
Abstract
Background
Extracellular vesicles (EVs) are amongst the most promising candidates for developing blood-based biomarkers. However, patient sample availability is a key barrier to translational research whilst most biobanks store samples of 1.5mL volume or less. To date, there is no consensus on the most suitable method of EV separation and current techniques frequently require large volumes of biofluids, complicated technology, technical expertise, or significant operating costs, which prevents their widespread adoption by less EV-focussed laboratories. Therefore, there is a need for an easy and reproducible method that separates representative EVs from clinically relevant 1mL volumes of plasma prior to subsequent biomarker identification.
Methods
In this study, EVs were separated from a clinically relevant 1mL volume of human plasma using four different separation techniques: size exclusion chromatography (SEC), differential ultracentrifugation, precipitation, and immunoaffinity magnetic bead capture. The EVs were characterised using several orthogonal techniques (protein quantification, nanoparticle tracking analysis, transmission electron microscopy, Western blot, single particle interferometric reflectance imaging sensing, and mass spectrometry-based proteomics) to comprehensively compare the separated samples.
Results
We provide examples of anticipated results highlighting that SEC-processed samples have greater protein quantification yield, greater particle yield of the expected size for EVs, and sufficient EV purity, which facilitates effective EV cargo assessment by proteomics. Moreover, we confirm significant overlap with known EV-related proteins within the Vesiclepedia database. Additionally, using single particle interferometric reflectance imaging sensing (Leprechaun®), we identify that SEC has the most representative surface tetraspanin distribution of the separated EV population compared to unprocessed plasma.
Discussion
Given that SEC requires minimal expertise, no complicated technology and can separate EVs within 90 min, this comparison reinforces SEC as a clinically relevant EV separation method from 1mL of plasma making it suitable for widespread implementation.
Similar content being viewed by others


Introduction
The development of less invasive blood-based “liquid biopsy” tests is being pursued for diverse applications [1] including oncology [2], neurology [3], cardiovascular disease [4], and obstetrics [5]. These liquid biopsies are looking to identify novel biomarkers to improve early detection [6], patient stratification [7], and treatment monitoring [8], with the hope of improving patients’ outcomes. One source of blood-based biomarkers, extracellular vesicles (EVs), has several distinct advantages over other analytes [9], including blood proteins, circulating nucleic acids, and circulating cells, due to the stability and relative abundance of EVs in the peripheral circulation [10].
EVs are lipid bilayer enclosed particles that are released by all living cells as a form of intercellular communication. Several EV subtypes have been identified, including exosomes (50-150 nm) and microvesicles (50-500 nm), that are differentiated based on their biogenesis [11]. However, the overlapping size, morphology, and composition makes it difficult to clearly differentiate EV subtypes and therefore it is recommended to report the isolated EVs by size [12, 13]. Small EVs (sEVs), normally defined as EVs < 100-200 nm, have the most evidence for their use as a liquid biopsy biomarker, however their size and density overlaps with lipoproteins which are present at six orders of magnitude higher concentration than EVs in plasma [14, 15] and requires effective depletion to optimise sEV-based biomarker identification.
Despite their potential and some promising early results [16], blood-based sEV biomarkers have yet to be translated into clinical practice. One challenge to clinical translation is the lack of standardisation in the methods used to separate EVs from other circulating proteins and particles, with several different techniques in frequent use in the scientific literature [17]. To fill this gap, several publications have sought to compare different EV separation techniques; however they typically (i) compared only a few methods [18], (ii) assessed multiple variations within a single EV separation methodological class [19], (iii) used less clinically relevant starting volumes [20], (iv) utilized serum as a starting biofluid [21] which is now thought to be a less useful source of EVs for biomarker investigation due to the increased co-separation of platelet-derived EVs [22, 23], (v) compared separation methods using conditioned cell culture media as the starting biofluid [24, 25], or (vi) a combination of all of these factors.
Traditionally, separation has been based on the physical characteristics of EVs. Ultracentrifugation-based techniques, which separate EVs based on density, have historically been the most frequently used EV separation methods [26]. Despite this familiarity, significant variation remains in the implementation of this procedure. Previous work [27] has demonstrated the need for an intermediate centrifugation of ~ 10,000 × g to remove cellular debris and larger apoptotic bodies, followed by a faster centrifugation of ~ 100,000 × g for > 70 min but < 4 h duration [28] to optimise EV pelleting and minimise the inclusion of soluble proteins, primarily albumin when starting with plasma. The addition of a washing step, involving resuspension in PBS and repeat ultracentrifugation at 100,000 × g to re-pellet EVs, can significantly reduce soluble protein contamination [29]. However, the inclusion of a filtration step remains contentious with advocates describing increased sample purity.
More recently, precipitation reagents using commercial kits are frequently employed when investigating small volume clinical samples. Precipitation reagents are extremely simple and work by binding water molecules thereby pushing EVs and other less soluble molecules out of solution to be pelleted by centrifugation. There are several proprietary kits available with similar effectiveness in separating EVs [19], and they frequently recommend the inclusion of proteinase K to remove the bulk of plasma proteins prior to EV separation. Compared to this untargeted approach, a more targeted separation of EVs can be achieved through immunoaffinity capture [30], although this has previously been limited by separation of specific EV subpopulations. The development of a phosphatidylserine binding protein TIM4 targeted antibody attached to a magnetic bead EV capture assay [31], has the benefit of targeting a marker that has been proposed as a near universal marker on all subpopulations of EVs [32] which would facilitate the separation of a purer sEV sample. However, the real situation is likely to be more complex, with evidence that phosphatidylserine negative-EVs are present and detectable in peripheral blood [33, 34].
In comparison to other EV separation techniques, size exclusion chromatography (SEC) is highly reproducible, requires minimal technical expertise, and is now one of the most frequently utilised methods to separate EVs, especially when using clinical samples [35]. SEC uses columns packed with a porous resin to separate EVs based on their size, with larger particles eluted first and smaller soluble proteins eluted later. A variety of column sizes, resin material, and pore sizes are available to optimise the SEC column for the separation of different sized particles [36, 37]. Whilst homemade columns are frequently used [38], commercial columns are now widely available, of which the IZON range is frequently used, and the 35 nm range is optimised for the separation of sEVs > 30 nm.
Given this variety of readily useable techniques, there is therefore a pressing need to identify appropriate EV separation methodologies from clinically relevant starting volumes of plasma as a necessary step for the subsequent clinical translation of any identified biomarkers. A standard blood test collects 4 mL of blood which results in 1–2 mL of plasma on average, whilst biobanks including the UK Biobank [39] typically store samples of 1.5 mL volume or less. Given the requirement to fully characterise the separated sample, as recommended by the International Society for Extracellular Vesicles [12, 13, 40,41,42], to demonstrate the enrichment of EVs and the depletion of common co-separated contaminants, a sufficient yield and purity from a limited volume is needed for downstream biomarker identification studies.
To highlight the suitability of SEC for EV separation from clinically relevant volumes of plasma and present representative data, we compare the simplicity, yield, purity, representativeness, and protein cargo of different EV separation methods from a clinically relevant starting sample of 1 mL of human plasma (Fig. 1). We also present the key methodological steps for all EV separation methods used in the comparison and the key EV characterisation steps.

Schematic summary of EV separation and downstream analyses. Following EV separation using five different methods, a comprehensive characterisation was performed by nanoparticle tracking analysis, Western blot, transmission electron microscopy, single particle interferometric reflectance imaging sensing (Leprechaun® analysis) and proteomics (mass spectrometry). Leprechaun® image reproduced with the permission of Unchained Labs (Pleasanton, Ca, USA)
Methods
Ethical approval and consent to participate
All experimental protocols were approved by the University of Sussex Research Ethics Committee (reference: ER/BSMS2229/1). Each participant provided written informed consent to take part in this research.
Plasma isolation - Timing: 45-60 minutes
The blood collection and processing protocol has been reported as per MIBlood-EV recommendations [41]. Venous plasma was obtained from three healthy non-fasting adult male volunteers (aged 25–35 years old) on three separate occasions. On each occasion, up to 40mL blood was drawn from the antecubital fossa using a 23-gauge butterfly needle (Prosum Medical Limited, Uxbridge, UK) into 4 mL K2EDTA spray tubes (Vacuette) and pooled for a total of nine distinct pooled samples (3 individuals, n = 3 repeats).
Processing was performed within 30 min using a validated method to reduce platelet activation [43]. Briefly, blood samples underwent sequential centrifugations at room temperature, first at 120 × g for 20 min to prepare platelet rich plasma and subsequently at 360 × g for 20 min, using a benchtop centrifuge with no brake applied (Universal 320R, Hettich). The resulting platelet depleted plasma samples were stored at -80 °C in 1 mL aliquots (Eppendorf).
EV separation
For all experiments, 1 mL starting volume of plasma was used and replicates were performed on separate days.
Differential ultracentrifugation (dUC) ± 0.22 μm filtration (dUC + f) - Timing: 4 hours
Samples were thawed on ice and centrifuged at 2000 × g for 5 min at 4 °C. The supernatant was collected and transferred to a clean ultracentrifuge tube (Beckman polypropylene centrifuge 25 × 89 mm tubes, Beckman Coulter Ltd., High Wycombe, UK) and diluted 1:9 with sterile-filtered PBS (Sigma-Aldrich). A 10,000 × g ultracentrifugation step was then performed for 30 min at 4 °C to remove cellular debris and larger EVs (Beckman optima LE 80-k ultracentrifuge, Beckman Type 70 Ti rotor, full dynamic braking, kadj = 15,638, Beckman Coulter Ltd., High Wycombe, UK). The supernatant was collected using a sterile 20 ml syringe (Fisher scientific) and transferred to a new ultracentrifuge tube directly or through a 0.22 μm filter (Sarstedt AG & Co. KG). To separate small EVs, a 100,000 × g ultracentrifugation step was performed for 90 min at 4 °C (Beckman optima LE 80-k ultracentrifuge, Beckman Type 70 Ti rotor, full dynamic braking, kadj = 494, Beckman Coulter Ltd., High Wycombe, UK). The EV pellet was washed with 10 mL sterile-filtered PBS and ultracentrifuged again at 100,000 × g for 90 min at 4 °C. The final pellet was resuspended in 100 μL sterile-filtered PBS and stored at -80 °C.
Precipitation (TEIR) - Timing: <90 minutes
Precipitation was performed using the Total Exosome Isolation Reagent (TEIR) (Invitrogen) as per the manufacturer’s instructions. Briefly, samples were thawed on ice and then centrifuged at 2000 × g for 20 min and the supernatant centrifuged at 10,000 × g for 20 min both at room temperature. The cleared supernatant was collected, diluted with sterile-filtered PBS (1:2) and, as per the manufacturers recommendations, proteinase K solution was added (1:20) and incubated at 37 °C for 10 min. Exosome precipitation reagent was added (1:5) and incubated at 8 °C for 30 min. The sample was centrifuged twice at 10,000 × g for 5 min at room temperature to pellet the EVs and the supernatant removed. The final pellet was resuspended in 100 μL sterile-filtered PBS and stored at -80 °C.
Size Exclusion Chromatography (SEC) - Timing: <90 minutes
Samples were defrosted on ice and then underwent centrifugation at 1500 × g for 10 min at room temperature and the supernatant was centrifuged at 10,000 × g for 10 min at room temperature to remove cellular debris. The cleared supernatant was collected and topped up to 1000 μL with sterile-filtered PBS. New SEC columns (IZON qEV1/35 nm) were drained and then flushed with sterile-filtered PBS. The sample was loaded followed by layering 3mL sterile-filtered PBS and the first 4mL buffer volume was collected. Further sterile-filtered PBS was added to the column (700 μL) and seven × 700 μL SEC fractions were collected. Samples were stored at -80 °C.
As per the manufacturer’s recommendations, and following initial characterisation (Supplementary Fig. 1), SEC fractions 2–5 were combined to obtain a sample balancing EV recovery with sample purity. The combined sample was concentrated to 100 μL using a 100kDa molecular weight ultrafiltration filter (Vivaspin 6, Sartorius) and centrifugation at 4000 × g (Mega Star 1.6R, VWR). Combined SEC fraction samples were stored at -80 °C.
Magnetic bead separation (Mag) - Timing 24 hours
Magnetic bead based EV separation was performed using the MagCapture™ Exosome Isolation Kit PS Ver.2 (Fujifilm Wako Pure Chemical Corporation) and a magnetic rack (MagnaRack, Invitrogen) as per the manufacture’’s instructions. Briefly, the magnetic beads were prepared by incubation with the biotin labelled exosome capture reagent. Samples were defrosted on ice and then underwent centrifugation at 1200 × g for 10 min at 4 °C and the supernatant was centrifuged at 10,000 × g for 30 min at 4 °C to remove cellular debris. Heparin (5 U/mL) to maintain anticoagulation and binding enhancer were added to the sample. The sample was subsequently added to the magnetic beads and incubated for 1 h at room temperature or overnight at 8 °C. The sample was washed three times with washing buffer and all the supernatant removed. EVs were eluted from the beads using 100 μL 2X elution buffer and stored at -80 °C.
Nanoparticle Tracking Analysis (NTA) - Timing: 20 minutes / sample
Particle concentration and size distribution were determined using a NanoSight NS300 with a blue laser (488 nm) and analysed using the Nanosight NTA 3.2 software (Malvern Technologies, Malvern, UK) as previously described [44]. Samples were diluted 1:100-1:1000 in sterile-filtered PBS and five videos of 30 s were captured per sample using a camera level set close to maximum (15 or 16) and a threshold of 5 was used for analysis.
Transmission Electron Microscopy (TEM) - Timing: <15 minutes
Particle morphology and size were visualised by Transmission Electron Microscopy as previously described [29]. Samples were diluted in particle free PBS to a particle concentration of 1 × 1010–8 × 1010/ml and 4μL was applied to a formvar/carbon film coated 200 mesh copper EM grid (Agar Scientific cat no. S162H) that had been previously treated with a glow discharger (PELCO easiGlow) for 1 min. The samples were incubated for 30 s then washed with PBS and stained with filtered 1% Uranyl Acetate for 30 s and allowed to dry. The grids were visualised using a JEOL JEM1400-Plus (120 kV, LaB6) microscope (JEOL Ltd., Welwyn Garden City, UK) equipped with a Gatan OneView 4K camera at × 10 k and × 30 k magnification. In total, 10–15 representative images per grid were taken.
Protein quantification - Timing: <3 hours
Protein quantification was performed using the micro BCA™ Protein Assay Kit (Thermo scientific). Briefly, 2 μL of EVs were lysed in 5 × RIPA buffer for 10 min in technical duplicate in a 96-well microplate. Subsequently, 100 μL of BCA™ reagent was added to each sample. Additionally, a standard BSA curve (serial dilutions from 10 μg to 0.078125 μg) and a RIPA control sample were incubated with 100 μL of BCA™ reagent. The plate was incubated at 37 °C in the dark for 2 h. Absorbance was measured at 560 nm (GloMax, Promega). Protein concentration was determined from the BSA standard curve and incorporated the influence of the RIPA buffer on the measured absorbance.
Western blot - Timing: 2 days
Protein expression of known EV markers and common contaminants were performed by Western Blot using standard protocols as described previously [29]. Briefly, equal amounts of EVs (5 μg or 40 μL) were lysed in 5 × Laemmli sample buffer (containing 1.5M Tris–HCl, Glycerol, 20% sodium dodecyl sulphate (SDS), 0.25% bromophenol blue and 1M dithiothreitol), boiled at 95 °C for 10 min, and resolved on SDS/PAGE gradient gels (4–12%). Transfer was performed using the iBlot 2 system (Invitrogen). Primary antibodies: anti-CD9 (1:1000, System Biosciences EXOAB-KIT-1), anti-CD63 (1:1000, System Biosciences EXOAB-KIT-1), anti-CD81 (1:1000, System Biosciences EXOAB-KIT-1), HSP70 (1:1000, System Biosciences EXOAB-KIT-1), anti-syntenin (1:1000, St John’s Laboratory STJ98556), anti-TSG101 (1:1000, St John’s Laboratory STJ96125), anti-GM130 (1:1000, Cell Signalling Technology #12480), anti-BSA (1:1000, Sigma 07-248-25UG), anti-apolipoprotein A1 (1:1000, St John’s Laboratory STJ96804), anti-apolipoprotein B (1:1000, St John’s Laboratory STJ22648), anti-apolipoprotein E (1:1000, St John’s Laboratory STJ91638). Secondary antibodies: Anti-rabbit IgG HRP linked (1:5000, Cell Signalling #7074) and Anti-mouse IgG HRP linked (1:5000, Cell Signalling #7072). Membranes were incubated with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo scientific) and imaged with a UVP Chemstudio instrument (Analytik Jena, London, UK) and VisionWorks software.
Single Particle Interferometric Reflectance Imaging Sensing (SP-IRIS) - Timing: 24 hours
Leprechaun® Exosome Human Plasma Kit were used with the Leprechaun® machine for surface marker analysis according to the manufacturer’s instructions (Unchained Laboratories, Pleasanton, Ca, USA). Briefly, chips were background scanned and incubated overnight at room temperature with 50 μL of sample diluted to a particle concentration of ~ 1 × 109/ml using 1X incubation solution (Mag required 0.5 × 109/ml whilst plasma required 10 × 109/ml for accurate particle quantification). Chips were washed, incubated with fluorescently labelled antibodies for 1 h (1:500 each of anti-CD9 CF488A, anti-CD81 CF555 and anti-CD63 CF647), and dried using the Luni Washer (Unchained Laboratories, Pleasanton, Ca, USA). Chips were scanned immediately after drying and analysed using Leprechaun® Analyser 1.0 software (Unchained Laboratories, Pleasanton, Ca, USA) and gated to the isotype control.
Proteomics - Timing: 2 days
The separated EV samples (10 μg) from each separation method were processed using in solution digestion in triplicate from individual repeats. Briefly, samples were lysed in 5X RIPA buffer and 50X protease inhibitor for 30 min, and concentration by Speedvac. The samples were reduced with 8 M Urea, 50 mM ammonium bicarbonate (pH 7.8) and 5 mM dithiothreitol at 37 °C for 1 h and subsequently alkylated with 15 mM iodoacetamide in the dark at room temperature for 30 min. The samples were diluted with three volumes of 50 mM ammonium bicarbonate (pH 7.8) and digested with trypsin (1:50) at 37 °C for 3 h. Proteins were further digested with a second round of trypsin (1:50) at 37 °C overnight. The peptides were acidified with 1% TFA, extracted using Pierce™ Peptide Desalting Spin Columns (Thermo Scientific) as per the manufacturer’s instructions, and dried by Speedvac. Samples were further purified by strong cation exchange using a Ziptip pipette tip (Millipore, Merck) as per the manufacturer’s instructions. Samples were processed through a Q Exactive hybrid Quadrupole-Orbitrap mass spectrometer coupled to a Dionex UltiMate 3000 RSLCnano system (Thermo Fisher Scientific, Waltham, MA, USA).
Proteomic data analysis - Timing: NA
Protein identification and label-free quantitation of proteins from mass spectrometry (MS) and MS/MS raw data were performed using the MaxQuant software (version 2.2.0.0) (Max Planck Institute of Biochemistry, Planegg, Germany) against a reference proteome database of Homo sapiens (Uniprot ID: UP000005640, accessed 12/01/2024). The following settings were used: trypsin digestion and oxidation (M) and acetyl (Protein N-term) set as fixed modifications. The minimum peptide length and minimum and maximum peptide length for unspecified search was 7, 8 and 25 amino acids respectively, maximum peptide mass was 4600 Da. The peptide and protein false discovery rate were both set at 0.01, whilst minimum razor and unique peptide was set to 1. The results were exported to Microsoft Office Excel for further processing. Obvious contaminants (e.g. keratins) were removed from the identified protein groups list. Protein groups were subsequently filtered and included only if they were present in at least 2 of the 3 replicates. In addition, the proteomic data were further deciphered by analysing the gene names identified from the MS data using the Functional Enrichment Analysis Tool platform (FunRich, accessed 15/01/2024) [45] for gene-enrichment analysis and comparison with Vesiclepedia [46]. The InteractiVenn (http://www.interactivenn.net) online software was used to make the Venn diagram [47].
Statistical analysis
Results are reported as the mean ± standard error of the mean (SEM). Welch’s unpaired t test was used to determine the significance of the observed differences between different techniques. Differences were considered statistically significant at p < 0.05 (ns not significant, * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001).
EV-track
We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase [48] (EV-TRACK ID: EV231013).
Results
The protein and particle yield from 1ml human plasma samples differs between separation techniques
The ability to perform subsequent investigations depends on the availability of sufficient quantity of separated EVs. However, without directly labelling of EVs prior to separation, the assessment of yield relies on the calculation of several surrogate markers, namely protein concentration as assessed by BCA™ assay and particle concentration as assessed by nanoparticle tracking analysis.
The total concentration of protein within the samples from the same starting volume (1mL) of human plasma was significantly less for all the samples compared to unprocessed plasma (Fig. 2a). There is a clear practical and statistically significant difference in the amount of protein obtained using different techniques. Precipitation produces the sample with greatest amount of protein from a 1mL plasma sample (384.6 μg) which was statistically greater than all other techniques, closely followed by SEC (184.1 μg), whilst magnetic bead capture (6.721 μg), dUC + 0.22 μm filtration (9.626 μg) and dUC (14.05 μg) obtain the smallest amount of protein with no statistical difference between methods.

Comparison of the yield from the five different plasma EV separation methods from a starting sample of 1mL plasma. A BCA™ assay identified the total protein level of the samples from the five different separation methods. b Nanoparticle tracking analysis identified the number of particles separated from the samples. c Sample purity as assessed by the particle to protein ratio. Data presented as mean ± SEM; n = 9 (3 per individual, individuals mean highlighted by symbol), p-values ns not significant, * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001
There is also a clear numeric and statistically significant difference in the particle concentration of the samples between the various separation methods as well as with unprocessed plasma (Fig. 2b). Similar to the differences seen in protein concentration yield, the precipitation samples had significantly more particles from 1mL plasma (3.18 × 1011) than the other separation techniques, followed by SEC (4.95 × 1010), whilst magnetic bead capture (1.92 × 109), dUC (1.50 × 109) and dUC + 0.22 μm filtration (1.15 × 109) samples had the fewest particles with no statistically significant difference between techniques. Unprocessed plasma had the highest particle count (9.87 × 1011), which is likely elevated due to the presence of lipoproteins and large protein complexes that are present in plasma at much greater concentrations than within the EVs [15].
By comparing the ratio of particle count to protein concentration (Fig. 2c), a measure of non-EV protein contamination described by Webber and Clayton [49], it is clear that all the different separation techniques samples are significantly enriched for particles compared to unprocessed plasma. Ultracentrifugation based techniques demonstrate the smallest enrichment, whilst precipitation demonstrated significantly greater enrichment.
EV enrichment and contaminant depletion varies between different separation techniques
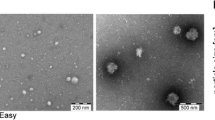
Comparison of the ability of the different separation methods to enrich the sample for EVs and deplete contaminants was performed using multiple assays. Firstly, TEM was performed to allow visualisation of the EVs within the samples s. Clearly identified particles, highlighted by the white arrows (Fig. 3a), can be seen in the images from each of the EV separation techniques. The precipitation and SEC samples have a much larger concentration of particles compared to the other EV separation methods, and the particles are generally smaller in the SEC and precipitation samples in keeping with the findings from the NTA. In addition, both TEIR and SEC have particles which seem to demonstrate lipoprotein-like morphology in addition to particles with EV-like morphology.

Assessment of EV enrichment and purity. Transmission electron microscopy (TEM) visualisation of the EVs separated from human plasma at a 10,000 × magnification (scale bar 500nm) and b 30,000 × magnification (scale bar 100nm), white arrow identifying a representative EV. c Ponceau and Western blot analysis of d EV surface markers (CD63, CD9 and CD81) and cargo markers (HSP70, TSG101 and syntenin), and e the most commonly co-separated contaminants (GM130, albumin, Apolipoprotein A1, Apolipoprotein B and Apolipoprotein E) following loading of 5 μg of sample from each separation method and unprocessed plasma. f Ponceau and g Western blot analysis of EV surface markers (CD63, CD9 and CD81) and lipoproteins (Apolipoprotein B and Apolipoprotein E) following loading of 40 μL of sample from precipitation (both with and without proteinase K (pK)) and size exclusion chromatrography plus unprocessed plasma (4 μL diluted 1:10). Representative images are shown
Additionally, analysis of common EV surface and cargo proteins, as well as common contaminant proteins was performed by Western blot. From the Ponceau stain analysis (Fig. 3c), there is a clear difference in the global protein expression despite loading equal amounts of protein highlighting the differential enrichment and depletion of proteins by distinct separation methods.
The most frequently used markers to assess for EV enrichment are the surface tetraspanins (CD9, CD63, and CD81) and cargo proteins such as HSP70, TSG101 and syntenin. As shown in Fig. 3d, CD9 is significantly enriched in ultracentrifugation-based techniques and magnetic bead capture compared to SEC. CD81 and CD63 are identified in both ultracentrifugation-based techniques and SEC separated sampled, although different isoforms of CD63 seem to be more prevalent between SEC and ultracentrifugation-based techniques. Interestingly, neither CD81 nor CD63 are identified following magnetic bead capture suggesting that this technique may not facilitate an unbiased enrichment of all EVs.
When loading a greater volume of sample for SEC (Fig. 3f,g), all three commonly assessed tetraspanins are identified demonstrating the effective separation of EVs. The inclusion of proteinase K, as recommended by the manufacturer, significantly reduces the overall protein content of the sample as expected and facilitates the identification of CD9 and CD81 in comparison to precipitation without proteinase K.
Similarly, the presence of EV cargo proteins differs between separation techniques. All three of the commonly used EV cargo markers are enriched in the dUC, dUC + 0.22 μm filtration and SEC samples, with reducing intensities respectively. The magnetic bead sample is clearly enriched for HSP70 but neither TSG101 nor syntenin are identified, again suggesting that a particular subgroup of EVs is separated when using this technique.
Through comparing commonly co-separated proteins (Fig. 3c) a measure of sample purity can be inferred. As expected, GM130, a marker of cellular debris, is either not identified or identified with much lower intensity in all the samples compared to plasma. This is expected given that the processing for all the separation methods includes performing a centrifugation step at 10,000 × g which is known to deplete cellular debris.
Albumin is the most abundant protein in plasma and a frequent co-separated protein in EV samples. As anticipated, there is an extremely strong band in the unprocessed plasma sample, whilst there is also a visible band for magnetic capture and ultracentrifugation-based techniques, and a fainter band just visible for SEC.
Lipoproteins are some of the most challenging contaminants to deplete when separating EVs from plasma due to their overlapping density (high density lipoproteins 1.063–1.210 g/mL) or size (chylomicrons 75-1200nm and very low density lipoproteins ~ 30-80nm) compared to extracellular vesicles (1.08–1.21g/ml and 30-1000nm respectively) [15]. Therefore, the relative lipoprotein contamination was also identified by the assessment of apolipoprotein A1 (APO A1, present primarily in high density lipoproteins), apolipoprotein B (APO B, present with different isoforms in chylomicrons, very low-density lipoproteins, intermediate-density lipoproteins, and low-density lipoproteins) and apolipoprotein E (APO E, present in chylomicrons, very low-density lipoproteins, intermediate-density lipoproteins and high density lipoproteins) levels present in the samples (Fig. 3c). The only technique which had detectable APO A1 presence was precipitation, whilst ultracentrifugation-based techniques identified the presence of APO E, albeit with low signal intensity. APO B is identified in ultracentrifugation and SEC based technique samples, although with greater intensity in SEC and with different isoforms. As expected, magnetic capture effectively depletes each of the apolipoproteins from the sample.
Different separation techniques affect the EV particle size distribution
EVs are known to have various sizes, with different size EVs associated with different subpopulations. The mean (Fig. 4a,d) and median (Fig. 4b) particle size was therefore assessed using several orthogonal methods to identify any size differences based on separation method.

a The mean and b the median particle size, and c size distribution of the separated particles as assessed by nanoparticle tracking analysis (NTA), and d the mean particle size as assessed by single particle interferometric reflectance imaging sensing (Leprechaun® analysis). Data presented as mean ± SEM; n = 9 (3 per individual, individuals mean highlighted by symbol) for NTA, n = 1 for Leprechaun® analysis, p-values ns not significant, * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001
By NTA, Plasma has the smallest mean and median particle size at 76 nm and 66 nm respectively, likely due to the presence of lipoproteins, cell debris, or protein aggregates. Of the samples, the precipitation reagent had the smallest mean particle size at 92 nm, then the SEC sample at 103 nm, followed by dUC + 0.22 μm filtration and dUC at 128 nm and 136 nm respectively, and then finally magnetic bead capture with the largest mean particle size at 150 nm. These findings were similar but with slightly smaller values when comparing median particle size by NTA with precipitation separating particles with a median size of 82 nm, followed by SEC with 95 nm, dUC + 0.22 μm filtration and dUC at 113.6 nm and 122.7 nm respectively, and magnetic bead capture with the largest median particle size at 132 nm.
This pattern was recapitulated in the mean size as assessed by the Leprechaun®, albeit at a lower mean size possibly due to the Leprechaun’s® improved resolution at smaller sizes compared to the Nanosight300. Interestingly, the mean particle size for plasma is now most similar to ultracentrifugation-based techniques, whilst there is also a smaller degree of variance between techniques. This finding is likely due to the exclusion of particles without tetraspanin surface markers when using the Leprechaun® and therefore may represent a more accurate picture of the true EV size distribution.
Additionally, the distribution of particle size as assessed by NTA (Fig. 4c) and by the Leprechaun® (Supplementary Fig. 2a,b) was compared between separation methods. For NTA, this identified a more prominent second peak of particles at ~ 150nm in size in the ultracentrifugation and magnetic bead capture samples compared to the precipitation reagent and SEC and likely explains the increase in mean and median particle size with these separation methods. Whilst for the Leprechaun®, ultracentrifugation-based techniques and SEC have the closest similarity to the EV size distribution from unprocessed plasma.
The representativeness of surface tetraspanin distribution differs amongst different techniques
Different subpopulations of EVs have different tetraspanin surface markers. Using the Leprechaun®, the proportion of EVs with specific tetraspanin marker positivity, including co-localisation of multiple tetraspanins on a single EV, can be analysed and compared to EVs captured directly from unprocessed plasma (Fig. 5 and Supplementary Fig. 3).

Comparison of the tetraspanin EV surface markers by single particle interferometric reflectance imaging sensing (Leprechaun® analysis) of particles captured on a CD63, b CD81, and c CD9 antibody chips and d the summed difference in values between plasma tetraspanin distribution and tetraspanin distribution using different EV separation techniques for the different capture spots and the mean difference of all capture spots
Our results demonstrated that precipitation and magnetic bead capture have the least representative pattern of tetraspanin positivity, which is surprising given their supposed less targeted mechanism of separation. Consistent with the Western blot results, magnetic bead capture has clearly separated an enriched population of CD9 positive EVs with less CD81 an CD63 positive EVs compared to plasma, suggesting that phosphatidylserine may not be equally distributed among different tetraspanin positive EV subtypes. Precipitation on the other hand seems to be enriching for single marker positive EVs at the expense of double or triple marker co-localised EVs. Whilst separating EVs with two or three co-localised markers is infrequent with all separation methods, and in unprocessed plasma, their omission clearly reduces the representativeness of the EVs separated using precipitation.
Both SEC and ultracentrifugation-based techniques have good representativeness of tetraspanin distribution on their separated EV population compared to plasma suggesting that they may be more suitable for downstream biomarker investigations. Similar to the previously highlighted findings, the addition of a 0.22 μm filtration step to differential ultracentrifugation has minimal impact on the tetraspanin distribution of the separated EVs. The ultracentrifugation-based techniques have the most similar tetraspanin distribution to unprocessed plasma when investigating the CD63 capture spot (Fig. 5a), whilst SEC has the most representative distribution when investigating the CD81 capture spot (Fig. 5b), with similar levels of representativeness using the CD9 capture spot (Fig. 5c). To identify the most indicative separation method, we compared the sum of the differences between the proportion of specific tetraspanin marker positivity using an individual separation method to that of unprocessed plasma (Fig. 5d), which favoured SEC closely followed by ultracentrifugation-based techniques.
Proteomic analysis of EV cargo
To further characterise the separated EVs and their cargo, MS based proteomic analysis was performed. A total of 379 unique proteins were identified with high confidence. Of the identified proteins, 105 (27.7%) were common to all EV separation techniques, 311 (82.0%) were identified in samples from at least two separation techniques, and 68 (17.9%) were unique to a single separation technique (Fig. 6a).

Mass spectrometry (MS) and bioinformatics-based proteomic analysis of EV cargo. a Venn diagram based on the proteins identified by MS hits. b Venn diagram of all proteins identified compared with proteins annotated in the Vesiclepedia database. c Gene enrichment analysis for ‘Cellular component’ was performed based on the MS hits identified from each EV separation method. d Pairwise comparison diagram showing the similarity between the proteome contents of the different EV separation methods and the identified EV-related proteins from the Vesiclepedia database
Bioinformatic analysis using FunRich [45] and the Vesiclepedia [46] database confirmed the presence of 304 known EV-related proteins out of all the identified proteins (80.2%) (Fig. 6b), whilst cellular component analysis identified ‘exosomes’ as the origin of ≥ 70% of the identified proteins for all the separation techniques (range: 70.0-75.8%) (Fig. 6c). When analysing the known EV-related proteins by EV separation method, the greatest number of known EV-related proteins was identified by UC (263) and then SEC (214), whilst TEIR (92) identified the lowest number of known EV related proteins (Fig. 6d), although this is impacted by the lower total number of proteins identified with precipitation-based separation.
Our results (Fig. 6d) demonstrate that the TEIR separated has the greatest difference in identified protein species compared to the other separation methods (32.1-42.5% protein species overlap), whilst SEC and ultracentrifugation-based techniques are the most similar separation methods (69.3-71.6% protein species overlap), in keeping with their similarities from the Leprechaun® analysis.
However, Vesiclepedia is an open-source repository containing results from studies employing different techniques with varying stringency in EV separation and characterisation. This increases the likely non-EV co-separated proteins in the database. Therefore, we also compared the common EV-associated proteins identified using the different separation methodology to those identified by Karimi et al. [50] who employed a stringent protocol of density gradient ultracentrifugation to separate EVs from 40-80 mL of healthy volunteer plasma (Table 1). Whilst comparison of the results from different MS protocols is challenging, this demonstrates that dUC has identified a greater number of common EV-associated proteins than the other methods, followed by SEC and magnetic bead capture, whilst precipitation identified the fewest number of common EV-associated proteins. This is in keeping with the findings when comparing to the full Vesiclepedia database.
The proteins identified by MS from the different separation methods can be used to assess the relative abundance of known EV associated proteins as well as residual lipoproteins and albumin. A panel of six proteins identified from both the Exocarta [51] and Vesiclepedia [46] top 20 EV-associated proteins, and strongly linked with the EV proteome [50, 52], were chosen to investigate differences in EV enrichment between the different separation methods. There is significant variation in whether individual methods enrich for EV-associated proteins (Fig. 7a), although consistently the dUC and to a lesser extent the SEC samples were able to identify all five EV-associated proteins.

Mass spectrometry label free quantification intensity comparing the relative expression of a proteins frequently associated with EVs (CD9, HSC70, HSP90AA1, β-actin, Cofilin 1, 14-3-3 proteinζ/δ) and b common co-separated proteins (albumin, apolipoprotein A1, apolipoprotein B, apolipoprotein E). Data presented as mean ± SEM; n = 3, p-values ns not significant, * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001
Magnetic bead capture results in significantly greater residual albumin abundance than the other separation methods from the equivalent starting protein concentration used for MS analysis, whilst SEC and precipitation has the lowest abundance numerically (Fig. 7b). However, due to the lower protein quantification yield with magnetic bead capture, the MS experiment utilised almost the entire eluted sample volume to facilitate loading 10 μg of protein compared to lower requirements for the other separation methods.
As would be expected, the pattern of residual apolipoproteins abundance reflects the separation methodology. Ultracentrifugation based techniques have the greatest relative residual abundance of apolipoprotein A1 (Fig. 7b), the major component of HDL cholesterol that is most similar in density to EVs, whilst SEC has significantly greater residual abundance of apolipoprotein B and apolipoprotein E (Fig. 7b), the major component of chylomicrons and LDL cholesterol both of which overlap in size with EVs.
Discussion
The ability to reliably separate representative EVs is a key requirement in the development of clinically validated blood-based EV liquid biopsy biomarkers [53]. To further advance the reproducibility and quality of EV related research, the International Society of Extracellular Vesicles has published [12] and recently updated guidelines [13] which further emphasised the key reporting requirements for EV related research. However, there is still no consensus regarding an optimal separation method, and they therefore recommended using an appropriate method for the planned downstream application.
Ultracentrifugation based techniques are frequently utilised [26], however the role of an additional filtration step is still uncertain. From our results presented above, we conclude that the addition of a 0.22 μm filtration step does not significantly improve the quality of the EV sample but does reduce the yield of the sample as assessed by multiple methods as described above. Given the low yield of differential ultracentrifugation already limits the number of downstream investigations that can be performed, the addition of an extra filtration step would not be recommended based on these results.
In this study, precipitation led to the greatest yield as assessed by protein quantification assay and nanoparticle tracking analysis, and both precipitation and SEC resulted in yields that would be sufficient for multiple downstream applications. Despite this much greater yield, the benefits of these techniques may be partially offset by a lower degree of EV enrichment as evidenced by the Western blot findings related to residual free protein and lipoprotein co-separation respectively. Additionally, when analysing the MS proteomics data, the greater protein quantification yield with precipitation is not translated into a greater number of identified protein sequences. This result is likely explained by the presence of residual co-isolating proteins, primarily albumin, within the separated sample despite the inclusion of proteinase K which substantially reduces the protein content of the precipitation sample. Whereas SEC-based separation seems to result in good yield of EV-related proteins and a representative tetraspanin distribution as demonstrated by the MS and Leprechaun® analysis respectively. The proteomic yield could potentially be optimised further if a greater starting protein concentration is used for MS analysis.
The SEC protocol used in this analysis does seem to co-separate a greater degree of apolipoproteins than other methods, particularly apolipoprotein B and apolipoprotein E. Given its size-based separation methodology it is expected that this is related to the co-separation of lipoproteins given that this column has a pore size of 35 nm to effectively separates particles which are larger than 30 nm and therefore overlaps with the size range of very low density lipoproteins and chylomicrons. A column with a larger pore size is commercially available, however this will separate particles larger than 70 nm, but not the smallest EVs, and so a balance between purity and completeness of EV separation must be struck. Additionally, the full interpretation of lipoprotein co-separation is further complicated by the identification of apolipoproteins as a significant component of the EV corona, proteins and particles which directly interact with the surface of EVs [52, 54, 55], and therefore further lipid focussed analyses such as enzyme linked immunosorbent assays would be useful to fully characterise the differences in co-separated lipoprotein for the various methods.
SEC also has the added benefit of scalability, with different sized columns available for different starting volumes. Whilst other separation methods can accommodate a range of starting volumes, they are typically limited either due to their lower yield with smaller volumes in the case of differential ultracentrifugation and magnetic bead capture or cost of reagents with larger volumes in the case of precipitation kits or magnetic bead capture. Meanwhile SEC columns can be purchased or designed for the required starting volume with no loss of yield and no excessive costs. Several groups are also investigating further optimisations to improve EV separation by SEC with dual- [56] and tri-mode [57] chromatography methods being described. However, these techniques are not widely practiced and remain the preserve of specialised laboratories.
As highlighted above, immunoaffinity capture can distinguish a highly pure EV sample; however, the yield was the lowest of all the techniques compared, thus limiting the number of downstream investigations that can be performed when a small volume of starting biofluid is present as in clinical samples. Additionally, the finding of CD9 enrichment in the EV samples, as demonstrated by the Western blot and Leprechaun® analyses, adds further evidence to the finding that phosphatidylserine is not ubiquitously expressed across all EVs but may be enriched in particular subgroups [33, 34]. Further work to define the lipid composition including phosphatidylserine of different EV subgroups would help elucidate this. Despite this clear enrichment of CD9 in the Western blot and Leprechaun results, CD9 expression was not identified in the MS data. This has been reported previously with prior publications [58] failing to identify CD9 via MS despite enrichment identified by Western blot due to intrinsic difficulties identifying tetraspanins by MS [59]. However, CD9 was identified in samples separated using other methodologies in this study, with less enrichment identified by Western blot, which raises the possibility that there may be some interference between the separation methodology and protein identification by MS.
One significant advancement of this study compared to previous studies is the ability to compare the separated EV population to the ‘ground truth’ EV population from unprocessed plasma using the Leprechaun®. This has demonstrated unexpected findings whereby the ‘non-targeted’ precipitation reagent and the magnetic bead capture targeting phosphatidylserine have the least representative pattern of tetraspanin marker positivity on their separated EVs suggestive of enrichment of subpopulations of EVs compared to those present in unprocessed plasma.
In this comparison, we have limited the separation methods to those that are frequently employed, without the requirement of complicated and expensive equipment, and therefore would be more easily adopted in clinical settings. Whilst novel methods to separate EVs continue to be developed, including asymmetric flow field flow fractionation [60], microfluidic based approaches [61] and advanced size exclusion chromatography techniques [57], they remain restricted to individual labs with significant expertise in their use. These techniques have shown promise for improving EV separation, but they are not currently suitable for widespread adoption due to high set up costs and the requirement for specialised equipment. Additionally, the samples all underwent different pre-processing steps, based on the different manufacturer’s instructions or standard practice for UC-based methods, with different initial centrifugation speeds and length of spins. The specific impact that this may have had on the results is unknown but could explain some of the variation. Finally, due to the limited protein quantification yield from some of the assays, only 10 μg of protein was used for MS analysis and so the sensitivity of protein identification from methods with greater protein quantification yield (precipitation- and SEC-based separation methods) has not been fully assessed.
Conclusion
In conclusion, the enrichment of EVs and depletion of different contaminants clearly varies according to separation method used. The suitability of the resulting EV enriched sample for downstream analyses is dependent on the separation method yielding enough EVs for the required analysis. Therefore, the optimal choice of separation method will be dependent on the requirement for different degrees of purity for the desired application. In this study, we have characterised the differences in EV enriched samples after using different EV separation techniques from a clinically relevant 1mL sample of human plasma. We have demonstrated differences in yield, required to allow multiple characterisation and investigative experiments, purity, and the representativeness of the separated EV population (Table 2).
From the results presented above, we highlight that SEC-based separation is a suitable EV separation technique from a clinically relevant 1mL plasma sample. The SEC-based separation sample results in a greater yield, that is sufficient to allow for both EV characterisation and biomarker identification analyses, whilst it also retains enough purity to be confident that the sample is EV enriched and has an EV tetraspanin distribution that is representative of EVs contained within unprocessed plasma. For these reasons, it is our preferred method and would be suitable for widespread adoption in less EV-specialised labs for clinical biomarker identification studies.
Availability of data and materials
The data that support the findings of this study can be viewed upon reasonable request to the corresponding authors.
Abbreviations
- EVs:
-
Extracellular Vesicles
- SEC:
-
Size exclusion chromatography
- dUC:
-
Differential ultracentrifugation
- dUC + f:
-
Differential ultracentrifugation + 0.22 μm filtration
- TEIR:
-
Total exosome isolation reagent
- Mag:
-
Magnetic bead separation
- NTA:
-
Nanoparticle tracking analysis
- TEM:
-
Transmission electron microscopy
- SP-IRIS:
-
Single particle interferometric reflectance imaging sensing
- MS:
-
Mass spectrometry
- SEM:
-
Standard error of the mean
References
Zhou B, Xu K, Zheng X, Chen T, Wang J, Song Y, et al. Application of exosomes as liquid biopsy in clinical diagnosis. Signal Transduct Target Ther. 2020;5(1):144.
Short SC, Noushmehr H. Unmet need for liquid biomarkers and the Brain-Liquid Biopsy Consortium. Neurooncol Adv. 2022;4(Suppl 2):ii1–3.
Raghav A, Singh M, Jeong GB, Giri R, Agarwal S, Kala S, et al. Extracellular vesicles in neurodegenerative diseases: a systematic review. Front Mol Neurosci. 2022;15:1061076.
Zhang X, Wu Y, Cheng Q, Bai L, Huang S, Gao J. Extracellular vesicles in cardiovascular diseases: diagnosis and therapy. Front Cell Dev Biol. 2022;10:875376.
Condrat CE, Varlas VN, Duică F, Antoniadis P, Danila CA, Cretoiu D, et al. Pregnancy-related extracellular vesicles revisited. Int J Mol Sci. 2021;22(8):3904.
Tatischeff I. Extracellular vesicle-DNA: the next liquid biopsy biomarker for early cancer diagnosis? Cancers (Basel). 2023;15(5):1456.
Reichert ZR, Morgan TM, Li G, Castellanos E, Snow T, Dall’Olio FG, et al. Prognostic value of plasma circulating tumor DNA fraction across four common cancer types: a real-world outcomes study. Ann Oncol. 2023;34(1):111–20.
Soffietti R, Bettegowda C, Mellinghoff IK, Warren KE, Ahluwalia MS, De Groot JF, et al. Liquid biopsy in gliomas: a RANO review and proposals for clinical applications. Neuro Oncol. 2022;24(6):855–71.
Irmer B, Chandrabalan S, Maas L, Bleckmann A, Menck K. Extracellular vesicles in liquid biopsies as biomarkers for solid tumors. Cancers (Basel). 2023;15(4):1307.
Pink RC, Beaman EM, Samuel P, Brooks SA, Carter DRF. Utilising extracellular vesicles for early cancer diagnostics: benefits, challenges and recommendations for the future. Br J Cancer. 2022;126(3):323–30.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–28.
Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
Welsh JA, Goberdhan DCI, O’Driscoll L, Buzas EI, Blenkiron C, Bussolati B, et al. Minimal information for studies of extracellular vesicles (MISEV2023): from basic to advanced approaches. J Extracell Vesicles. 2024;13(2):e12404.
Vergauwen G, Tulkens J, Pinheiro C, Avila Cobos F, Dedeyne S, De Scheerder MA, et al. Robust sequential biophysical fractionation of blood plasma to study variations in the biomolecular landscape of systemically circulating extracellular vesicles across clinical conditions. J Extracell Vesicles. 2021;10(10):e12122.
Simonsen JB. What are we looking at? Extracellular vesicles, lipoproteins, or both? Circ Res. 2017;121(8):920–2.
Cilibrasi C, Simon T, Vintu M, Tolias C, Samuels M, Mazarakis NK, et al. Definition of an inflammatory biomarker signature in plasma-derived extracellular vesicles of glioblastoma patients. Biomedicines. 2022;10(1):125.
De Sousa KP, Rossi I, Abdullahi M, Ramirez MI, Stratton D, Inal JM. Isolation and characterization of extracellular vesicles and future directions in diagnosis and therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2023;15(1):e1835.
Wei R, Zhao L, Kong G, Liu X, Zhu S, Zhang S, et al. Combination of size-exclusion chromatography and ultracentrifugation improves the proteomic profiling of plasma-derived small extracellular vesicles. Biol Proced Online. 2020;22:12.
Helwa I, Cai J, Drewry MD, Zimmerman A, Dinkins MB, Khaled ML, et al. A comparative study of serum exosome isolation using differential ultracentrifugation and three commercial reagents. PLoS One. 2017;12(1):e0170628.
Veerman RE, Teeuwen L, Czarnewski P, GüclülerAkpinar G, Sandberg A, Cao X, et al. Molecular evaluation of five different isolation methods for extracellular vesicles reveals different clinical applicability and subcellular origin. J Extracell Vesicles. 2021;10(9):e12128.
Brennan K, Martin K, FitzGerald SP, O’Sullivan J, Wu Y, Blanco A, et al. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci Rep. 2020;10(1):1039.
Zhang X, Takeuchi T, Takeda A, Mochizuki H, Nagai Y. Comparison of serum and plasma as a source of blood extracellular vesicles: Increased levels of platelet-derived particles in serum extracellular vesicle fractions alter content profiles from plasma extracellular vesicle fractions. PLoS One. 2022;17(6):e0270634.
Dhondt B, Pinheiro C, Geeurickx E, Tulkens J, Vergauwen G, Van Der Pol E, et al. Benchmarking blood collection tubes and processing intervals for extracellular vesicle performance metrics. J Extracell Vesicles. 2023;12(5):e12315.
Kamei N, Nishimura H, Matsumoto A, Asano R, Muranaka K, Fujita M, et al. Comparative study of commercial protocols for high recovery of high-purity mesenchymal stem cell-derived extracellular vesicle isolation and their efficient labeling with fluorescent dyes. Nanomedicine. 2021;35:102396.
Patel GK, Khan MA, Zubair H, Srivastava SK, Khushman M, Singh S, et al. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci Rep. 2019;9(1):5335.
Gardiner C, Di Vizio D, Sahoo S, Théry C, Witwer KW, Wauben M, et al. Techniques used for the isolation and characterization of extracellular vesicles: results of a worldwide survey. J Extracell Vesicles. 2016;5:32945.
Livshits MA, Khomyakova E, Evtushenko EG, Lazarev VN, Kulemin NA, Semina SE, et al. Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci Rep. 2015;5:17319.
Cvjetkovic A, Lötvall J, Lässer C. The influence of rotor type and centrifugation time on the yield and purity of extracellular vesicles. J Extracell Vesicles. 2014;3:23111.
Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;Chapter 3:Unit 3.22.
Ströhle G, Gan J, Li H. Affinity-based isolation of extracellular vesicles and the effects on downstream molecular analysis. Anal Bioanal Chem. 2022;414(24):7051–67.
Nakai W, Yoshida T, Diez D, Miyatake Y, Nishibu T, Imawaka N, et al. A novel affinity-based method for the isolation of highly purified extracellular vesicles. Sci Rep. 2016;6:33935.
Zaborowski MP, Balaj L, Breakefield XO, Lai CP. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience. 2015;65(8):783–97.
Connor DE, Exner T, Ma DD, Joseph JE. The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb Haemost. 2010;103(5):1044–52.
Matsumoto A, Takahashi Y, Ogata K, Kitamura S, Nakagawa N, Yamamoto A, et al. Phosphatidylserine-deficient small extracellular vesicle is a major somatic cell-derived sEV subpopulation in blood. iScience. 2021;24(8):102839.
Williams S, Jalal AR, Lewis MP, Davies OG. A survey to evaluate parameters governing the selection and application of extracellular vesicle isolation methods. J Tissue Eng. 2023;14:20417314231155110.
KrušićAlić V, Malenica M, Biberić M, Zrna S, Valenčić L, Šuput A, et al. Extracellular vesicles from human cerebrospinal fluid are effectively separated by sepharose cl-6b-comparison of four gravity-flow size exclusion chromatography methods. Biomedicines. 2022;10(4):785.
Liu DSK, Upton FM, Rees E, Limb C, Jiao LR, Krell J, et al. Size-Exclusion Chromatography as a Technique for the Investigation of Novel Extracellular Vesicles in Cancer. Cancers (Basel). 2020;12(11):3156.
Lane RE, Korbie D, Trau M, Hill MM. Optimizing size exclusion chromatography for extracellular vesicle enrichment and proteomic analysis from clinically relevant samples. Proteomics. 2019;19(8):e1800156.
Conroy MC, Lacey B, Bešević J, Omiyale W, Feng Q, Effingham M, et al. UK Biobank: a globally important resource for cancer research. Br J Cancer. 2023;128(4):519–27.
Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913.
Lucien F, Gustafson D, Lenassi M, Li B, Teske JJ, Boilard E, et al. MIBlood-EV: minimal information to enhance the quality and reproducibility of blood extracellular vesicle research. J Extracell Vesicles. 2023;12(12):e12385.
Bordanaba-Florit G, Royo F, Kruglik SG, Falcón-Pérez JM. Using single-vesicle technologies to unravel the heterogeneity of extracellular vesicles. Nat Protoc. 2021;16(7):3163–85.
Best MG, In ‘t Veld S, Sol N, Wurdinger T. RNA sequencing and swarm intelligence-enhanced classification algorithm development for blood-based disease diagnostics using spliced blood platelet RNA. Nat Protoc. 2019;14(4):1206–34.
Lane R, Simon T, Vintu M, Solkin B, Koch B, Stewart N, et al. Cell-derived extracellular vesicles can be used as a biomarker reservoir for glioblastoma tumor subtyping. Commun Biol. 2019;2:315.
Pathan M, Keerthikumar S, Chisanga D, Alessandro R, Ang CS, Askenase P, et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracell Vesicles. 2017;6(1):1321455.
Chitti SV, Gummadi S, Kang T, Shahi S, Marzan AL, Nedeva C, et al. Vesiclepedia 2024: an extracellular vesicles and extracellular particles repository. Nucleic Acids Res. 2024;52(D1):D1694–8.
Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16(1):169.
Van Deun J, Mestdagh P, Agostinis P, Akay Ö, Anand S, Anckaert J, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14(3):228–32.
Webber J, Clayton A. How pure are your vesicles? J Extracell Vesicles. 2013;2(1):19861.
Karimi N, Cvjetkovic A, Jang SC, Crescitelli R, HosseinpourFeizi MA, Nieuwland R, et al. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell Mol Life Sci. 2018;75(15):2873–86.
Keerthikumar S, Chisanga D, Ariyaratne D, Al Saffar H, Anand S, Zhao K, et al. ExoCarta: a web-based compendium of exosomal cargo. J Mol Biol. 2016;428(4):688–92.
Hallal S, Tűzesi Á, Grau GE, Buckland ME, Alexander KL. Understanding the extracellular vesicle surface for clinical molecular biology. J Extracell Vesicles. 2022;11(10):e12260.
Robinson SD, de Boisanger J, Pearl FMG, Critchley G, Rosenfelder N, Giamas G. A brain metastasis liquid biopsy: Where are we now? Neurooncol Adv. 2024;6(1):vdae066.
Tóth E, Turiák L, Visnovitz T, Cserép C, Mázló A, Sódar BW, et al. Formation of a protein corona on the surface of extracellular vesicles in blood plasma. J Extracell Vesicles. 2021;10(11):e12140.
Wolf M, Poupardin RW, Ebner-Peking P, Andrade AC, Blöchl C, Obermayer A, et al. A functional corona around extracellular vesicles enhances angiogenesis, skin regeneration and immunomodulation. J Extracell Vesicles. 2022;11(4):e12207.
Van Deun J, Jo A, Li H, Lin HY, Weissleder R, Im H, et al. Integrated dual-mode chromatography to enrich extracellular vesicles from plasma. Adv Biosyst. 2020;4(12):e1900310.
Ter-Ovanesyan D, Gilboa T, Budnik B, Nikitina A, Whiteman S, Lazarovits R, et al. Improved isolation of extracellular vesicles by removal of both free proteins and lipoproteins. Elife. 2023;12:e86394.
de Menezes-Neto A, Saez MJ, Lozano-Ramos I, Segui-Barber J, Martin-Jaular L, Ullate JM, et al. Size-exclusion chromatography as a stand-alone methodology identifies novel markers in mass spectrometry analyses of plasma-derived vesicles from healthy individuals. J Extracell Vesicles. 2015;4:27378.
Andre M, Le Caer JP, Greco C, Planchon S, El Nemer W, Boucheix C, et al. Proteomic analysis of the tetraspanin web using LC-ESI-MS/MS and MALDI-FTICR-MS. Proteomics. 2006;6(5):1437–49.
Zhang H, Lyden D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat Protoc. 2019;14(4):1027–53.
Cheung LS, Sahloul S, Orozaliev A, Song YA. Rapid detection and trapping of extracellular vesicles by electrokinetic concentration for liquid biopsy on chip. Micromachines (Basel). 2018;9(6):306.
Acknowledgements
The authors acknowledge the Electron microscopy imaging centre at the School of Life Sciences, University of Sussex and want to thank Pascale Schellenberger for her technical support in performing the electron microscopy analysis.
Funding
Dr Stephen Robinson is funded by a University Hospitals Sussex NHS Foundation Trust Medical Doctoral Fellowship.
Author information
Authors and Affiliations
Contributions
Stephen David Robinson: Conceptualisation; funding acquisition; methodology; investigation; validation; formal analysis; data curation; writing – original draft. Mark Samuels: Conceptualisation; methodology; validation; writing – review & editing. William Jones: Conceptualisation; methodology; validation; writing – review & editing. Nicolas Stewart: Investigation; writing – review & editing. Murat Eravci: Formal analysis; writing – review & editing. Nektarios Mazarakis: Supervision; writing – review & editing. Duncan Gilbert: Supervision; funding acquisition; writing – review & editing. Giles Critchley: Supervision; writing – review & editing. Georgios Giamas: Conceptualization; funding acquisition; methodology; validation; supervision; writing – review & editing.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experimental protocols were approved by the University of Sussex Research Ethics Committee (reference: ER/BSMS2229/1). Each participant provided written informed consent to take part in this research.
Consent for publication
Not applicable.
Competing interests
Georgios Giamas is the founder and chief scientific officer of Stingray Bio. The rest of the authors declare that they have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Robinson, S.D., Samuels, M., Jones, W. et al. Confirming size-exclusion chromatography as a clinically relevant extracellular vesicles separation method from 1mL plasma through a comprehensive comparison of methods. BMC Methods 1, 7 (2024). https://doi.org/10.1186/s44330-024-00007-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s44330-024-00007-2