence bound is, from Eq. 31, the sum of these two terms. In order to add up to zero, as expected by the FDA guidances (4,5), the expression within the square brackets should be 1.00. Its apparent deviation will be discussed later.
The ABEL Approach
The method of ABEL (average bioequivalence with expanding limits) treats s W as if it was a constant. RSABE is established if
with 90% confidence. The lower and upper bounds of the 90% confidence interval around Ȳ T − Ȳ R are
Here again S.E. is the standard error of the difference between the means, t is the central, Student’s t distribution, evaluated at the 0.05 level with df = n − s where s is the number of sequences. RSABE is declared if the L and U confidence intervals are within the regulatory limits:
Experimental: Simulations
The simulations were performed on a desktop PC with an Intel Core i5-2500K processor and 8 Gb RAM. A program was written in the R language (22) for the simulation of paired (two-period crossover), parallel, three-period, three-sequence partial replicate and four-period, two-sequence full replicate crossover studies. Under each condition, 25,000 simulations were performed. For crossover design studies, we assumed zero period and sequence effects. If otherwise not stated, then we used the following default values: n = 24, s WT, s WR, s T, and s R = 0.4. The simulated random variables followed a normal distribution, but to keep the conventions, we report them as if we initially simulated lognormal variables with a given geometric mean ratio. That is, in the simulations, the true deviation between the means was set at various values starting from zero (indicating actual bioequivalence) and gradually rising to increasing deviations from true bioequivalence, but we report this process as if the simulated GMR values were between 1.00 and 1.60.
The simulated bioequivalence studies were evaluated with the ‘lm’ function of R according to the standard linear model:
In this model, the formulation effect corresponds to the difference between the treatment means and, to be in line with the theoretical section, we refer to it in this way.
The within-subject standard deviations were computed from the residual errors obtained from all data or their subsets. But this approach was not possible in a case of the partial replicate design. Therefore, we used here the lme function from the nlme library (23) to estimate the s WT/s WR ratio. The following R code snippet provides all parameters which are needed to evaluate RSABE from a three-period partial replicate bioequivalence study:
The FDA (5) and EMA guidelines (6) allow to set several additional constraints beside the RSABE criterion. The HVD criterion means that RSABE to demonstrate bioequivalence can be used only if CVWR > 30% (s WR > 0.294) and also the estimated GMR must be between 0.80 and 1.25. Neither of these additional constraints was applied. Finally, the US and European authorities recommend different regulatory values of θ for RSABE. The US suggestion is more liberal and recommends θ = 0.893 (1,5) while the corresponding regulatory cutoff in the EU is 0.760 (6). Statistical methods can be compared if all other parameters are the same; therefore, we used the FDA criterion in all simulations. In any case, we could have selected any θ, including the value used in the EU, because the conclusions are independent of the selection of the regulatory constant.
RESULTS
We compared the performances of the Exact method to evaluating RSABE with the other regulatory recommendations: Hyslop’s method from the FDA (5,8) and the ABEL method recommended by the EMA EU (6).
Correction of the Bias of d
The straightforward, plug-in estimator for reference-scaled difference, (μ T − μ R)/σ, when the individual estimates are inserted into the formula, is the (Ȳ T − Ȳ R)/s W ratio (Eq. 1). But Hedges (16) showed that this naïve estimator is biased, and to get the correct value, a correction factor should be applied. The bias depends only on the number of degrees of freedom of the sampling distribution of s W. To illustrate the prediction of the bias, four-period, two-sequence bioequivalence trials were simulated with different numbers of subjects. The estimated (Ȳ T − Ȳ R)/s W, the scaled difference from each simulation, was divided by the true value. Figure 1 illustrates how the ratio of naïve estimate/true value depends on the sample size. The simulated ratios are represented by symbols and the curve is the predicted value according to Hedges (16).

Correction for the bias of the scaled difference between the means. Reference-scaled differences, (Y T − Y R)/s R were estimated from 25,000 simulated four-period crossover trials by standard ANOVA. The plug-in estimator for reference-scaled difference is the ratio of the corresponding least-squares estimates of Y T − Y R and s R. The estimated scaled difference was divided by the true value used in the simulations. The squares in the figure represent the means of the simulations; the continuous curve is the value predicted by Eqs. 9a and 9b. Simulation conditions: n = 40, s R and s T = 0.4, GMR = 1
Figure 1 shows a perfect match between simulated and predicted values. Figure 1 illustrates that the bias is quite severe when n is low and remains noticeable when n is high. It depends only on n. An unbiased estimate can still be obtained with the correction factor introduced by Hedges (Eq. 8).
Parallel Bioequivalence Study
Parallel bioequivalence studies with different GMRs were simulated by assuming that both s R and s T were 0.4. Figure 2 shows the dependence of the power on the number of subjects (n) in each arm.

Power curves for methods assessing RSABE. The dependence of the percentage of accepted BE studies is shown at various ratios of the geometric means (GMR) of the two formulations. Parallel design was assumed. The standard deviation for both products was 0.4 which corresponds to CV = 41.65%. The regulatory constant was set to 0.89 according to the FDA requirements. Bioequivalence was evaluated with the method of Hyslop et al. (2) as recommended by the FDA (4,8), the ABEL method as recommended by the EMA (6), and the Exact approach as described in the “METHODS” section
As explained in the “METHODS” section, the reference-scaled difference follows a noncentral t distribution. Based on this observation, there are two possible ways to construct an equivalence test for RSABE. The first, as described by Steiger and Fouladi (13), is to construct confidence intervals around the scaled difference and reject RSABE if the confidence interval overlaps the regulatory cutoff. This is analogous to the classical confidence interval approach for ABE. The second way is the TOST approach.
Figure 2 shows that the confidence interval approach (C.I.) and the exact method using TOST are not the same, in fact C.I. is always worse.
There is substantial difference between the performances of the FDA and Exact methods, particularly when the power is low. Even though this difference gradually diminishes as the overall power increases, the order between the methods remains the same, the Exact method is always better than that based on the approximation, the FDA method.
This is not true for ABEL, the relative performance of which changes with n. It is slightly worse than the Exact method when n = 10 but slightly better in all other cases. But the consumer risk with ABEL also rises as n increases and it will be slightly above the nominal 5% when n = 20 and 30. This is illustrated in Table I. The power is largest when there is no difference between the Test and Reference products. This situation corresponds in the simulations to the condition when there is no difference between μ T and μ R. In general, the power recorded by the Exact method is closer to that observed with the ABEL than with the FDA approaches (Table I, Fig. 2).
A second interesting case is when the simulation parameters are set to the boundary condition. If (μ T − μ R)/σ = θ, then the passing rate, the observed consumer risk, should be equal to or below the nominal error rate of 5%. As Table I shows, ABEL slightly exceeds this limit when n is 20 and 30. Thus, among the RSABE tests which keep the consumer risk below the 5% level, the Exact method is the best.
Crossover Design with Equal Within-Subject Standard Deviations
(s WT = s WR, Homoscedastic Case)
Three sets of bioequivalence trials were simulated with identical parameters but different designs. The three designs were as follows: three-period, three-sequence partial replicate design (RRT-RTR-TRR); three-period, two-sequence replicate design (TRT-RTR); and four-period, two-sequence replicate design (TRTR-RTRT). Regardless of the design, all other simulation parameters were the same: 24 subjects in each trial and s W = s WR = s WT = 0.4. Bioequivalence was evaluated with the FDA, ABEL, and Exact methods. The corresponding power curves are shown in Fig. 3. The confidence interval method (C.I.) was not investigated since we showed previously that it was worse than the TOST procedure. As Fig. 3 illustrates, there is a very slight difference among the powers of the three methods. The exact method always has a slight advantage over the FDA method (Table II).

RSABE evaluated from simulated crossover bioequivalence studies with different study designs. The designs were as follows: four-period, two-sequence replicate design (RTRT-TRTR); three-period, two-sequence replicate design (TRT-RTR); and three-period, three-sequence partial replicate design (RRT-RTR-TRR). The dependence of the percentage of accepted BE studies is shown at various ratios of the geometric means (GMR) of the two formulations. It was assumed that the true within-subject standard deviations for both products were 0.4, and 24 subjects received the Test and Reference formulations in the simulated trials
Design had an important effect on the power. The four-period design was clearly better than either of the three-period designs. The three-period partial replicate design had an advantage over the TRT-RTR design which is understandable because the number of degrees of freedom is larger. Under almost all conditions, the power noted by the Exact method was closer to that recorded with the ABEL than with the FDA approaches (Table II, Fig. 3).
Table II shows that the consumer risk with the FDA and Exact methods was below the nominal 5% level while ABEL was slightly nonconservative.
Crossover Design with Unequal Within-Subject Standard Deviations
(s WT ≠ s WR, Heteroscedastic Case)
The simulations with crossover designs were repeated, but it was assumed that the within-subject variation of the Test formulation was either half or double of that of the Reference product. The test formulation s WT was set to either 0.25 or to 0.50 and the corresponding s WR to 0.50 or 0.25. The results are summarized in Fig. 4 and Table III. The large difference between the powers in the upper and lower panels is easy to understand with the ABEL method. ABEL is essentially an average bioequivalence (ABE) approach except that the cutoff values are proportional to s WR. Therefore, when s WR was 0.50, then the average ABE cutoff was exp (0.89*0.50) = 1.56 whereas when s WR was 0.25, then the average ABE cutoff was 1.25.

Effect of heteroscedasticity. It was assumed that the Test and Reference formulations have different within-subject standard deviations. The SD ratio is the s WT/s WR ratio. When the SD ratio was 0.5, then the simulation parameters were s WT = 0.25 and s WR = 0.5. When the SD ratio was 2, the simulation parameters were switched and s WT was 0.5 and s WR = 0.25. For other notations, see Fig. 3
Just as in the other scenarios, the power with the Exact method was higher in all but one case than that of the FDA approach. But when the within-subject deviations are not equal, then the Exact method is not true in the mathematical sense because the mathematical assumptions behind the method are not true. Heteroscedasticity is such an assumption (11) and, as Table III shows, in some cases, the consumer risks with the Exact method were slightly above the nominal 5%. Note, however, that for the partial replicate design, the FDA method also had a higher than nominal consumer risk (Table III).
Bias of the FDA Method
As noted earlier, FDA expects, following the approach of Hyslop et al. (2), that the upper 95% bound for the squared, linearized form of the RSABE model (Eqs. 1, 2, 27, and 31) should be zero or negative in order to be able declare bioequivalence (4,5). RSABE would be rejected if positive values are estimated following the application of Eq. 31.
However, Eqs. 36a and 36b demonstrate that a positive bias is incurred when Eq. 31 is used. The bias is small but meaningful (Table IV). As expected, the bias decreases when the number of subjects, and thereby the number of degrees of freedom, increases. The bias is very slightly larger in three-sequence than two-sequence studies.
RSABE for NTI Drugs
Features of the Exact method of determining RSABE were evaluated for BE studies of drug products having a narrow therapeutic index (NTI). NTI drugs typically have a low within-subject variance (10); therefore, we set s WT and s WR to either 0.05 or 0.10. Following the FDA draft recommendations (8,9), only the TRTR-RTRT design was considered and the regulatory θ was set to log(1.11)/0.10. Figure 5 and Table V compare the passing rates of the FDA, Exact, and ABEL methods under conditions typical for NTI drugs. The overall patterns were very similar to those observed for the previous HVD/P simulation conditions. In terms of power, the Exact method was marginally better than the current FDA recommendation and both the FDA and Exact methods kept the consumer risk below the 5% nominal level. This was not true for ABEL where the actual error rate was slightly above the 5% nominal value.

RSABE for Narrow Therapeutic Index drugs. The draft FDA guideline for warfarin (8) recommends that for NTI drugs, the regulatory constant (θ) should be set to log(1.111)/0.10. Four-period, two-sequence studies were simulated with 24 subjects with different s WT/s WR ratios. Simulation conditions from left to right: s WT = 0.05, s WR = 0.1; s WT = 0.1, s WR = 0.1; s WT = 0.1, s WR = 0.05
DISCUSSION
The theoretical framework and algorithmic details of a new, numerically exact method were provided for the determination of RSABE. A close parallel was drawn between RSABE and the estimation of effect size.
For bioequivalence studies with parallel design, direct connection was shown between RSABE and effect size. For parallel designs, results published mostly in psychology publications (12,16) were directly applied (Figs. 1 and 2, Table II). However, for crossover studies, the theory of equivalence tests of effect sizes had to be developed further. Based on the theoretical results of Hedges (16), a new, exact algorithm was developed to asses RSABE. We call our algorithm Exact to emphasize the difference from the FDA’s algorithm which is based on numerical approximation.
Merits of the Exact Procedure
Compared to FDA’s draft proposals, the Exact procedure has two attractive features:
-
1. Simplicity
The computer code of the proposed Exact method is literally a one-liner (Eqs. 17a and 17b).
It requires nothing but just calculating the quantiles of the noncentral t distribution which is a built-in function in professional statistical software packages. The formula of the computations contains two constants. The first constant is a correction factor introduced by Hedges (Eq. 8). The second is a design-dependent constant. By giving the three most important cases, we showed that it can be computed in a simple way (Eq. 22). But computation is needed only for unbalanced designs; otherwise, only the presented design-dependent constants need to be inserted. The great advantage of the simple computer code is that it makes the computer program easy to transport to other software platforms and it facilitates regulatory assessment.
-
2. Power
Theory predicts (15) that the Exact method is the most powerful test at a given consumer risk. We confirmed this theoretical prediction (Tables II, III, and V). The gain in power is very modest when the power is close to 100% but can be more substantial at lower levels of power (Fig. 2).
Biases of the SABE and RSABE Models and of the FDA Approach
It is noted for clarification that two kinds of biases are discussed in this manuscript. The first is that of the SABE and RSABE model (Eqs. 1 and 11) which was established and corrected by Hedges (16). The effectiveness of this correction is demonstrated in Fig. 1. The second kind of bias arises from the approach of FDA which is applied for the evaluation of RSABE (5,8). The small but meaningful bias inherent in the use of the FDA method (Eq. 36, Table IV), could be one of the reasons for the comparatively low power exhibited by this approach. In any case, FDA currently expects that RSABE is accepted only if the estimated 95% upper bound is negative or zero for the squared and linearized model. The consequence of the bias is that some bioequivalent drug products will be rejected. In effect, this bias is (or should be) the bioequivalence limit, instead of zero, for the transformed RSABE model. It would be useful if this effect will be taken into account in the future.
Consequences of Heteroscedasticity
We could derive the exact formula for the RSABE test only if homoscedasticity was assumed, i.e., only if we assumed s WR = s WT. It must be stressed that homoscedasticity from a biopharmaceutical viewpoint is a reasonable assumption. Nevertheless, we checked the robustness of the three methods when the assumption of homogenous variability was violated.
Table III illustrates the effects of the unequal within-subject variations on the power at GMR = 1.0. The power is high when the Test product has a lower intrasubject variation than the Reference formulation. Low power is seen with the opposite relationship. These considerations have direct consequences on the sample size required for a study. Smaller samples are needed when the variation of the Test preparation is smaller than that of the Reference product than with the opposite relationship. These conclusions are expected to apply both in the highly variable and NTI regions.
Table III shows that the partial replicate design is particularly sensitive to the assumption of homogenous variability. In this case, each method can have a higher than nominal error rate. The special sensitivity of the partial replicate design to the violation of the variance homogeneity assumption calls for additional investigations. Theoretical considerations also suggest that the statistical model behind these tests is not valid if the variance homogeneity condition is violated and the numbers of observations in the Test and Reference groups are different (24). We are not aware of any publication which investigated this aspect of replicate design bioequivalence studies in detail. Commonly used simulation algorithms (25) in these special cases provide grossly inaccurate results (26). That is why we sampled individual observations from normal distributions and used regression methods to estimate the parameters. The alternative fast algorithms (9,25,26) are based on assumed sampling distributions of the parameters. This is theoretically not justifiable in the case of heteroscedasticity.
It appears to be a disadvantage that the proposed Exact method requires to estimate the s WT/s WR ratio while the FDA method does not. But, from a regulatory viewpoint, the s WT/s WR ratio is of interest; therefore, requiring to compute the s WT/s WR may not be disadvantageous. The s WT/s WR ratio can be estimated in bioequivalence studies with replicate designs, even from studies with the partial replicate design. The estimation is a straightforward, simple procedure for full replicate designs, but it is not with the partial replicate design. We have provided an R script to perform the computations. However, we have found that the code frequently requires a change of the “options” settings. For this reason, the FDA algorithm is the preferred method for the partial replicate design.
Other Comments
The other alternative for all designs is the ABEL approach which can be applied very easily. But as reported earlier, this method has a higher than 5% consumer risk (27). This observation was confirmed (28,29). As Table II shows, the consumer risk was between 6 and 7% even when the assumption of variance homogeneity was correct. From a statistical point of view, this could be of concern but much less so in regulatory practice. The ABEL method is utilized in the EU with a regulatory constant (0.76) (6) which is much more stringent that the regulatory constant (0.893) used by the FDA (1,5); for the sake of comparability, the latter values were always applied in the present study. Furthermore, the EU Guideline (6) places a number of other restrictions, including a cap on the maximum widening (69.84–143.19%) of the bioequivalence limits. None of these constraints was applied in the presented simulations.
The Exact approach and the FDA procedure have many similar features. Therefore, it is expected that the sample sizes required for the determination of BE by the two methods would be similar.
We investigated only a single aspect of the rather complex statistical procedures of the FDA draft guidelines (4,8), even though there are other potentially important details in these drafts. For example, the statistical models still include the so-called subject-by-formulation interaction, with a published SAS code (8), even though this term is estimated with a serious bias (30,31). The effect of this biased estimation on the power and consumer risk is unknown. This and other modeling and procedural options call for additional investigations.
CONCLUSION
A simple algorithm was developed to evaluate the test for RSABE in bioequivalence studies. The algorithm is based on the understanding that there is a close connection between the concepts of effect size and scaled bioequivalence. The algorithm is straightforward and is more powerful, even if slightly, than the currently recommended approach in the draft FDA guidelines (1,8). It could be considered as an alternative to the current procedure in the FDA draft proposal, particularly with full replicate design studies.
Regarding the partial design, we noted above the nominal consumer risk in heteroscedastic simulation conditions. The increased risk may not be a real concern because we applied in our simulations only a subset of the regulatory constraints. But it certainly warrants the reappraisal of the design recommendations in this regard (5).
Abbreviations
- ABEL:
-
Average bioequivalence with expanding limits
- BE:
-
Bioequivalence
- C:
-
Contrast matrix
- C.I.:
-
Confidence interval
- C m, C s :
-
Confidence limits of E m, E s
- cr(df):
-
Correction factor of Hedges for bias of d
- CV:
-
Coefficient of variation
- D:
-
Estimated index of scaled average bioequivalence
- d’:
-
Difference of means divided by the corresponding standard error
- df:
-
Degrees of freedom
- E m, E s :
-
Components of the squared, linearized RSABE model
- EMA:
-
European Medicines Agency
- FDA:
-
Food and Drug Administration
- HV:
-
Highly variable
- HVD/P:
-
Highly variable drugs and drug products
- K :
-
Constant related to sample size
- L, L’:
-
Lower limit of confidence interval
- L m, L s :
-
Squared length of confidence interval of E m, E s
- M :
-
Number of subjects in an arm of parallel design studies
- n :
-
Number of subjects
- NTI:
-
Narrow therapeutic index
- Prob:
-
Probability
- R :
-
Reference product
- RSABE:
-
Reference-scaled average bioequivalence
- s :
-
Estimated standard deviation number of sequences
- SABE:
-
Scaled average bioequivalence
- SE:
-
Standard error
- t :
-
t distribution, t statistic
- T :
-
Test product
- Tnc:
-
Noncentral t distribution
- TOST:
-
Two one-sided tests
- U, U’:
-
Upper limit of confidence interval
- y :
-
Logarithmic pharmacokinetic variable
- Ȳ :
-
Estimated mean, logarithmic
- W :
-
Within subjects
- z :
-
Ratio of within-subject standard deviations, σ WT/σ WR
- δ :
-
True, population index of scaled average bioequivalence
- ε :
-
Error
- μ :
-
Mean, logarithmic
- χ 2 :
-
Chi-square, chi-square distribution
- σ 2 :
-
Variance
- θ :
-
Regulatory criterion
References
Haidar SH, Davit BM, Chen M-L, Conner D, Lee LM, Li QH, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res. 2008;25:237–41.
Hyslop T, Hsuan F, Holder DJ. A small sample confidence interval approach to assess individual bioequivalence. Stat Med. 2000;19(20):2885–97.
Tothfalusi L, Endrenyi L, Arieta AG. Evaluation of bioequivalence for highly variable drugs with scaled average bioequivalence. Clin Pharmacokinet. 2009;48:725–43.
FDA. Draft guidance for industry: statistical approaches to establishing bioequivalence. Rockville: Center for Drug Evaluation and Research (CDER); 2001.
FDA. Draft guidance for industry: bioequivalence recommendations for progesterone oral capsules. Silver Spring: Center for Drug Evaluation and Research (CDER); 2011.
European Medicines Agency. Guideline on the investigation of bioequivalence. London, United Kingdom; 2010.
Tothfalusi L, Endrenyi L. Limits for the scaled average bioequivalence of highly variable drugs and drug products. Pharm Res. 2003;20(3):382–9.
FDA. Draft guidance on warfarin sodium. Silver Spring: Center for Drug Evaluation and Research (CDER); 2012.
Jiang W, Makhlouf F, Schuirmann DJ, Zhang X, Zheng N, Conner D, et al. A bioequivalence approach for generic narrow therapeutic index drugs: evaluation of the reference-scaled approach and variability comparison criterion. AAPS J. 2015;17(4):891–901.
Yu LX, Jiang W, Zhang X, Lionberger R, Makhlouf F, Schuirmann DJ, et al. Novel bioequivalence approach for narrow therapeutic index drugs. Clin Pharmacol Ther. 2015;97(3):286–91.
Algina J, Keselman HJ, Penfield RD. Confidence intervals for an effect size when variances are not equal. J Modern Appl Stat Meth. 2006;5(1)
Steiger JH. Beyond the F test: effect size confidence intervals and tests of close fit in the analysis of variance and contrast analysis. Psychol Methods. 2004;9(2):164–82.
Steiger JH, Fouladi RT. Noncentrality interval estimation and the evaluation of statistical model. In: Harlow LL, Mulaik SA, Steiger JH, editors. What if there were no significance tests? Hillsdale: Erlbaum; 1997.
Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15(6):657–80.
Wellek S. Testing statistical hypotheses of equivalence and noninferiority. Boca Raton: Chapman & Hall/CRC; 2003.
Hedges LV. Distribution theory for Glass’s estimator of effect size and related estimator. J Educ Stat. 1981;6(2):107–28.
Milliken GA, Johnson DE. Analysis of messy data. Boca Raton: CRC Press; 2009.
Cleophas TJ. Statistics applied to clinical studies. New York: Springer; 2012.
Kulinskaya E, Staudte RG. Interval estimates of weighted effect sizes in the one-way heteroscedastic ANOVA. Br J Math Stat Psychol. 2006;59:97–111.
Ratkowsky DA, Alldredge JR, Evans MA. Cross-over experiments: design, analysis, and application. New York: Marcel Dekker; 1993.
Howe W. Approximate confidence limits on the mean of X + Y where X and Y are two tabled independent random variables. J Am Stat Assoc. 1974;69:2885–97.
R Core Team. R: a language and environment for statistical computing. In: R Foundation for Statistical Computing. 2015.
Pinheiro JBD, DebRoy S, Sarkar D, R Core Team. Linear and nonlinear mixed effects models. R package version 3. 2015.
Dannenberg O, Dette H, Munk A. An extension of Welch’s approximate t-solution to comparative bioequivalence trials. Biometrika. 1994;81(1):91–101.
Zheng C, Wang J, Zhao L. Testing bioequivalence for multiple formulations with power and sample size calculations. Pharm Stat. 2012;11(4):334–41.
Labes D, Schuetz H. PowerTOST: power and sample size based on two one-sided t-tests (TOST) for (bio)equivalence studies. R package version 1.2-06. 2015. http://CRAN.R-project.org/package=PowerTOST.
Endrenyi L, Tothfalusi L. Regulatory and study conditions for the determination of bioequivalence of highly variable drugs. J Pharm Pharm Sci. 2009;12(1):138–49.
Labes D. ‘Alpha’ of scaled ABE. Bioequivalence and bioavailability forum. BEBAC Consultancy Services for Bioequivalence and Bioavailability Studies, Vienna, Austria; 2013. http://forum.bebac.at/mix_entry.php?id=10202.
Wonnemann M, Fromke C, Koch A. Inflation of the type I error: investigations on regulatory recommendations for bioequivalence of highly variable drugs. Pharm Res. 2015;32(1):135–43.
Endrenyi L, Taback N, Tothfalusi L. Properties of the estimated variance component for subject-by-formulation interaction in studies of individual bioequivalence. Stat Med. 2000;19(20):2867–78.
Endrenyi L, Tothfalusi L. Subject-by-formulation interaction in determinations of individual bioequivalence: bias and prevalence. Pharm Res. 1999;16(2):186–90.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tothfalusi, L., Endrenyi, L. An Exact Procedure for the Evaluation of Reference-Scaled Average Bioequivalence. AAPS J 18, 476–489 (2016). https://doi.org/10.1208/s12248-016-9873-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-016-9873-6