
Abstract
Metformin, a biguanide drug, is the most commonly used first-line medication for type 2 diabetes mellites due to its outstanding glucose-lowering ability. After oral administration of 1 g, metformin peaked plasma concentration of approximately 20–30 μM in 3 h, and then it mainly accumulated in the gastrointestinal tract, liver and kidney. Substantial studies have indicated that metformin exerts its beneficial or deleterious effect by multiple mechanisms, apart from AMPK-dependent mechanism, also including several AMPK-independent mechanisms, such as restoring of redox balance, affecting mitochondrial function, modulating gut microbiome and regulating several other signals, such as FBP1, PP2A, FGF21, SIRT1 and mTOR. On the basis of these multiple mechanisms, researchers tried to repurpose this old drug and further explored the possible indications and adverse effects of metformin. Through investigating with clinical studies, researchers concluded that in addition to decreasing cardiovascular events and anti-obesity, metformin is also beneficial for neurodegenerative disease, polycystic ovary syndrome, aging, cancer and COVID-19, however, it also induces some adverse effects, such as gastrointestinal complaints, lactic acidosis, vitamin B12 deficiency, neurodegenerative disease and offspring impairment. Of note, the dose of metformin used in most studies is much higher than its clinically relevant dose, which may cast doubt on the actual effects of metformin on these disease in the clinic. This review summarizes these research developments on the mechanism of action and clinical evidence of metformin and discusses its therapeutic potential and clinical safety.
Similar content being viewed by others


Introduction
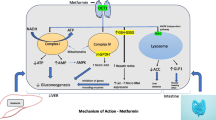
Metformin, derivated from biguanide, is able to effectively lower plasma glucose level by inhibiting hepatic gluconeogenesis (HGP) and improving insulin-resistance with high cost-effectiveness, but nearly has no hypoglycemia side effects [1, 2]. Therefore, since it was synthesized in the 1920s, metformin has been the recommended first-line medication for type 2 diabetes mellites (T2DM) [3]. Metformin is not metabolized and is eliminated unchanged through renal excretion, and this drug is widely distributed into various organs, including intestinal, liver, kidney, brain and so on. After oral administration, metformin is first absorbed in the intestine, which is mediated by plasma membrane monoamine transporter (PMAT) or organic cation transporter 3 (OCT3) on the luminal side of enterocytes [4, 5]. Then, metformin leaves the enterocytes and is transferred into the portal vein through OCT1 on the basolateral membrane. Next, metformin is delivered to the liver and absorbed via OCT1/OCT3, which is expressed on the basolateral membrane of hepatocytes [6, 7], and is excreted from the liver to the circulation via multidrug and toxin extrusion 1(MATE1) [8]. Last, metformin in the circulation is absorbed into renal epithelial cells, which is mediated by OCT2 on the basolateral membrane in the renal tubules [9], and further eliminated into urine through MATE1 and MATE2-K on the apical membrane of the renal proximal tubule cells [10, 11] (Fig. 1).

Pharmacokinetics of Metformin. Following oral dosing of 1 g metformin, the uptake of metformin in intestinal, liver and kidney is mediated by PMAT/OCT3, OCT1/3 and OCT2, respectively, and the excretion of metformin in intestinal, liver and kidney is mediated by OCT1, MATE1 and MATE1/2, respectively. The plasma concentrations of metformin are between 20–30 μM, and the concentrations of metformin in the portal vein are 60–90 μM
Accordingly, following oral dosing of 1 g metformin, a prescribed dose for T2DM treatment in the clinic, the plasma concentrations of metformin are between 20–30 μM, and the concentrations of metformin in the portal vein are roughly estimated to be threefold higher. Therefore, following a therapeutic dose, the hepatic exposure to metformin ranges from 60–90 μM [12]. To explore the clinically relevant doses of metformin in preclinical studies, Madiraju et al. compared the hepatic exposures in rats following different oral ingestions of metformin, and he found that the hepatic exposure to metformin (approximately 50–100 μM) is similar between oral ingestions of 50–100 mg/kg metformin in rats and oral ingestions of 1 g metformin in humans. And the oral dosing of ≥ 250 mg/kg metformin results in > 1 mM hepatic exposure to metformin [13, 14].
Recently, researchers have further explored the underlying mechanisms of action mediated by metformin. One of the most studies mechanisms is the activation of AMP-activated protein kinase (AMPK) [15, 16], a key regulator of various pathways involved in glucose, lipid and energy metabolism. For example, the blockade of AMPK signaling significantly influences the efficiency of metformin for T2DM and atherosclerosis [17, 18]. Besides, metformin also plays roles in changing the pathogenesis of diseases by restoring redox balance, affecting mitochondrial function, activating protein phosphatase 2 (PP2A), releasing fibroblast growth factor 21(FGF21) and so on [19,20,21,22,23]. Moreover, metformin even enables the modulation of gut microbiota [24, 25].
Due to the board mechanisms of action, despite of T2DM, new applications of this old drug have been investigated, such as decreasing cardiovascular events and anti-obesity [26,27,28,29]. In addition, evidence is accumulating that metformin also has potential benefits for cancer [30,31,32], neurodegenerative disease [33, 34], metabolic syndrome [35, 36], polycystic ovary syndrome (PCOS) [37,38,39], aging [40,41,42], coronavirus disease 2019 (COVID-19) [43,44,45]and so on. However, metformin also results in some adverse effects, such as gastrointestinal complaints, lactic acidosis, vitamin B12 deficiency and neurodegenerative disease [46,47,48]. Recently, it has even been reported that metformin treatment during the sperm development increased the risk of birth defects in offspring [49].
However, compared to clinical concentration for treating T2DM patients, much higher metformin concentrations are widely used in beforementioned studies. Hence, although multiple pharmacological effects and clinical evidences have been reported, the mechanisms of action and new applications of this most commonly antidiabetic drug remains only partially elucidated and controversial, especially the metformin dosage in researches, its clinical use is now still limited to diabetic patients. Here, we will summarize and analyze recent research developments on the mechanism of action and clinical evidence of metformin, helping to better understand and repurpose metformin.
Mechanism of action
Metformin is reported to have a number of targets, the first and studied most is AMPK-signaling. Then, researchers found metformin can still affect cells in the absence of AMPK through targeting redox states, mitochondria, and some other signaling, such as FGF21, PP2A, and mTOR. Furthermore, metformin also modulates the gut microbiome to indirectly regulate the human homeostasis. In this part, we will first discuss these mechanisms of action mediated by metformin.
Metfromin exerts its effect in an AMPK-dependent manner
Numerous literatures have demonstrated that metformin exerts its effect through AMPK activation (Fig. 2). AMPK is a heterotrimeric complex, consisting of the α catalytic subunit, scaffold protein β subunit and regulatory γ noncatalytic subunit [50]. The activation of AMPK is initiated by the binding of adenosine monophosphate (AMP) to the γ-subunit, which can lead to structural changes in AMPK and then induce the phosphorylation of the α subunit at Thr172. Based on this mechanism, metformin may mediate AMPK activation by increasing the AMP/ATP (adenosine triphosphate). Interestingly, it has been reported that metformin could also directly bind to the γ subunit of AMPK, however, it is still unclear that whether this interaction between metformin and the γ-subunit can directly activate AMPK, such as AMP [51]. In addition, following glucose starvation, low-dose metformin (5–30 μM) also could activate AMPK through binding with the presenilin enhancer (PEN2) to inhibit the lysosomal proton v-ATPase, while the phosphorylation of AMPK could be suppressed by imidazole propionate, a microbial metabolite, via activating the p38g/AKT (also known as protein kinase B or PKB) pathway [52, 53].

AMPK-dependent mechanism of action mediated by metformin treatment. Depending on AMPK activation, metformin exerts its effects by regulating Glp1r/Pka pathway, mTOR/autophagy pathway, NLRP3, eNOS, STAT3,COX-2, iNOS, Smad3, FOX3, IRS, GCRP and PD-L1 and ACE2 signals
Because of its role in the reduction of acetyl-CoA carboxylase (ACC) activity and lipogenic enzymes and the induction of fatty acid oxidation, AMPK is reported to be as a key regulator in lipid and glucose metabolism [54]. Furthermore, AMPK is also involved in a number of pathways, such as mammalian target of rapamycin complex1(mTORC1) signaling, peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) signaling and signal transducer and activator of transcription 3 (STAT3) signaling [55,56,57]. Consequently, the various effects of AMPK may be partly responsible for metformin’s the wide effects on homeostasis and diseases. As a classical antidiabetic drug, metformin was discovered to lower plasma glucose levels by reducing hepatic glucose production (HGP) and alleviating insulin resistance. Cao et al. reported that 80 μM metformin, a therapeutic metformin concentration in the portal vein, is enough to decrease glucose production and the mRNA levels of glucose-6-phosphatase catalytic (G6pc) and phosphoenolpyruvate carboxykinase 1 (Pck1) in primary hepatocytes in an AMPK-dependent way [58]. Furthermore, Frank et al. demonstrated that a clinically relevant dose of metformin treatment (50 mg/kg) in rats could initiate the AMPK- glucagon like peptide 1 receptor (Glp1r)- protein kinase A (PKA) pathway in the duodenal mucosa, and then enhance the HGP inhibitory effect of metformin depending on the gut-brain-liver neuronal network [59, 60]. Besides, pharmacological metformin concentration (75 μM) treatment of hepatocytes was reported to improve mitochondrial respiratory activity and increase ATP levels by increasing mitochondrial oxidative enzymes and promoting mitochondrial fission through AMPK/mitochondrial fission factor (Mff) signaling. As it has been widely accepted that impaired mitochondrial respiratory activity is a key inducer for the development of insulin resistance, it is reasonable that a pharmacological dose of metformin can improve insulin resistance by activating AMPK [61].
Apart from benefits for T2DM, metformin is also able to improve cardiovascular diseases through reducing cardiovascular end points, not just because of its glucose-lowering effect. The role of AMPK in this metformin-mediated cardiovascular protective action has been elucidated in number of literatures. After treating streptozotocin-induced diabetic cardiomyopathy (DCM) mice with 200 mg/kg metformin and high glucose-treated cardiomyocytes with 2 mM metformin, Fan et al. found that metformin improves autophagy and then alleviates the pyroptosis in DCM by inhibiting the AMPK/mTOR pathway [62]. This AMPK/mTOR-mediated inhibition of autophagy also drives neuroprotection against focal cerebral ischemia after acute preconditioning with a subtherapeutic dose of metformin (10 mg/kg, i.p.) [63]. Moreover, by activating the AMPK/mTOR pathway, metformin is a potential therapeutic for other neurological diseases, such as Parkinson’s disease (PD) and Huntington’s disease, through enhancing neuronal bioenergetics, protecting nerve repair and reducing toxin protein aggregates [64].
Moreover, metformin is also reported to be beneficial for patients with inflammatory diseases. Metformin inhibits the inflammatory response through activating the AMPK/ NLR family pyrin domain containing 3(NLRP3) or AMPK/ endothelial nitric oxide synthase (eNOS) pathway, thus protecting the myocardial from ischemia–reperfusion [65, 66]. In addition, the antiatherosclerosis role of metformin has also been documented, relying on AMPK activation. A therapeutic dose of metformin (100 mg/kg) inhibits monocyte-to-macrophage differentiation and proinflammatory cytokine production via sequentially decreasing STAT3 phosphorylation and attenuating Angiotensin (Ang)-II-induced atheromatous plaque formation and aortic aneurysm in an atherosclerosis mice model [17]. Researches have also pointed out that T2DM-linked neurodegenerative disease (ND), such as Alzheimer’s disease (AD), is related to advanced glycosylation end product (AGE)-caused neuronal impairment via the inflammatory response, and metformin (1 mM) could rescue this inflammation-induced impairment through upregulating of ACC and inhibitory kappa B kinase (IKK), accompanied by restoring inducible nitric oxide synthase (iNOS) and cyclooxygenase-2(COX-2) in an AMPK-dependent way [67]. As metformin plays its cardiovascular protective and neuroprotective roles by exerting an inflammatory inhibitory effect, researchers have further investigated the role of metformin in inflammatory diseases. By utilizing a partial medical meniscectomy (DMM) model, Chen et al. found that metformin (205 mg/kg) inhibits cartilage degradation and limits osteoarthritis development and progression in an AMPK-dependent way [68]. Besides, it has also been illustrated that with the activation of AMPK, metformin (10 mM) reduces transforming growth factor beta (TGF-β)-induced renal fibroblast collagen type I production via inhibiting Smad3-driven connective tissue growth factor (CTGF) expression, and this mechanism may provide a potential role for metformin in the treatment of chronic kidney disease (CKD) through suppressing renal interstitial fibrosis [69].
Accordingly, the AMPK-dependent effect of metformin has been implied to be beneficial for many other pathogeneses, such as cancer [70]. For the critical role of cancer stem/initiating cells in tumorigenesis and cancer development, researchers explored whether metformin affects cancer initiating cells, and found that metformin (1 mM) activates hexaribonucleotide-binding protein 3(FOX3) via AMPK activation, which is sufficient to promote the differentiation of glioma-initiating cells into nontumorigenic cells [71]. Moreover, growing evidences have indicated that metformin has direct therapeutic potential for cancers, whether as a sole drug or in combination with other regimens. AMPK activation by metformin (up to10 mM for cells and 300 mg/kg for mice models) induces autophagy through inhibiting mTOR signaling or the immune response, and thus downregulates programmed death-ligand 1 (PD-L1) expression in a variety of cancer types, such as lymphoma, breast cancer, pancreatic cancer, non-small cell lung cancer, eventually, the growth or metastasis of cancer cells is inhibited [72,73,74,75,76]. It has also been demonstrated that following activation of AMPK, metformin (5 mM for cells and 250 mg/kg for mice models) inhibits pancreatic cancer growth by disrupting the insulin receptor signaling (IRS) or G protein coupled receptor systems (GPCRs) [77,78,79]. Moreover, metformin (100 μM-10 mM for cells and 300–500 mg/kg for mice models) also amplifies its therapeutic effects and enhances cancer patient survival beneficial in an AMPK-dependent way when combined with radiotherapy or chemotherapy [80, 81].
As AMPK is widely expressed in the ovary and testes, so the role of metformin, an AMPK activator, in the reproductive system has also attracted much interest from researchers. The results showed that through activating AMPK-cyclic AMP (cAMP) signaling, metformin (10 mM) has a positive effect on polycystic ovary syndrome (PCOS), a disease associated with reproductive and metabolic abnormalities, by inhibiting steroidogenic enzymes and decreasing androstenedione production [82,83,84,85]. To add, it is well documented that the inhibition of testicular AMPK is an important contributor to the impairment of spermatogenesis and steroidogenesis, so there is reason to believe that in the patients with T2DM or other metabolic disorders, metformin’s restorative role in male reproductive dysfunction is mainly through normalizing of AMPK in testes [86,87,88,89].
Coronavirus disease 2019 (COVID-19), a currently leading threat to public health and the economy, is caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). The SARS-CoV-2 has strong binding affinity to the angiotensin covering enzyme 2(ACE2) receptor of pneumocystis and enterocytes, which is essential for virus entry into cells and leads to its rapid spread throughout the world [90]. On the other hand, ACE2 signaling protects against from COVID-19 complications by regulating the renin–angiotensin–aldosterone system (RAAS) to exert antihypertensive and anti-inflammatory effects [91,92,93]. Zhang et al. demonstrated that the activation of AMPK could cause the Ser-680 phosphorylation of ACE2, thus resulting in inhibiting of the binding of ACE2-viral spike protein, extending the half-life and increasing stability of ACE2 [94, 95]. Taken together, the possible molecular basis for the beneficial role of metformin in COVID-19 complications is also associated with metformin-mediated AMPK activation. However, the underlying mechanisms and theoretical potential for metformin as a treatment for COVID-19 need to be further investigated and confirmed.
However, AMPK activation may also lead adverse effects on homeostasis or disease. Researches have suggested that AMPK signaling, simulated by maternal hyperglycemia-induced embryo oxidative stress, could disrupt embryonic gene expression, so it may cause neural tube defects. To confirm this hypothesis, Loeken and his colleagues investigated whether AMPK activator metformin has a similar adverse effect on embryos, however, the results indicated that due to the lack of the metformin transporters, MATE1 and OCT3, metformin (120 mg/kg) has no influence on AMPK activation in embryos, and did not cause consequent neural tube defects during pregnancy [96]. In Other researches, the evidence implied that an over-dose (2 mM) of metformin indeed has adverse effects; for example, AMPK activation inhibits MIN6 pancreatic β cells proliferation and promotes apoptosis in vitro, which is the underlying mechanism of metformin-induced acute pancreatitis in patients with renal insufficiency [97]. Overactivation of AMPK by metformin (100 μM -1 mM) also inhibits axon growth, impairs neuronal polarization, and even dendritic spine loss, which is related to the early stage of AD [98, 99] (Table 1).
Metformin exerts its effect in an AMPK-independent manner
Metformin exerts its effects by restoring redox balance
In addition to AMPK-dependent manner, it has also been reported that metformin elicits pleiotropic effects in an AMPK-independent way, such as restoring the cellular redox balance (Fig. 3a). Redox homeostasis is a balance between reactive oxygen species (ROS) and the antioxidant system, which is involved in diverse biological and pathological processes, such as metabolism, aging and cancer [130, 131]. Madiraju et al. uncovered that although chronic metformin treatment increased the phosphorylation of AMPK, acute metformin treatment does not lead to the activation of AMPK, it failed to increase the phosphorylation of ACC, a generally accepted signal for AMPK activation. The antihyperglycemic effect of metformin (50 mg/kg) is achieved by increasing the cytosolic redox state and decreasing the mitochondrial redox state, as determined by the ratio of NADH to NAD+, thus the G2PD activity and glycerophosphate dehydrogenase are inhibited, which results in blockade of lactate and glycerol entry into glucose, eventually, HGP is limited [13, 14]. Additionally, substantial evidence indicated that the remodeling redox status of metformin is relevant to different types of cancer. The apoptosis of acute myeloid leukemia (AML) cells is observed after treatment with metformin, which is mediated by reducing ROS via downregulation of oxidative phosphorylation (OXPHOS) [100]. The proliferation of pancreatic cells and osteosarcoma is also inhibited by metformin-mediated ROS downregulation [101, 102]. Moreover, metformin also enhances the sensitivity of esophageal squamous cell carcinoma and colorectal cancer to cisplatin in a redox-dependent way [103, 104]. Of note, the concentrations of metformin used in these cancer researches ranged from 1 to 10 mM, and they were all much higher than the clinically relevant metformin dose.

AMPK-independent mechanisms of action mediated by metformin treatment. a Metformin-induced restoration of redox balance. b Metformin-induced changes in mitochondria. c Metformin-induced modulation of gut microbiome. d metformin-induced regulation of signals, including FBP1, PP2A, FGF21, SIRT1 and mTOR
Besides, the significance of redox homeostasis on aging may offer an explanation for the metformin’s role in aging. Studies with erythrocytes confirmed the hypothesis that metformin could maintain redox homeostasis by reducing aging-related oxidative stress and strengthening antioxidant machinery to improve heme function [105,106,107]. In healthy older people, the ROS in CD4+ T cells could produce a Th17 inflammation profile, however, Bharath and his colleagues indicated that by increasing autophagy and improving mitochondrial function, the metformin(100 μM)-restored redox balance is able to alleviate this inflammation profile [42]. Furthermore, some severity and fatality cases of COVID-19, a currently pandemic disease, are likely relevant to elevated IL-6 levels [132], Previous studies revealed that the reduction of ROS by metformin(1 mM for cells and 150 mg/kg for mice models) is capable of inhibiting calcium release-activated channels(CRAC)-mediated Ca2+ release from the endoplasmic reticulum, consecutively, inhibiting interleukin 6(IL-6) release [108, 133]. Based on these results, the impediment of ROS/Ca2+/IL-6 pathway may be another explanation for the beneficial role of metformin in COVID-19 (Table 1).
Metformin exerts its effects in a mitochondria-dependent way
Since it was mentioned in 1950, it has been generally accepted that metformin has an inhibitory effect on mitochondrial biological function (Fig. 3b), based on convincing data from various cellular models, including rat, mouse and human hepatocytes [134]. The major function of mitochondria is producing ATP through oxidative phosphorylation, which is mediated by respiratory chain complex I. It has been reported that as a noncompetitive inhibitor, metformin enables binding to the Cys39-containing matrix loop of the mitochondrial complex I subunit ND3, however, data from bovine heart mitochondria indicated that the metformin is only a weaker inhibitor of complex I with an IC50 value of 19.4 ± 1.4 mM [135].
As the gluconeogenesis is highly dependent on energy production, consuming six ATP molecules per one glucose molecule synthesized, the metformin-mediated inhibition of mitochondrial biological function, which further results in a decrease in cellular ATP production, which may be another mechanism for its role in HGP reduction. With an AMPKα1/2 knockout mice model, Foretz et al. found that high-dose (≥ 250 μM) metformin treatment still inhibits HGP by decreasing ATP and increasing AMP [109]. In addition, by suppressing the mitochondrial electron transport chain, metformin upregulates the ratio of AMP to ATP, and the increased AMP subsequently inhibits adenylate cyclase to abrogate cAMP production, which further lowers PKA and fructose-2,6-bisphosphate1. These metformin-induced events eventually lead to a decrease in HGP. They further confirmed that in the AMPK-deficient mice and hepatocytes, metformin is still able to block the cAMP accumulation [110]. Apart from its effect on glucose metabolism, the effect of metformin on mitochondria is also plays a role in lipid metabolism. By increasing the biogenesis of mitochondria in brown adipose tissue, a tissue with a vast number of mitochondria, metformin (250 mg/kg) suppresses fatty acid uptake and promotes thermogenesis, exerting anti-obesity effects [111].
Besides its role in energy metabolism, metformin also affects cancer by regulating mitochondrial biogenesis. Dan et al. found that AMPK signaling changes could not fully explain the anticancer effect of metformin, and NAD + /NADH homeostasis and aspartate are also involved. As NAD + /NADH homeostasis and aspartate biosynthesis were previously reported to be critical for cancer cell proliferation, they indicated that metformin (1 mM for cells and 500 mg/kg for mice models) could suppress the proliferation of cancer cells by inducing the loss of NAD + /NADH homeostasis and downregulating aspartate biosynthesis levels through inhibiting mitochondrial complex I, which is also called NADH dehydrogenase [20, 112]. Moreover, the suppression of mitochondrial complex I by metformin (500 μM) also results in the enhanced glycolysis and a reduced citric acid cycle; subsequently, the cancer cells become energetically inefficient and their proliferation is inhibited [113].
Accordingly, mitochondria also participate in many other protective actions of metformin. In AMPK-deficient mice, the metformin treatment could still reduce infract size following ischemia reperfusion in an AKT-dependent way. This cardioprotective effect of metformin (0.05–2 mM) is executed by inhibiting mitochondrial complex I, consecutively, suppressing the attenuation of mitochondrial permeability pore (mPTP) opening [114, 115]. Besides, by utilizing a PD worm model established by knocking down bcat-1, a recent research reported that PD-like features are closely correlated with “mitochondrial hyperactivity”, and metformin(50 μM) could improve neuronal activity and motor function by reducing this “mitochondrial hyperactivity” [116]. Furthermore, it is well known that mitochondrial function can be affected by its morphology and that metformin can affect the morphology of mitochondria [136]. Increasing evidence suggests that mitochondrial abnormalities might be a key contributor to the generation of the Down syndrome(DS) phenotype, and some chromosome 21(Has21) genes also affect mitochondrial function and morphology [137,138,139,140]. Lucio et al. pointed out that metformin(500 μM) corrects the mitochondrial dysfunctions of human fibroblasts from DS foeti by restoring the mitochondria to a branched and elongated tubular morphology in a PGC-1-dependent way, thus, metformin presented a promising role in improving DS-associated pathologies [117].
However, the inhibition of mitochondrial complex I or G3P dehydrogenase by metformin blocks pyruvate carboxylase and promotes glycolysis, resulting in an increase in lactate production and a decrease in lactate metabolism. Therefore, if the patient has chronic kidney disease, which may impair the metformin excretion, and circulatory dysfunction and chronic liver disease, which may impair lactate clearance, metformin treatment increases the risk of lactic acidosis, a low-incidence but serious adverse effect of metformin [141, 142] (Table 1).
Metformin exerts its effects via the modulation of gut microbiome
Notably, the bioavailability of metformin in the gut is 300 times higher than that in the plasma. Accumulating evidence from preclinical studies has uncovered the role of metformin in gut microbiome modulation, including increasing the proportion of parts of the microbiome, such as A. muciniphila, Bacteroides, Butyricimonas, Megasphaera, and Prevotella, and decreasing the proportion of parts of the microbiome such as Anaerotruncus, Lactococcus, and Parabacteroides [143, 144]. Indeed, substantial data have demonstrated that gut microbiome dysbiosis puts contribution to various diseases, such as glucose metabolism, cancer, aging, and even acquired immunodeficiency syndrome(AIDS) [145,146,147,148], and increasing evidence indicated that the modulatory action of metformin on the gut microbiome is another mechanism that accounting for the pleiotropic effects of this drug (Fig. 3c).
According to previous studies, there is four gut microbiome-involved mechanisms exerting metformin’s glucose-lowering effects: (1) Increasing glucagon-like peptide-1(GLP-1) release. Pretreatment with metformin (200 mg/kg) recoveried the Lactobacillu abundance in the upper intestine and prompted sodium-glucose cotransporter-1 (SGLT1) sensing, a critical negative signal for glucose absorption in the upper intestine, which caused the increased GLP-1 secretion, eventually lowering HGP [24, 149,150,151,152]. (2) Promotion of short-chain fatty acids (SCFAs) production. SCFAs, including butyrate, acetate, propionate and lactate, execute protective roles against insulin resistance by multiple pathways, such as G protein-coupled receptors -41(GPR-41) and GPR-43 [153, 154]. Metformin (200 mg/kg) could upregulate the levels of SCFAs by increasing the abundance of the SCFA-producing gut microbiome, such as Butyricimonas spp, Allobaculum [118, 155, 156]. (3) Reducing gut permeability. The association of glucose metabolism and gut permeability has been clarified in several studies, and increased gut permeability results in insulin resistance through increasing the lipopolysaccharide (LPS) levels and causing chronic inflammation [157, 158]. According to previous data, metformin is capable of increasing the proportion of A. muciniphila in the gut which could reduce the gut permeability by stimulating mucin production or tight-junction protein expression [119, 159,160,161]. (4) Modulating inflammation. As the close relationships between glucose metabolism and inflammation have been studied, a hypothesis attracted researchers’ interest that metformin might elicit glucose-lowering effect through modulating inflammation in a gut microbiome-dependent way. Lee and his colleagues have shown that metformin treatment (250 mg/kg) increases abundance of Bacteroides and Butyricimonas in the gut, and then inhibits IL-6 levels or IL-1β expression, which are negative contributors to lowering the glucose process [25].
In terms of the effect on modulating gut microbiome, the most studied cancer type influenced by metformin is colorectal cancer [162]. In summary, the underlying mechanisms, which is mainly regulated by gut microbiome or its catabolite and metabolite, could be divided into four categories: (1) suppressing inflammation through Toll-like receptors 1(TLR1)/TLR4 pathway, or pro-inflammatory cytokinc, such as IL-6, IL-17a, IL-18 [163]; (2) increasing anticarcinogenic metabolites, such as SCFAs, or decreasing carcinogenic metabolites, such as hydrogen sulphide [164]; (3) inhibiting genotoxins production, such as B.frigilis toxin, CDT [24, 165]; (4) regulating not only innate immune by P38 map kinase-1 (PMK-1)/P38, receptor for advanced glycation endproducts (RAGE) ligands pathway and cytokines, such as interferon–γ (IFN-γ) of natural killer (NK) cells, IL-12 of dendritic cells [166,167,168], but also adaptive immune by T cells infiltration [169, 170]. Of note, the metformin doses used in these studies are all much higher. Besides, with the Lox-Stop-Lox Kras G12D/ + mice model, Eibl et al. confirmed that metformin ( approximately 200 mg/kg) is able to reduce the incidence of pancreatic ductal adenocarcinoma by changing the duodenal microbiome in the mice models treated with high-fat diet [120]. Furthermore, as there has been convincing evidence that the immune checkpoint inhibitors (ICI) therapy and metformin exposure both increase the abundance of A. muciniphila and Bifidobacterium spp in the mice models and humans [171, 172], it is plausible to speculate that metformin-mediated modulation of the gut microbiome is capable of improving effectiveness of immunotherapy on cancers, which has been confirmed by a large number of prospective and retrospective studies [173,174,175,176].
Accordingly, the gut microbiome is closely associated with human life span, and gut microbiome dysbiosis plays an important role in aging development via affecting multiple processes [177,178,179]. For instance, the data from studying the African killifish model showed that the natural gut microbiome from young individuals has a life-extended impact on vertebrate models through inducing long-lasting systemic advantages. Hematopoietic development and terminal myeloid differentiation are also regulated by microbiome-inducible inflammation. Lucas et al. found that the percentage of resident T cells in the secondary lymphoid organ, which increases with age, is affected by gut microbiome [121, 180, 181]. As mentioned above, metformin has a profound influence on gut microbial composition and metabolism, taken together, it is plausible that the action of metformin in improving aging-related pathology and extending life span is relevant to its modulatory action on gut microbiome. This hypothesis is also consistent with the research conducted by Cabreiro and his colleagues with the C.elegans models, who presented that metformin (50 mM) can specifically prolong the life span of C.elegans by inhibiting the microbial folate cycle and reducing methionine [121]. Besides, as the increased gut permeability is also linked with inflammation in older adults, a risk factor for aging-related morbidities and mortalities, Yadav et al. found the metformin-regulated (100 mg/kg) gut microbiome has a protective role in aging by decreasing gut permeability and inflammation [119] (Table 1).
Noteworthy, studies have found a role of the gut microbiome modulated by metformin in inhibiting human immunodeficiency virus (HIV)-related inflammation [182]. The underlying mechanisms involved in the activation of tryptophan pathway are mediated by influencing tryptophan-catabolizing bacteria, and the improvement of the gut epithelial barrier mediated by Akkermansia muciniphila or other SCFA producing bacteria [183, 184].
Metformin exerts its effects by regulating several other signals
In addition to above mentioned mechanisms of action, studies have reported several other signaling which also affected by metformin, including FB1, PP2A, FGF21, SIRT1 and mTOR (Fig. 3d). Generally accepted as a rate-controlling enzyme in gluconeogenesis, fructose-1,6-bisphosphatase 1(FBP1) is able to catalyze the irreversible hydrolysis of fructose-1,6-bisphosphate (F-1,6-P2) to fructose- 6-phosphate (F6P), which can be inhibited by AMP and F-2,6-P2. In a mice model with a point mutation in FBP1, Roger and his colleagues uncovered that the glucose-lowering effect of metformin (250 mg/kg) is blunted, even though it still leads to the activation of AMPK. They concluded that FBP1 is a key regulator for the HGP inhibition of metformin, but does not depend on AMPK activation [122].
As high-glucose simulates cardiomyocyte apoptosis, metformin(500 μM) can exert its cardioprotective role by activating PP2A, thus reducing ROS production and inhibiting the proinflammatory response [123]. Furthermore, with the intermittent fasting model, Minucci and his colleagues found that the combination of hypoglycemia and metformin (200 mg/kg) could inhibit tumor growth by activating PP2A, a tumor suppressor, in the absence of AMPK. Mechanistically, metformin activates the PP2A-GSK3β-MCL-1 pathway by inhibiting cancerous Inhibitor Of PP2A (CIP2A), a PP2A inhibitor, and hypoglycemia upregulates the B56δ, a PP2A regulatory subunit, eventually, the active PP2A-B56δ has higher affinity for GSK3β [124].
It has been well accepted that fibroblast growth factor 21 (FGF21) is a critical regulator of glucose and lipid metabolism. Consistent with its function, some studies have found that the anti-obesity effects and glucose-lowering effects of metformin are also exerted by FGF21. Metformin suppresses adipocyte differentiation by increasing FGF21 expression in the liver and white adipocytes in an AMPK-independent way, thus eliciting its therapeutic effect on T2DM, obesity and fatty liver [125, 185].
Sirtuin 1 (SIRT1), an NAD + -dependent deacetylase, leads to an anti-inflammatory effect by suppressing NF-κB signaling through deacetylation of its p65 subunit. Song et al. showed that the SIRT1 can be activated in an AMPK-dependent manner [186]. It has been shown that metformin ameliorates inflammation of circulating peripheral blood mononuclear cells in patients with carotid artery atherosclerosis by inducing SIRT1 [187, 188]. In addition, the anticancer effect of metformin may also be relevant to SIRT1, as the metformin-induced SIRT1 is able to reduce the Th17 population and IL-17 levels in tumors by deacetylating STAT3 [126].
By utilizing an AMPKα1/α2 double-knockout MEF model, Kalender et al. discovered that the inhibition of mTORC1 by metformin is independent of AMPK, but in a Rag GTPase-dependent manner [127]. Additionally, in the AMPK-deficient MEFs, it was reported that metformin inhibited phosphorylation of RTKs and AKT/mTOR way [128]. Metformin suppresses the proliferation of the AMPK-deleted glioma by activating mTOR signaling [189]. Besides, metformin is reported to reduce the anticancer efficiency of cisplatin in an AKT-dependent manner, but not an AMPK-dependent manner, as metformin failed to further increase cisplatin-induced AMPK activation [129] (Table1).
Clinical study
Based on the various of underlying mechanisms, through which metformin can affect some diseases, including diabetes mellitus, cardiovascular diseases, neurodegenerative diseases, reproductive disease, aging, cancer and COVID-19, researches considered that metfomin is possible to have therapeutic potential for these disease in the chilic, so they further conducted serious clinical studies and analyzed the outcomes of these diseases when treatment with or without metformin to explore the possibility for repurposing this old drug (Fig. 4).

Summary of metformin in different diseases and the major related mechanism. Based on its mechanisms, metformin has potential beneficial for diabetes mellitus, cancer, neurodegenerative disease, aging, cardiovascular disease, reproductive disease, COVID-19, and even Down syndrome and AIDS, however, it is also companied with some adverse effect, including gastrointestinal complaints, vitamin B12 deficiency, lactic acidosis, offspring impairment and neurodegenerative disease
Clinical efficacy of metformin
Diabetes mellitus
Metformin monotherapy effectively improves blood glucose control and lipid concentration in patients with T2DM and was approved in the USA in 1994 [190]. Since then, metformin has been widely used as a first-line oral glucose-lowering medication for the management of T2DM in the clinic [2]. Accumulating studies and clinical trials demonstrate that metformin-based regimens are effective in the curation of T2DM [191]. Recent evidence indicated that the regimen of metformin in combination with other antihyperglycemic drugs, including troglitazone, dipeptidyl peptidase 4 (DPP4) inhibitors, glibenclamide, insulin, glucagon-like peptide 1 (GLP1) receptor agonists, and sodium-dependent glucose transporters 2 (SGLT2) inhibitors, presents a better therapeutic effect on controlling plasma glucose levels than metformin alone. So, compared to using metformin alone, the combined use of metformin and glibenclamide exhibited a better glucose-lowering effect [192]. For instance, in a 16-week, randomized,double-blind study, the data showed that the fasting plasma glucose(FPG) is signicficantly lower in the metformin/ glibenclamide group (9.4 mmol/l, 2.5 mg/500 mg) than in metformin alone group (13.0 mmol/l, 500 mg [193]. Similarly, the combination of metformin with troglitazone reduced the production of endogenous glucose and promoted the metabolism of peripheral glucose, consequently, presenting better control of the plasma glucose level in T2DM patients [194]. Moreover, adding metformin to insulin therapy was reported to have better therapeutic efficiency for T2DM patients [195]. Although the efficiency of the combination strategy on glucose-lowering is evident by these studies, more studies, especially on the side effects, should be conducted (Table 2).
Cardiovascular diseases
In 1988, the randomized UK Prospective Diabetes Study (UKPDS) trial with 1704 type 2 diabetes patients confirmed that metformin has cardiovascular protective effects. After a median follow-up of 10.7 years, this study confirmed that metformin treatment significantly reduces the risk of cardiovascular events in newly diagnosed T2DM patients compared to conventional therapy (diet control), and a 39% reduction in the risk of myocardial infarction was reported (P = 0.01) [196]. The follow-up for 10 years of the UKPDS intervention study found that the metformin has continuous cardiovascular benefits and reduces the risk of myocardial infarction by 33% (P = 0.005) [197]. Another randomized trial of 304 patients with T2DM demonstrated that there were a significantly fewer cardiovascular events for metformin than for sulfonylurea after 5 years of treatment [198]. A study by Larsen et.al showed that 3 months of metformin treatment (target dose of 1000 mg bid) in diabetic chronic heart failure patients with reduced ejection fraction (HFrEF) patients significantly reduced myocardial oxygen uptake by 17% (P = 0.01). Moreover, patients with higher plasma concentrations of metformin (> 1268 ng/ml) had better myocardial efficiency [199]. Recently, a large-scale clinical endpoint study of metformin for patients with chronic heart failure-Danish Heart Failure Study (DANHEART) is ongoing. This is a multicenter, randomized, double-blind, placebo-controlled study of 1500 patients with T2DM and heart failure. The dose of metformin was 2000 mg/d (1000 mg/d when eGFR was 35–60 ml/min/1.73 m2), and the follow-up period was expected to be 4 years. The primary endpoint is a composite of death, hospitalization for worsening heart failure, acute myocardial infarction or stroke. The results are expected to be published in 2023 [200]. Although, a large number of studies supported the improved clinical outcomes of patients with heart failure treated with metformin, especially HFpEF, it is not yet enough for the approval of metformin in treating heart failure, we believe its effect and mechanism deserve further exploration (Table 2).
Neurodegenerative diseases
Neurodegenerative diseases (ND) mainly include Alzheimer's disease (AD) and Parkinson's disease (PD). Accordingly, neurodegenerative diseases are characterized by misfolded and aggregated proteins in neurons, such as mutated α-conucleoprotein, tau protein, β-amyloid and Huntington's protein. These proteins are toxic to neurons because of their role in changing neuronal connectivity and plasticity and even activating of cell death signaling pathways. Besides, it is well documented that aging is a main risk factor for neurodegenerative diseases [226].
The effects of metformin on ND are controversial, and in this part, we focused on the beneficial effects. Currently, preclinical and clinical evidence mostly reveals that metformin seems to be a prime candidate for a clinical trial that aims to target AD. A meta-analysis described that metformin reduced the risk for developing AD in patients with T2DM [201]. Similarly, a prospective observational study found that metformin improves cognitive performance in elderly patients with diabetes [202]. Regarding PD, a retrospective study revealed that metformin can reduce the risk of PD in T2DM patients in a Taiwanese population [227]. Overall, current evidence mostly supports that metformin improvs cognitive performance and decreases the risk of AD, preventing AD (Table 2).
Reproductive disease
Worldwide, approximately 5–20% of reproductive-aged women worldwide are affected by PCOS worldwide, and most of them is characterized by hyperandrogenism, ovulatory dysfunction, insulin resistance and so on [228]. Researchers found that metformin treatment improved the pregnancy rate probabilities for women with PCOS [229, 230]. A randomized, double-blinded, placebo-controlled trial (PregMet2) showed a lower rate of miscarriage and preterm birth of women with PCOS treated with metformin (OR = 0.50, 95% CI, 0.22–1.08; p = 0.08), and no significant serious adverse events in either mothers or offspring were considered drug-related [203]. Also, several systemic studies have suggested that metformin improved assisted reproductive technique outcomes by lowering the rate of ovarian hyperstimulation syndrome (OHSS) during the treatment of PCOS [231, 232], however, a randomized placebo-controlled trial concluded that a short-term of metformin use did not reduce OHSS in a gonadotropin-releasing hormone antagonist cycle for patients with PCOS (p = 0.66) [204].
In addition, metformin can reduce the secretion of antiangiogenic factors from the placenta in a dose-dependent manner and mitigate endothelial dysfunction, thereby potentially promoting vasodilation in whole maternal omental blood vessels in patients with preeclampsia. A randomized controlled trial that included 357 obese pregnant women reported that using metformin during gestation can prevent preeclampsia (OR = 0.17, 95% CI 0.10–0.41) [205]. Another trial including 180 women with preterm preeclampsia showed that the median prolongation of gestation in the metformin group was 17.5 days compared with 7.9 days in the placebo group [206]. Despite its effects on the female reproductive system, metformin is also presented to improve semen parameters in obese males [233] (Table 2).
Aging
Although aging is an inevitable process for lives, researchers never stop exploring the mystery of aging and continue to devote themselves to extend lifespan. Given its anti-inflammation and restoration of redox balance effects, metformin was chosen to be investigated for its effect on the aging improvement. Some epidemiological studies have described that metformin can delay aging and reduce all-cause mortality in age-related diseases. Importantly, existing evidence has shown that metformin could extend life and health spans by acting as a geroprotective agent in diabetic patients and nondiabetic patients [234]. Besides, a randomized crossover trial has revealed that metformin affects both metabolic and nonmetabolic processes associated with aging in the 70-year-old participants [235]. Recently, an observational study demonstrated that metformin improves the overall survival of older diabetic patients compared with controls without diabetes [236]. It is also speculated that the glucose-lowering effects of metformin are a contributor to reduced risks of aging-related diseases, and thereby extending lifespan [237]. Of note, the randomized clinical trials, TAME (Targeting Aging with Metformin) and MILES (Metformin In Longevity Study), are investigating the potential of metformin as an anti-aging drug. To date, the results from MILES suggested that although metformin is possibly involved in antiaging transcriptional changes, its protective role in those subjects free of diseases remains controversial [238].The TAME trial proposed blood-based biomarkers of underlying biologic aging hallmarks: IL-6, TNFα-receptor I or II, C-reactive protein (CRP), growth differentiation factor15 (GDF15), insulin, IGF1, cystatin C, N-terminal pro brain natriuretic peptide (NT-proBNP), and hemoglobin A1c. Future trials to discover and validate future biomarkers were warranted [239] (Table 2).
Cancer
Based on the mechanisms of action, the clinical studies on the role of metformin on cancer, a fatal threat to humans, is continues increasing. Epidemiological studies have indicated that metformin can decrease the risk of developing cancer and reduce the cancer-related mortality. A series of clinical trials evaluating the anticancer effects of metformin in various solid cancers of different stages are ongoing. In these trials, metformin is used as a monotherapy, or in combination with chemotherapy, radiotherapy and immunotherapy. These trials mainly investigated the effects of metformin on survival outcomes, and the evalution marker includs overall survival (OS), progression free survival (PFS), and recurrence free survival, pathological response rate, and cancer proliferation markers. Also, limited trials have evaluated the maximum tolerance of metformin in specific tumors. Published data from these completed clinical trials showed promising results.
Several studies have estimated the role of metformin in the cancer prevention. Compared with the control group, the risk of adenomas was 0.60 (95% CI 0.39, 0.92) in individuals with a history of colorectal adenoma treated with a low dose of metformin (250 mg/day) for 1 year [240, 241]. While a similar population was given metformin at an escalating dose, from 500 to 2000 mg/day for 12 weeks, there was no significant difference in the primary endpoint and pS6K levels [242]. Metformin also displayed the consistent results in the prevention of endometrial cancer. Among patients with endometrioid endometrial cancer, administration of 850 mg/day metformin resulted in decreased cell proliferation (an 11.75% reduction in Ki-67, P = 0.008) [243, 244]. Besides, metabolic disorders, including obesity and T2DM, are related to the high risk and poor survival of pancreatic ductal adenocarcinoma (PDAC) [245, 246].Most epidemiologic studies have found that metformin treatment reduces the incidence of in PDAC patients with T2DM [247]. Taken together, the correlation between metformin treatment and cancer prevention indicates that cancer may benefit from metformin through the effect of metofmrin on the high risk factors of cancer (T2DM, obesity, etc.).
Besides, completed trials have demonstrated the role of metformin monotherapy before surgery [248]. In metformin-treated patients with breast cancer, Ki67 and homeostatic model assessment (HOMA)) were significantly reduced, and TdT-mediated dUTP nick end labeling (TUNEL) levels were also increased in the metformin-treated group [249]. Interestingly, TUNEL staining is higher in cancer patients without insulin resistance, while individuals who have insulin resistance show converse results [250]. Importantly, the estimation of the pAMPK change may depend on the cancer type. In breast cancer, pAMPK (P = 0.04) is significantly upregulated and pAKT is downregulated (P = 0.043). Ki67 and cleaved caspase-3 (P = 0.044) were obviously decreased compared with the control group [251]. Conversely, another phase-II trial found that pAMPK, pS6, pAKT, p-4E-BP-1 and ER expression were reduced after metformin treatment [243]. In prostate cancer, pAMPK showed no difference between the arm group and the experimental group with metformin monotherapy [252]. These biomarker changes revealed that metformin exerts anticancer effects in pleiotropic pathways.
Given the proposed preclinical data of metformin and cytotoxic reagents, the combination of chemotherapy and metformin has also been explored in clinical trials. Metformin in combination with established cytotoxic chemotherapy accounts for the majority of ongoing clinical trials of cancer treatment [253]. The results of in combination of metformin with anticancer agents are expected. As a adjuvant agent, metformin benefits the CSS (HR 0.58, CI 0.39–0.86) and OS (HR 0.69, CI 0.58–0.83) of patients with colorectal cancer. A meta-analysis has suggested that metformin, as a useful adjuvant agent, benefits the survival of patients with prostate cancer, particularly those after radical radiotherapy, however, in breast and urothelial cancer, no significant benefits were observed [254,255,256]. Meanwhile, in the adjuvant setting, a phase I study exhibited that metformin combined with chemotherapy had a lower rate of defined dose-limiting toxicities (DLTs) (6.1%) compared to those who received only chemotherapy (7.8%). AMPK phosphorylation increased by 4–6 folds, 46% showed stable disease and 28% of the patients who had quantifiable tumor markers showed favorable changes [207]. The other randomized, phase 2 clinical trial evaluated the efficacy of doxorubicin and cyclophosphamide versus chemotherapy alone plus metformin in nondiabetic patients with metastatic breast cancer. Moreover, it is found that insulin-resistant patients with HER2-negative metastatic breast cancer (HOMA ≥ 2.5) have significantly shorter PFS than those without insulin resistance. Metformin as a potential chemotherapeutic drug or effective adjuvant agent exerts an affordable, well-tolerated, and beneficial anticancer effects. However, another phase 2 trial showed that metformin showed no significant effect on RR, PFS or OS of chemotherapy plus metformin versus placebo in non-diabetic patients with metastatic breast cancer [208]. The inconsistent responses to adjuvant metformin therapy is attributed to the insulin status of patients with cancer. This suggests that the positive potential of metformin as a chemotherapeutic drug depends on the patients’ status and the simultaneous management of diabetes and cancer is necessary.
The impacts of metformin also vary by the tumor stage. Metformin decreased cancer-specific mortality rates and prolonged the survival of localized resectable PDAC patients with T2DM [257, 258].In contrast, a double-blind, randomized study of patients with advanced PDAC did not benefit from metformin when combined with gemcitabine and erlotinib [259]. Two meta-analyses described that metformin prolonged the survival of patients with local disease but not those with metastatic PDAC [260, 261]. Metformin had contradictory results in the survival outcome of cancer patients in the local and metastatic stages, indicating the importance of the cancer stage in the studies of metformin for cancer treatment.
Collectively, the existing studies showed inconsistent marker expression and survival outcomes with metformin use as an anticancer agent in different settings. Variation in study design and potential bias, especially time-dependent confounders affected by previous treatment, make it complex to explain the different results [262]. Besides, there is no enough evidence to analyze the impact of metformin dose and duration. Future randomized, controlled trials to elevate the dose and duration and the efficacy of metformin anticancer agents as are warranted.
Furthermore, considering the immunomodulatory properties of metformin, metformin has been combined with immunotherapy, in particular programmed death-1 (PD-1)/ PD-L1 immune checkpoint inhibitors [263]. A phase II clinical trial showed that low-dose metformin treatment (250 mg/day) to reprograms and activates the tumor immune microenvironment and may be a suitable immune response modifier for patients with esophageal squamous cell carcinoma [209]. An active tumor immune microenvironment is the foundation for checkpoint inhibitors to enhance the immune response. A study of 40 patients with solid tumors suggested that the combination of nivolumab and metformin is safe. Adverse events (AEs) occurred in 75% of patients (mainly fatigue, pruritus, rash, and asthenia). Grade 3 AEs occurred in only 20% of cases; no grade 4 AEs were observed. There is a statistically significant correlation between higher doses of metformin (> 1,000 mg daily) and longer PFS and OS [210]. Overall, low-dose metformin treatment is a tolerated and efficacious pretreatment/combination option to boost the effectiveness of checkpoint inhibitors. However, both patients with and without diabetes and tumors are heterogeneous.
Therefore, it might be rational to elevate the anticancer activity of metformin and survival outcomes according to the insulin resistance status and various stages of cancer of participants in future clinical trials. Further investigations on a possible synergistic effect of checkpoint inhibitors and metformin are recommended (Table 2).
COVID-19
Since it was first reported in 2019, the COVID-19 has spread throughout the world. According to data from World Health Organization COVID-19 dashboard on August 28, 2022, the cumulative number of cases is 596,873,121, including 6,459,684 deaths, so it is urgent to develop effective preventive and therapeutic methods. Apart from developing new drugs, researchers are also engaging in repurposing the old drugs to treat COVID-19. As discussed above, based on its effects on multiple pathogeneses, it is reasonable to speculate that metformin has therapeutic potential in COVID-19 treatment, and clinical data also support this hypothesis. Retrospective studies reported a significant metformin treatment-associated reduction in COVID-19 infection-related mortality in patients with T2DM [211,212,213]. A meta-analysis study, including 32 cohort studies with 2,916,231 diabetic COVID-19 patients, showed that metformin is significantly relevant to lower mortality with a pooled adjusted odds ratio (OR) of 0.78 (95% CI, 0.69–0.88) [264].
Moreover, clinical trials are also undergoing to reaffirm the beneficial effect of metformin on COVID-19 patients. Data from ClinicalTrials.gov [as of August 28, 2022; primary search keyword (condition/disease): COVID-19; secondary search keyword (other terms): metformin] exhibited only 3 clinical trials that are investigating the role of metformin in COVID-19 treatment. Among these, COVID-OUT, a phase 3, randomized, double-blind, placebo-controlled trial, has reported its result and showed that compared with the primary composite endpoint (hypoxemia, emergency department visit, hospitalization, or death) in nonhospitalized patients with COVID-19 between 663 patients receiving metformin and 660 patients not receiving metformin, the adjusted OR was 0.84 (95% CI, 0.66–1.09; P = 0.19), and there was no significant benefit for COVID-19-related primary events. However, through further analysis, it indicated that metformin has the potential to prevent the more severe components, including emergency department visits, hospitalization or death, as the adjusted OR was 0.58 (95% CI, 0.35–0.94) [214].
Adverse effects of metformin
Besides efficacy and benefits, the safety of a drug needs to be fully considered. Due to its pleiotropic mechanism of action, metformin is not only beneficial to various diseases, conversely, it also results in several adverse effects, including gastrointestinal complaints, vitamin B12 deficiency, lactic acidosis, offspring impairment, and neurodegenerative diseases. When the patients are treated with metformin, clinicians need to closely monitor these adverse effects, especially those with fatal and irreversible harm. Generally, the most common adverse effect caused by metformin treatment is gastrointestinal complaints, which occurr in 2–63% of T2DM patients, and the complaints are diarrhea, nausea/vomiting, abdominal pain, flatulence, retching, dysgeusia. Although severe symptoms may lead to discontinuation in 5%-10% of metformin users [141, 265], the harm of gastrointestinal complaints is usually not fatal and irreversible, so we will mainly review the other adverse effects here (Table 2).
Vitamin B12 deficiency
Since it was first reported in 1969 [266], metformin-related vitamin B12 deficiency is prevalent in T2DM patients, the reported incidence varies from 5 to 40% [267,268,269]and decreased serum vitamin B12 levels vary from 14 to 30% [270, 271] in different studies. To date, although the mechanism by which metformin causes vitamin B12 deficiency is still unclear, clinical data have provided largely significant related-factors about the metformin-induced vitamin B12 deficiency. Ting et al. conducted a nested case–control study and found that the risk of vitamin B12 deficiency is dependent on the dose and duration of metformin use, for each 1 g/d metformin dose, the OR for developing vitamin B12 deficiency increased by 2.88 (95% CI, 2.15–3.87; P < 0.001). Compared with those receiving metformin for less than 3 years, among those using metformin for 3 years or more, the adjusted OR was 2.39 (95% CI, 1.46–3.91; P = 0.001) [215]. A hypothesis speculated that the mechanism responsible for metformin-mediated vitamin B12 deficiency is that metformin interferes with the calcium-dependent ileal membrane, which is responsible for the absorption of vitamin B12. Thus, Bauman et al. investigated the effect of calcium use on metformin-induced vitamin B12 deficiency, and the results confirmed that oral calcium supplementation reverses the decreased metformin-induced serum vitamin B12 level [272].
In addition, for the clinical manifestations of vitamin B12 deficiency mainly presenting as neurological and hematological symptoms, researchers further investigated the link between vitamin B12 deficiency and anemia or neuropathy. Regarding anemia, the RCTs, A Diabetes Outcome Progression Trial (ADOPT; n = 3,967) and UK Prospective Diabetes Study (UKPDS; n = 1,473), and an observational study, Genetics of Diabetes Audit and Research in Tayside Scotland (GoDARTS) population (n = 3,485), all give a similar result that the metformin-induced vitamin B12 deficiency is relevant to a higher risk of anemia [216]. Regarding to neuropathy, based on the severity of peripheral neuropathy (using the Toronto Clinical Scoring System (TCSS)) in both metformin users and non-metformin users, Singh et al. found an association of clinical neuropathy with metformin in T2DM patients (5.72 ± 2.04 in the metformin-exposed group versus 4.62 ± 2.12 in the metformin-unexposed group, P = 0.0064) [217]. Consequently, it is prudent to monitor vitamin B12 levels in metformin users, especially those with anemia or neuropathy manifestations. Because vitamin B12-caused neuropathy may be arrested with vitamin B12 or calcium supplementation, but diabetic neuropathy cannot (Table 2).
Lactic acidosis
According to clinical data, the lactate concentrations were 0.34 mmol/L higher in patients receiving metformin treatment [273], so metformin may increase the risk of lactic acidosis, especially in patients with kidney, liver and heart comorbidities. Interestingly, a recent retrospective study in a cohort of 1213 hospitalized diabetic COVID-19 patients displayed that metformin treatment is significantly relevant to a higher incidence of acidosis, especially in cases with severe COVID-19 complications [218]. Metformin-associated lactic acidosis (MALA), diagnosed by blood pH < 7.35, arterial lactate level > 5.0 mmol/L and metformin level > 5 mg/L [274], is an extremely rare event with an estimated incidence ≤ 10 events per 100,000 patients. However, its associated-mortality rates are up to 30–50% [46, 275], and a meta-analysis encompassing 177 patients with MALA from 44 studies showed an overall mortality of 36.2% (95% CI, 29.6–43.94%) with a median pH of 7.02 mmol/l and lactate of 14.45 mmol/l [276].
For this reason, a significant number of T2DM patients, who have a higher risk of MALA, were deprived of the benefits of metformin, but there is a debate in terms of the use of metformin in these patients.
Several studies have pointed out that the metformin would not increase the risk of MALA even in patients with eGFR 30–45 ml/min/1.73/m2, so FDA 2016 relaxed the renal restriction of metformin, recommending not to start using metformin if the eGFR is < 45 ml/min/1.73/m2 (CKD stage 3a) and not to continue using metformin if the eGFR is < 30 ml/min/1.73/m2 (CKD stage 3b) [277,278,279]. Over the years, new studies have reaffirmed the metformin safety in CKD patients with an eGFR ≥ 30 ml/min/1.73/m2. A community-based cohort study of 75,413 T2DM patients in the Geisinger health system showed that metformin treatment is only relevant to increased risk of MALA when the eGFR is < 30 ml/min/1.73/m2 (adjusted HR = 2.07, 95% CI, 1.33–3.22), and the results can be replicated by analyzing 67,578 new metformin users from 350 private US health systems [280]. A retrospective nested case‒control study in 2020 reported a consistent conclusion, by analyzing data from 320,882 diabetic CKD patients from the national VA Corporate Data Warehouse. Metformin exposure prior 3 months in patients with CKD stage 3a to 5 was associated with an elevated adjusted hazard of MALA (HR = 3.09, 95% CI 2.19–4.35 in CKD stage 3a; HR = 3.34, 95% CI 1.95–5.72 in CKD stage 3b; HR = 7.87, 95% CI 3.51–17.61 in CKD stage 4&5), but no association was evident in patients with CKD stage 1 or 2 (HR = 1.05, 95% CI 0.71–1.57) [219].
Although there have been a number of studies supporting the criteria of metformin use in CKD patients, further studies that test the precise criteria of tolerability and effectiveness of metformin in heart failure and chronic liver disease, even COVID-19, are still needed (Table 2).
Offspring impairment
Studies considering the long-term effects of metformin use during or before pregnancy in offspring demonstrated conflicting results. Despite of the reported benefits, several studies also found that metformin may cause offspring impairment. Two randomized trials considered 4- to 9-year-old metformin-exposed children of mothers with gestational diabetes (GDM) or PCOS to acquire some long-term metabolic programming effects such as higher BMI and increased prevalence of overweight or obesity [220, 221].On the other hand, a study including 1,996 children exposed to metformin during the fetal period and 1,932 treated with insulin showed no differences in either child growth or neurodevelopment between both the groups [222]. Consequently, the role of metformin-exposure to pregnant women in offspring need to be further confirmed.
Although it has been reported that metformin is able to reduce serum testosterone levels [281], but in March 2022, Eisenberg and his colleagues proposed a surprising result regarding of the deleterious effect of metformin on offspring: preconception metformin treatment in fathers is associated with an elevated risk for major birth defects, particularly genital birth defects in boys. In this nationwide prospective registry-based cohort study, data from newborns and parents (1997–2016) through Denmark were collected, by analyzing sex and frequencies of major birth defects in offspring whose fathers used metformin during the development of fertilizing sperm. This research indicated that offspring exposed to metformin (n = 1451) had an increased birth defect frequency (aOR = 1.40, 95% CI, 1.08–1.82). For metformin-exposed offspring, genital birth defects were increased compared with the cohort (0.90% vs. 0.24%; aOR = 3.39, 95% CI, 1.82- 6.30), and more than 99% of genital birth defects occur in boys [49]. This is the first study to suggest that metformin use in fathers may be linked to birth defects; however, it is not sufficient to make any clinical changes to offer new medication advice for men with T2DM of reproductive age. More clinical studies are warranted to confirm these results, and further preclinical research is needed to explore the underlying mechanism of this phenomenon (Table 2).
Neurodegenerative disease
Although researches have shown a beneficial effect of metformin on ND, including AD and PD, the role of metformin in ND is still quite controversial, given some preclinical or clinical studies have reported that long-term metformin use may increase the risk of ND. In preclinical studies, when the C57BL/L mice received chronic metformin treatment, they exhibited impaired spatial memory and visual acuity, which indicated that metformin may have deleterious effects on the central nervous system [282]. Angela et al. further showed that metformin treatment causes enhanced gliosis in the ApoE-/- mice, a mice model of tauopathy that is usually used to study ND, and increases tau phosphorylation, resulting in elevated lipogenesis to aggravate the neurodegenerative process [283]. Moreover, some clinical studies further reaffirmed that metformin treatment is positively associated with ND. Regarding AD, scholars have reported that when the duration of metformin use is less than 3 years, the metformin may increase the risk of AD. A nested case‒control study of cases with diabetes diagnosed ≥ 3 years before AD diagnosis (n = 7552) and controls who received metformin (n = 14,528) at least once is conducted by Sluggett et al., the results showed that taking metformin 1–3 years increases AD risk [223]. Also, by studying data from 70,860 persons from the United Kingdom–based General Practice Research Database (GPRD), Imfeld et al. found that long-term users of metformin (≥ 60 prescriptions) had an increased risk of developing AD, compared to other antidiabetic drug users and nonusers (adjusted OR = 1.71, 95% CI, 1.12–2.60) [224]. Regarding PD, similar result was also observed. A cohort study by using Taiwan’s National Health Insurance Research Database to collect data from 4651 metformin users and an equal number of non-metformin users was performed by Kuan et al., through 12-year follow-up, and they pointed out that the metformin cohort presented a higher risk of PD than the non-metformin cohort (HR = 2.27, 95% CI, 1.68–3.07) [284]. Recently, a meta-analysis including 19 studies with 285,966 participants also supported that compared to non-metformin users or glitazone users, metformin monotherapy exhibits a significantly elevated risk of PD (OR = 1.66, 95% CI, 1.14–2.42) [34].
Furthermore, to explore the factors related to the adverse effect of metformin on ND, Moore et al. recruited participants from the Primary Research in Memory (PRIME) clinics study, the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging and the Barwon region of southeastern Australia, they suggested that this phenomenon is possibly associated with another adverse effect of metformin, vitamin B12 deficiency. For this reason, a low serum vitamin B12 level (< 250 ρmol/L) is associated with ND, and taking vitamin B12 and calcium [225] which could promote vitamin B12 absorption supplements may alleviate metformin-associated ND outcomes [285]. Considering the controversial role of metformin in NDs, it is recommended to monitor the ND complications of T2DM patients when treated with metformin, and the serum vitamin B12 level could be a candidate biomarker (Table 2).
Conclusion
After being used in T2DM treatment for more than 50 years, metformin, an old drug with magic effects, has attracted much interest in recent years because of its potential for repurposing. Metformin exerts various effects through pleiotropic mechanisms of action, including AMPK-dependent mechanism and AMPK-independent mechanism. Based on these diverse mechanisms, metformin has various influences on types of tissue including, but not limited, liver, gut, adipose, heart, vascular, brain, ovary, semen and even cancers.
The most involved mechanism of metformin’s action is AMPK activation. Emerging evidence has indicated that AMPK is not only a key effector of glucose and lipid metabolism, but involved in the regulation of various pathway, including: (1)Glp1r/Pka pathway and mitochondria, reducing HGP and improving insulin resistance; (2) mTOR/autophagy pathway, driving the cardiovascular protection and neuroprotection; (3)NLRP3, eNOS, STAT3, COX-2, iNOS or Smad3 pathways, mediating anti-inflammation; (4) FOX3, IRS, GCRP and PD-L1, executing anticancer; (5)cAMP pathway, improving reproductive system; (6)ACE2, probably protecting against COVID-19. Recently, researchers found when in the absence of AMPK, metformin is still able to exert its effect by restoring redox balance, affecting mitochondrial function, modulating gut microbiome and regulating several other signals. The investigation of these mechanisms has led us to further move toward understanding the role of metformin’s protective actions, for example, inhibiting HGP and cancers, anti-obesity, improving complications of COVID-19 and Down syndrome, cardioprotection and neuroprotection. It is worth mentioning that studies have pointed out by restoring the redox balance, metformin is able to improve aging via alleviating the related inflammation. What about other aspects of pharmacogenetics of metformin? The evidence summarized in this review suggests that due to its importance for human health, the gut microbiome is partly responsible for the beneficial effect of metformin. Through modulating the gut microbiome, which results in increased GLP-1 release and SCFAs production, reduced gut permeability, suppressed inflammation, decreased genotoxins production, and an improved immune system, metformin exerts glucose-lowering, anticancer, antiaging, and even anti-HIV effects. However, besides its beneficial roles, metformin also has some adverse effects, and the underlying mechanism is still unclear. Accordingly, the activation of AMPK may participate in the deleterious process of metformin, such as acute pancreatitis and AD. The promotion of glycolysis by metformin also increased the risk for lactic acidosis through increasing lactate production and decreasing lactate metabolism.
Under these circumstances, the roles of metformin in human homeostasis and disease are reaffirmed from a clinical standpoint. Consistent with the mechanism of metformin discussed above, the researches, whether observation studies or RCT studies, we reviewed here suggested that metformin has therapeutic potential for T2DM, cancer, cardiovascular disease, aging, COVID-19. However, clinical data also showed some adverse effects associated with metformin treatment, such as gastrointestinal complaints, vitamin B12 deficiency and lactic acidosis. Of note, the most interesting aspect of metformin is its controversial role in the reproductive system and nervous system. Regarding reproductive system, it is suggested that metformin is not only improves PCOS in reproductive aged women, but also increase the risk of birth defects via affecting the development of sperm. Regarding nervous system, clinical and preclinical evidences have confirmed the protective role of metformin in both AD and PD; on the other hand, some clinical studies have presented that metformin treatment may be a contributor to the cognitive impairment, and may be involved in the development of AD and PD. We considered that these different influences of metformin may be caused by the features of disease itself and the treatment duration of metformin.
However, despite the extensive preclinical and clinical data highlighting the potential therapeutic effect on an enormous spectrum of diseases, including T2DM, cardiovascular diseases, neurodegenerative diseases, reproductive disease, aging, cancer and COVID-19, for now, metformin is approved only for T2DM treatment in the clinic. We supposed that it is related to the metformin dose used in most researches, which is always much higher than its clinically relevant dose, and the debate about this huge dosage gap between plasma concentrations in the clinic and supraphysiological conditions is ongoing. According to the recommended dose, the chinically relevant dose of metformin is 50–100 μM for cells and 50–100 mg/kg for mice models. However, the doses of metformin used in most studies are more than 500 μM for in vitro researches or 250 mg/kg for in vivo researches (Table 1). Besides, the different dose of metformin also may lead to controversial effects; for example, in terms of mitochondria, a low-dose of metformin (75 μM) improves mitochondrial respiration through AMPK-mediated mitochondrial fission, in contrast, a high-dose of metformin (no less than 5 mM) inhibits mitochondrial respiration chain complex I. Therefore, the dose of metformin used in studies is critical for investigating its mechanism and it repurpose it in other indications.
Hence, to repurpose the metformin, in-depth mechanisms of action and more clinical evidence remain to be elucidated. Additionally, considering that in most diseases, only supra-pharmacologic doses of metformin work, which is possible to cause serious adverse effects and even toxicities, the application of metformin in other indications should be more prudent and further researches are needed to establish safer criteria for metformin use.
Availability of data and materials
Not applicable.
References
King P, Peacock I, Donnelly R. The UK prospective diabetes study (UKPDS): clinical and therapeutic implications for type 2 diabetes. Br J Clin Pharmacol. 1999;48(5):643. https://doi.org/10.1046/j.1365-2125.1999.00092.x.
Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60(9):1586–93. https://doi.org/10.1007/s00125-017-4336-x.
Bailey CJ. Metformin: historical overview. Diabetologia. 2017;60(9):1566–76. https://doi.org/10.1007/s00125-017-4318-z.
Zhou M, Xia L, Wang J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab Dispos. 2007;35(10):1956–62. https://doi.org/10.1124/dmd.107.015495.
Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenetics Genomics. 2012;22(11):820. https://doi.org/10.1097/fpc.0b013e3283559b22.
Chen L, Pawlikowski B, Schlessinger A, More SS, Stryke D, Johns SJ, et al. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenetics Genomics. 2010;20(11):687. https://doi.org/10.1097/fpc.0b013e32833fe789.
Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Investig. 2007;117(5):1422–31. https://doi.org/10.1172/JCI30558.
Tsuda M, Terada T, Ueba M, Sato T, Masuda S, Katsura T, et al. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J Pharmacol Exp Ther. 2009;329(1):185–91. https://doi.org/10.1124/jpet.108.147918.
Takane H, Shikata E, Otsubo K, Higuchi S, Ieiri I. Polymorphism in human organic cation transporters and metformin action. Pharmacogenomics. 2008;9(4):415–22. https://doi.org/10.2217/14622416.9.4.415.
Tsuda M, Terada T, Mizuno T, Katsura T, Shimakura J, Inui K. Targeted disruption of the multidrug and toxin extrusion 1 (mate1) gene in mice reduces renal secretion of metformin. Mol Pharmacol. 2009;75(6):1280–6. https://doi.org/10.1124/mol.109.056242.
Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, et al. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther. 2011;89(6):837–44. https://doi.org/10.1038/clpt.2011.36.
LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocr Rev. 2021;42(1):77–96. https://doi.org/10.1210/endrev/bnaa023.
Madiraju AK, Erion DM, Rahimi Y, Zhang X-M, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–6. https://doi.org/10.1038/nature13270.
Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang X-M, Zhang D, et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat Med. 2018;24(9):1384–94. https://doi.org/10.1038/s41591-018-0125-4.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Investig. 2001;108(8):1167–74. https://doi.org/10.1172/JCI13505.
Duca FA, Côté CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, et al. Metformin activates a duodenal Ampk–dependent pathway to lower hepatic glucose production in rats. Nat Med. 2015;21(5):506–11. https://doi.org/10.1038/nm.3787.
Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64(6):2028–41. https://doi.org/10.2337/db14-1225.
Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51(7):2074–81. https://doi.org/10.2337/diabetes.51.7.2074.
Batandier C, Guigas B, Detaille D, El-Mir M, Fontaine E, Rigoulet M, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr. 2006;38(1):33–42. https://doi.org/10.1007/s10863-006-9003-8.
Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. elife. 2014;3:e02242. https://doi.org/10.7554/eLife.02242.
Gunton JE, Delhanty PJ, Takahashi S-I, Baxter RC. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J Clin Endocrinol Metab. 2003;88(3):1323–32. https://doi.org/10.1210/jc.2002-021394.
Mannucci E, Ognibene A, Cremasco F, Bardini G, Mencucci A, Pierazzuoli E, et al. Effect of metformin on glucagon-like peptide 1 (GLP-1) and leptin levels in obese nondiabetic subjects. Diabetes Care. 2001;24(3):489–94. https://doi.org/10.2337/diacare.24.3.489.
Chakraborty A, Chowdhury S, Bhattacharyya M. Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res Clin Pract. 2011;93(1):56–62. https://doi.org/10.1016/j.diabres.2010.11.030.
Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24(12):1919–29. https://doi.org/10.1038/s41591-018-0222-4.
Lee H, Lee Y, Kim J, An J, Lee S, Kong H, et al. Modulation of the gut microbiota by metformin improves metabolic profiles in aged obese mice. Gut Microbes. 2018;9(2):155–65. https://doi.org/10.1080/19490976.2017.1405209.
Lamanna C, Monami M, Marchionni N, Mannucci E. Effect of metformin on cardiovascular events and mortality: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2011;13(3):221–8. https://doi.org/10.1111/j.1463-1326.2010.01349.x.
Han Y, Xie H, Liu Y, Gao P, Yang X, Shen Z. Effect of metformin on all-cause and cardiovascular mortality in patients with coronary artery diseases: a systematic review and an updated meta-analysis. Cardiovasc Diabetol. 2019;18(1):1–16. https://doi.org/10.1186/s12933-019-0900-7.
Group DPPR. Impact of intensive lifestyle and metformin therapy on cardiovascular disease risk factors in the diabetes prevention program. Diabetes Care. 2005;28(4):888–94. https://doi.org/10.2337/diacare.28.4.888.
Golay A. Metformin and body weight. Int J Obes. 2008;32(1):61–72. https://doi.org/10.1038/sj.ijo.0803695.
Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66(1):17–29. https://doi.org/10.1146/annurev-med-062613-093128.
Chen YC, Li H, Wang J. Mechanisms of metformin inhibiting cancer invasion and migration. Am J Trans Res. 2020;12(9):4885.
Ma R, Yi B, Riker AI, Xi Y. Metformin and cancer immunity. Acta Pharmacol Sin. 2020;41(11):1403–9. https://doi.org/10.1038/s41401-020-00508-0.
Rotermund C, Machetanz G, Fitzgerald JC. The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol. 2018;9:400. https://doi.org/10.3389/fendo.2018.00400.
Ping F, Jiang N, Li Y. Association between metformin and neurodegenerative diseases of observational studies: systematic review and meta-analysis. BMJ Open Diabetes Res Care. 2020;8(1):e001370. https://doi.org/10.1136/bmjdrc-2020-001370.
Orchard TJ, Temprosa M, Goldberg R, Haffner S, Ratner R, Marcovina S, et al. The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: the Diabetes Prevention Program randomized trial. Ann Intern Med. 2005;142(8):611–9. https://doi.org/10.7326/0003-4819-142-8-200504190-00009.
Negrotto L, Farez MF, Correale J. Immunologic effects of metformin and pioglitazone treatment on metabolic syndrome and multiple sclerosis. JAMA Neurol. 2016;73(5):520–8. https://doi.org/10.1001/jamaneurol.2015.4807.
Nestler JE. Metformin for the treatment of the polycystic ovary syndrome. N Engl J Med. 2008;358(1):47–54. https://doi.org/10.1056/nejmct0707092.
Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ. 2003;327(7421):951. https://doi.org/10.1136/bmj.327.7421.951.
Facchinetti F, Orru B, Grandi G, Unfer V. Short-term effects of metformin and myo-inositol in women with polycystic ovarian syndrome (PCOS): a meta-analysis of randomized clinical trials. Gynecol Endocrinol. 2019;35(3):198–206. https://doi.org/10.1080/09513590.2018.1540578.
Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 2020;32(1):15–30. https://doi.org/10.1016/j.cmet.2020.04.001.
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metabol. 2016;23(6):1060–5. https://doi.org/10.1016/j.cmet.2016.05.011.
Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metabol. 2020;32(1):44-55. e6. https://doi.org/10.1016/j.cmet.2020.04.015.
Scheen A. Metformin and COVID-19: from cellular mechanisms to reduced mortality. Diabetes Metabol. 2020;46(6):423–6. https://doi.org/10.1016/j.diabet.2020.07.006.
Sharma S, Ray A, Sadasivam B. Metformin in COVID-19: a possible role beyond diabetes. Diabetes Res Clin Pract. 2020;164:108183. https://doi.org/10.1016/j.diabres.2020.108183.
Luo P, Qiu L, Liu Y, Liu X-l, Zheng J-l, Xue H-Y, et al. Metformin treatment was associated with decreased mortality in COVID-19 patients with diabetes in a retrospective analysis. Am J Trop Med Hygiene. 2020;103(1):69. https://doi.org/10.4269/ajtmh.20-0375.
DeFronzo R, Fleming GA, Chen K, Bicsak TA. Metformin-associated lactic acidosis: Current perspectives on causes and risk. Metabolism. 2016;65(2):20–9. https://doi.org/10.1016/j.metabol.2015.10.014.
De Jager J, Kooy A, Lehert P, Wulffelé MG, Van der Kolk J, Bets D, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. Bmj. 2010;340. https://doi.org/10.1136/bmj.c2181.
Bouchoucha M, Uzzan B, Cohen R. Metformin and digestive disorders. Diabetes Metabol. 2011;37(2):90–6. https://doi.org/10.1016/j.diabet.2010.11.002.
Wensink MJ, Lu Y, Tian L, Shaw GM, Rizzi S, Jensen TK, et al. Preconception antidiabetic drugs in men and birth defects in offspring: a nationwide cohort study. Ann Intern Med. 2022;175(5):665–73. https://doi.org/10.7326/M21-4389.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62. https://doi.org/10.1038/nrm3311.
Zhang Y, Wang Y, Bao C, Xu Y, Shen H, Chen J, et al. Metformin interacts with AMPK through binding to γ subunit. Mol Cell Biochem. 2012;368(1–2):69–76. https://doi.org/10.1007/s11010-012-1344-5.
Ma T, Tian X, Zhang B, Li M, Wang Y, Yang C, et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. 2022;603(7899):159–65. https://doi.org/10.1038/s41586-022-04431-8.
Koh A, Mannerås-Holm L, Yunn NO, Nilsson PM, Ryu SH, Molinaro A, et al. Microbial Imidazole Propionate Affects Responses to Metformin through p38γ-Dependent Inhibitory AMPK Phosphorylation. Cell Metab. 2020;32(4):643-53. e4. https://doi.org/10.1016/j.cmet.2020.07.012.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. https://doi.org/10.1172/jci13505.
Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, et al. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017;25(2):463–71. https://doi.org/10.1016/j.cmet.2016.12.009.
Zhang Q, Liang XC. Effects of Mitochondrial Dysfunction via AMPK/PGC-1 α Signal Pathway on Pathogenic Mechanism of Diabetic Peripheral Neuropathy and the Protective Effects of Chinese Medicine. Chin J Integr Med. 2019;25(5):386–94. https://doi.org/10.1007/s11655-018-2579-0.
Gong H, Tai H, Huang N, Xiao P, Mo C, Wang X, et al. Nrf2-SHP Cascade-Mediated STAT3 Inactivation Contributes to AMPK-Driven Protection Against Endotoxic Inflammation. Front Immunol. 2020;11:414. https://doi.org/10.3389/fimmu.2020.00414.
Cao J, Meng S, Chang E, Beckwith-Fickas K, Xiong L, Cole RN, et al. Low Concentrations of Metformin Suppress Glucose Production in Hepatocytes through AMP-activated Protein Kinase (AMPK)*♦. J Biol Chem. 2014;289(30):20435–46. https://doi.org/10.1074/jbc.M114.567271.
Duca FA, Côté CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, et al. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat Med. 2015;21(5):506–11. https://doi.org/10.1038/nm.3787.
Lam TK. Neuronal regulation of homeostasis by nutrient sensing. Nat Med. 2010;16(4):392–5. https://doi.org/10.1038/nm0410-392.
Wang Y, An H, Liu T, Qin C, Sesaki H, Guo S, et al. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep. 2019;29(6):1511-23. e5. https://doi.org/10.1016/j.celrep.2019.09.070.
Yang F, Qin Y, Wang Y, Meng S, Xian H, Che H, et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int J Biol Sci. 2019;15(5):1010–9. https://doi.org/10.7150/ijbs.29680.
Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L, et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. 2014;171(13):3146–57. https://doi.org/10.1111/bph.12655.
Demaré S, Kothari A, Calcutt NA, Fernyhough P. Metformin as a potential therapeutic for neurological disease: mobilizing AMPK to repair the nervous system. Expert Rev Neurother. 2021;21(1):45–63. https://doi.org/10.1080/14737175.2021.1847645.
Zhang J, Huang L, Shi X, Yang L, Hua F, Ma J, et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging (Albany NY). 2020;12(23):24270–87. https://doi.org/10.18632/aging.202143.
Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57(3):696–705. https://doi.org/10.2337/db07-1098.
Chung MM, Nicol CJ, Cheng YC, Lin KH, Chen YL, Pei D, et al. Metformin activation of AMPK suppresses AGE-induced inflammatory response in hNSCs. Exp Cell Res. 2017;352(1):75–83. https://doi.org/10.1016/j.yexcr.2017.01.017.
Li J, Zhang B, Liu WX, Lu K, Pan H, Wang T, et al. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann Rheum Dis. 2020;79(5):635–45. https://doi.org/10.1136/annrheumdis-2019-216713.
Lu J, Shi J, Li M, Gui B, Fu R, Yao G, et al. Activation of AMPK by metformin inhibits TGF-β-induced collagen production in mouse renal fibroblasts. Life Sci. 2015;127:59–65. https://doi.org/10.1016/j.lfs.2015.01.042.
Ding J, Zhu Y-T, Yang L, Tang J, Wang Y-Y, Chen Y, et al. 14-3-3zeta is involved in the anticancer effect of metformin in colorectal carcinoma. Carcinogenesis. 2018;39(3):493–502. https://doi.org/10.1093/carcin/bgy008.
Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med. 2012;1(11):811–24. https://doi.org/10.5966/sctm.2012-0058.
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012;3(3):e275. https://doi.org/10.1038/cddis.2012.13.
Faria J, Negalha G, Azevedo A, Martel F. Metformin and breast cancer: molecular targets. J Mammary Gland Biol Neoplasia. 2019;24(2):111–23. https://doi.org/10.1007/s10911-019-09429-z.
Hampsch RA, Wells JD, Traphagen NA, McCleery CF, Fields JL, Shee K, et al. AMPK Activation by Metformin Promotes Survival of Dormant ER (+) Breast Cancer Cells. Clin Cancer Res. 2020;26(14):3707–19. https://doi.org/10.1158/1078-0432.Ccr-20-0269.
Yue W, Yang CS, DiPaola RS, Tan XL. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev Res (Phila). 2014;7(4):388–97. https://doi.org/10.1158/1940-6207.Capr-13-0337.
Lu T, Li M, Zhao M, Huang Y, Bi G, Liang J, et al. Metformin inhibits human non-small cell lung cancer by regulating AMPK-CEBPB-PDL1 signaling pathway. Cancer Immunol Immunother. 2022;71(7):1733–46. https://doi.org/10.1007/s00262-021-03116-x.
Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res. 2010;16(9):2505–11. https://doi.org/10.1158/1078-0432.CCR-09-2229.
Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein–coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69(16):6539–45. https://doi.org/10.1158/0008-5472.CAN-09-0418.
Ding J, Li C, Tang J, Yi C, Liu J-Y, Qiu M. Higher expression of proteins in IGF/IR axes in colorectal cancer is associated with type 2 diabetes mellitus. Pathology & Oncology Research. 2016;22(4):773–9. https://doi.org/10.1158/1078-0432.Ccr-10-2243.
Rocha GZ, Dias MM, Ropelle ER, Osório-Costa F, Rossato FA, Vercesi AE, et al. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin Cancer Res. 2011;17(12):3993–4005. https://doi.org/10.1158/1078-0432.Ccr-10-2243.
Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013;108(10):2021–32. https://doi.org/10.1038/bjc.2013.187.
Diri H, Bayram F, Simsek Y, Caliskan Z, Kocer D. Comparison of finasteride, metformin, and finasteride plus metformin in PCOS. Acta Endocrinologica (Bucharest). 2017;13(1):84. https://doi.org/10.4183/aeb.2017.84.
Kurzthaler D, Hadziomerovic-Pekic D, Wildt L, Seeber BE. Metformin induces a prompt decrease in LH-stimulated testosterone response in women with PCOS independent of its insulin-sensitizing effects. Reprod Biol Endocrinol. 2014;12(1):1–6. https://doi.org/10.1186/1477-7827-12-98.
Attia GR, Rainey WE, Carr BR. Metformin directly inhibits androgen production in human thecal cells. Fertil Steril. 2001;76(3):517–24. https://doi.org/10.1016/S0015-0282(01)01975-6.
Tosca L, Chabrolle C, Uzbekova S, Dupont J. Effects of metformin on bovine granulosa cells steroidogenesis: possible involvement of adenosine 5′ monophosphate-activated protein kinase (AMPK). Biol Reprod. 2007;76(3):368–78. https://doi.org/10.1095/biolreprod.106.055749.
Martin-Hidalgo D, Hurtado de Llera A, Calle-Guisado V, Gonzalez-Fernandez L, Garcia-Marin L, Bragado MJ. AMPK function in mammalian spermatozoa. Int J Mol Sci. 2018;19(11):3293. https://doi.org/10.3390/ijms19113293.
Calle-Guisado V, de Llera AH, Martin-Hidalgo D, Mijares J, Gil MC, Alvarez IS, et al. AMP-activated kinase in human spermatozoa: identification, intracellular localization, and key function in the regulation of sperm motility. Asian J Andrology. 2017;19(6):707. https://doi.org/10.4103/1008-682X.185848.
Liu C-Y, Chang T-C, Lin S-H, Wu S-T, Cha T-L, Tsao C-W. Metformin ameliorates testicular function and spermatogenesis in male mice with high-fat and high-cholesterol diet-induced obesity. Nutrients. 2020;12(7):1932. https://doi.org/10.3390/nu12071932.
Morgante G, Tosti C, Orvieto R, Musacchio MC, Piomboni P, De Leo V. Metformin improves semen characteristics of oligo-terato-asthenozoospermic men with metabolic syndrome. Fertility Sterility. 2011;95(6):2150–2. https://doi.org/10.1016/j.fertnstert.2010.12.009.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–3. https://doi.org/10.1126/science.abb2507.
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/angiotensin-(1–7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev. 2017;98(1):505–53. https://doi.org/10.1152/physrev.00023.2016.
Malhotra A, Hepokoski M, McCowen KC, Shyy JY. ACE2, metformin, and COVID-19. Iscience. 2020;23(9):101425. https://doi.org/10.1016/j.isci.2020.101425.
Samuel SM, Varghese E, Büsselberg D. Therapeutic potential of metformin in COVID-19: reasoning for its protective role. Trends Microbiol. 2021;29(10):894–907. https://doi.org/10.1016/j.tim.2021.03.004.
Zhang J, Dong J, Martin M, He M, Gongol B, Marin TL, et al. AMP-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am J Respir Crit Care Med. 2018;198(4):509–20. https://doi.org/10.1164/rccm.201712-2570OC.
Plattner F, Bibb JA. Serine and threonine phosphorylation. In: Basic Neurochemistry. Elsevier; 2012. 467–92. https://doi.org/10.1016/b978-0-12-374947-5.00025-0.
Lee HY, Wei D, Loeken MR. Lack of metformin effect on mouse embryo AMPK activity: implications for metformin treatment during pregnancy. Diabetes Metab Res Rev. 2014;30(1):23–30. https://doi.org/10.1002/dmrr.2451.
Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X, et al. Metformin plays a dual role in MIN6 pancreatic β cell function through AMPK-dependent autophagy. Int J Biol Sci. 2014;10(3):268–77. https://doi.org/10.7150/ijbs.7929.
Williams T, Courchet J, Viollet B, Brenman JE, Polleux F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci U S A. 2011;108(14):5849–54. https://doi.org/10.1073/pnas.1013660108.
Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron. 2013;78(1):94–108. https://doi.org/10.1016/j.neuron.2013.02.003.
Valiulienė G, Vitkevičienė A, Skliutė G, Borutinskaitė V, Navakauskienė R. Pharmaceutical drug metformin and MCL1 inhibitor S63845 exhibit anticancer activity in myeloid leukemia cells via redox remodeling. Molecules. 2021;26(8):2303. https://doi.org/10.3390/molecules26082303.
Li B, Zhou P, Xu K, Chen T, Jiao J, Wei H, et al. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int J Biol Sci. 2020;16(1):74. https://doi.org/10.7150/ijbs.33787.
Cheng G, Lanza-Jacoby S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem Biophysical Res Communications. 2015;465(1):41–6. https://doi.org/10.1016/j.bbrc.2015.07.118.
Li PD, Liu Z, Cheng TT, Luo WG, Yao J, Chen J, et al. Redox-dependent modulation of metformin contributes to enhanced sensitivity of esophageal squamous cell carcinoma to cisplatin. Oncotarget. 2017;8(37):62057. https://doi.org/10.18632/oncotarget.18907.
Zhang P, Zhao S, Lu X, Shi Z, Liu H, Zhu B. Metformin enhances the sensitivity of colorectal cancer cells to cisplatin through ROS-mediated PI3K/Akt signaling pathway. Gene. 2020;745:144623. https://doi.org/10.1016/j.gene.2020.144623.
Tripathi SS, Singh AK, Akhtar F, Chaudhary A, Rizvi SI. Metformin protects red blood cells against rotenone induced oxidative stress and cytotoxicity. Arch Physiol Biochem. 2021;127(2):102–11. https://doi.org/10.1080/13813455.2019.1620288.
Garg G, Singh S, Singh AK, Rizvi SI. Metformin alleviates altered erythrocyte redox status during aging in rats. Rejuvenation Res. 2017;20(1):15–24. https://doi.org/10.1089/rej.2016.1826.
Li X, Wang X, Snyder MP. Metformin affects heme function as a possible mechanism of action. G3: Genes, Genomes, Genetics. 2019;9(2):513–22.https://doi.org/10.1534/g3.118.200803.
Menendez JA. Metformin and SARS-CoV-2: mechanistic lessons on air pollution to weather the cytokine/thrombotic storm in COVID-19. Aging (Albany NY). 2020;12(10): 8760. https://doi.org/10.18632/aging.103347.
Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Investig. 2010;120(7):2355–69. https://doi.org/10.1172/JCI40671.
Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–60. https://doi.org/10.1038/nature11808.
Karise I, Bargut TC, Del Sol M, Aguila MB, Mandarim-de-Lacerda CA. Metformin enhances mitochondrial biogenesis and thermogenesis in brown adipocytes of mice. Biomedicine Pharmacotherapy. 2019;111:1156–65. https://doi.org/10.1016/j.biopha.2019.01.021.
Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 2016;24(5):716–27. https://doi.org/10.1016/j.cmet.2016.09.006.
Andrzejewski S, Gravel S-P, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metabol. 2014;2(1):1–14. https://doi.org/10.1186/2049-3002-2-12.
Mohsin AA, Chen Q, Quan N, Rousselle T, Maceyka MW, Samidurai A, et al. Mitochondrial complex I inhibition by metformin limits reperfusion injury. J Pharmacol Exp Ther. 2019;369(2):282–90. https://doi.org/10.1124/jpet.118.254300.
Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, et al. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol. 2008;103(3):274–84.
Mor DE, Sohrabi S, Kaletsky R, Keyes W, Tartici A, Kalia V, et al. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc Natl Acad Sci. 2020;117(42):26438–47. https://doi.org/10.1073/pnas.2009838117.
Izzo A, Nitti M, Mollo N, Paladino S, Procaccini C, Faicchia D, et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum Mol Genetics. 2017;26(6):1056–69. https://doi.org/10.1093/hmg/ddx016.
Zhang X, Zhao Y, Xu J, Xue Z, Zhang M, Pang X, et al. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci Rep. 2015;5(1):1–10. https://doi.org/10.1038/srep14405.
Ahmadi S, Razazan A, Nagpal R, Jain S, Wang B, Mishra SP, et al. Metformin reduces aging-related leaky gut and improves cognitive function by beneficially modulating gut microbiome/goblet cell/mucin axis. The Journals of Gerontology: Series A. 2020;75(7):e9-e21.https://doi.org/10.1093/gerona/glaa056.
Dong TS, Chang H-H, Hauer M, Lagishetty V, Katzka W, Rozengurt E, et al. Metformin alters the duodenal microbiome and decreases the incidence of pancreatic ductal adenocarcinoma promoted by diet-induced obesity. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2019;317(6): G763-G72. https://doi.org/10.1152/ajpgi.00170.2019.
Cabreiro F, Au C, Leung K-Y, Vergara-Irigaray N, Cochemé HM, Noori T, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153(1):228–39. https://doi.org/10.1016/j.cell.2013.02.035.
Hunter RW, Hughey CC, Lantier L, Sundelin EI, Peggie M, Zeqiraj E, et al. Metformin reduces liver glucose production by inhibition of fructose-1–6-bisphosphatase. Nat Med. 2018;24(9):1395–406. https://doi.org/10.1038/s41591-018-0159-7.
Cheng G, Li L. High-glucose-induced apoptosis, ROS production and pro-inflammatory response in cardiomyocytes is attenuated by metformin treatment via PP2A activation. J Biosci. 2020;45(1):1–11. https://doi.org/10.1007/s12038-020-00096-5.
Elgendy M, Ciro M, Hosseini A, Weiszmann J, Mazzarella L, Ferrari E, et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3β-MCL-1 axis. Cancer Cell. 2019;35(5):798–815. e5. https://doi.org/10.1016/j.ccell.2019.03.007.
Kim EK, Lee SH, Jhun JY, Byun JK, Jeong JH, Lee S-Y, et al. Metformin prevents fatty liver and improves balance of white/brown adipose in an obesity mouse model by inducing FGF21. Mediators Inflammation. 2016;2016:5813030.https://doi.org/10.1155/2016/5813030
Limagne E, Thibaudin M, Euvrard R, Berger H, Chalons P, Végan F, et al. Sirtuin-1 activation controls tumor growth by impeding Th17 differentiation via STAT3 deacetylation. Cell Rep. 2017;19(4):746–59. https://doi.org/10.1016/j.celrep.2017.04.004.
Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11(5):390–401. https://doi.org/10.1016/j.cmet.2010.03.014.
Quinn BJ, Dallos M, Kitagawa H, Kunnumakkara AB, Memmott RM, Hollander MC, et al. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma IGF-I and diminished receptor tyrosine kinase signaling. Cancer Prev Res. 2013;6(8):801–10. https://doi.org/10.1158/1940-6207.CAPR-13-0058-T.
Janjetovic K, Vucicevic L, Misirkic M, Vilimanovich U, Tovilovic G, Zogovic N, et al. Metformin reduces cisplatin-mediated apoptotic death of cancer cells through AMPK-independent activation of Akt. Eur J Pharmacol. 2011;651(1–3):41–50. https://doi.org/10.1016/j.ejphar.2010.11.005.
Menon SG, Goswami PC. A redox cycle within the cell cycle: ring in the old with the new. Oncogene. 2007;26(8):1101–9. https://doi.org/10.1038/sj.onc.1209895.
Le Gal K, Schmidt EE, Sayin VI. Cellular Redox Homeostasis. Antioxidants (Basel). 2021;10(9). https://doi.org/10.3390/antiox10091377.
Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan. China Intensive care medicine. 2020;46(5):846–8. https://doi.org/10.1007/s00134-020-05991-x.
Kuperman Y, Weiss M, Dine J, Staikin K, Golani O, Ramot A, et al. CRFR1 in AgRP neurons modulates sympathetic nervous system activity to adapt to cold stress and fasting. Cell Metabol. 2016;23(6):1185–99. https://doi.org/10.1016/j.cmet.2016.04.017.
Feng J, Wang X, Ye X, Ares I, Lopez-Torres B, Martínez M, et al. Mitochondria as an important target of metformin: The mechanism of action, toxic and side effects, and new therapeutic applications. Pharmacol Res. 2022:106114. https://doi.org/10.1016/j.phrs.2022.106114.
Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462(3):475–87. https://doi.org/10.1042/BJ20140620.
Du Y, Zhu Y-J, Zeng B, Mu X-L, Liu J. Super-resolution quantification of T2DM-induced mitochondrial morphology changes and its implications in pharmacodynamics of metformin and sorafenib. Front Pharmacology. 2022:2449. https://doi.org/10.3389/fphar.2022.932116.
Pallardó FV, Lloret A, Lebel M, d’Ischia M, Cogger VC, Le Couteur DG, et al. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: Ataxia-Telangiectasia, Down Syndrome. Fanconi Anaemia and Werner Syndrome Biogerontology. 2010;11(4):401–19. https://doi.org/10.1007/s10522-010-9269-4.
Piccoli C, Izzo A, Scrima R, Bonfiglio F, Manco R, Negri R, et al. Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum Mol Genet. 2013;22(6):1218–32. https://doi.org/10.1093/hmg/dds529.
Izzo A, Manco R, Bonfiglio F, Calì G, De Cristofaro T, Patergnani S, et al. NRIP1/RIP140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in Down syndrome. Hum Mol Genetics. 2014;23(16):4406–19. https://doi.org/10.1093/hmg/ddu157.
Gardiner KJ. Pharmacological approaches to improving cognitive function in Down syndrome: current status and considerations. Drug Des Dev Ther. 2015;9:103. https://doi.org/10.2147/DDDT.S51476.
Bolen S, Feldman L, Vassy J, Wilson L, Yeh H-C, Marinopoulos S, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Int Med. 2007;147(6):386–99. https://doi.org/10.7326/0003-4819-147-6-200709180-00178.
Piel S, Ehinger J, Elmer E, Hansson M. Metformin induces lactate production in peripheral blood mononuclear cells and platelets through specific mitochondrial complex I inhibition. Acta physiologica. 2015;213(1):171–80. https://doi.org/10.1111/apha.12311.
Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23(7):850–8. https://doi.org/10.1038/nm.4345.
Lee CB, Chae SU, Jo SJ, Jerng UM, Bae SK. The relationship between the gut microbiome and metformin as a key for treating type 2 diabetes mellitus. Int J Mol Sci. 2021;22(7):3566. https://doi.org/10.3390/ijms22073566.
Shin N-R, Lee J-C, Lee H-Y, Kim M-S, Whon TW, Lee M-S, et al. An increase in the Akkermansia spp population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63(5):727–35. https://doi.org/10.1136/gutjnl-2012-303839.
Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33(4):570–80. https://doi.org/10.1016/j.ccell.2018.03.015.
Nagpal R, Mainali R, Ahmadi S, Wang S, Singh R, Kavanagh K, et al. Gut microbiome and aging: Physiological and mechanistic insights. Nutr Health Aging. 2018;4(4):267–85. https://doi.org/10.3233/NHA-170030.
Dillon SM, Frank DN, Wilson CC. The gut microbiome and HIV-1 pathogenesis: a two-way street. AIDS (London, England). 2016;30(18):2737. https://doi.org/10.1097/QAD.0000000000001289.
Gorboulev V, Schürmann A, Vallon V, Kipp H, Jaschke A, Klessen D, et al. Na+-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes. 2012;61(1):187–96. https://doi.org/10.2337/db11-1029.
Kuhre RE, Frost CR, Svendsen B, Holst JJ. Molecular mechanisms of glucose-stimulated GLP-1 secretion from perfused rat small intestine. Diabetes. 2015;64(2):370–82. https://doi.org/10.2337/db14-0807.
Bauer PV, Duca FA, Waise TZ, Rasmussen BA, Abraham MA, Dranse HJ, et al. Metformin alters upper small intestinal microbiota that impact a glucose-SGLT1-sensing glucoregulatory pathway. Cell Metabol. 2018;27(1):101–17. e5. https://doi.org/10.1016/j.cmet.2017.09.019.
Rooj AK, Kimura Y, Buddington RK. Metabolites produced by probiotic Lactobacilli rapidly increase glucose uptake by Caco-2 cells. BMC Microbiol. 2010;10(1):1–10. https://doi.org/10.1186/1471-2180-10-16.
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein–coupled receptor FFAR2. Diabetes. 2012;61(2):364–71. https://doi.org/10.2337/db11-1019.
Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19(1):55–71. https://doi.org/10.1038/s41579-020-0433-9.
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–17. https://doi.org/10.2337/db08-1637.
Li X, Wang E, Yin B, Fang D, Chen P, Wang G, et al. Effects of Lactobacillus casei CCFM419 on insulin resistance and gut microbiota in type 2 diabetic mice. Beneficial Microbes. 2017;8(3):421–32. https://doi.org/10.3920/BM2016.0167.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. https://doi.org/10.2337/db06-1491.
Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50(11):2374–83. https://doi.org/10.1007/s00125-007-0791-0.
Liu Y, Wang C, Li J, Li T, Zhang Y, Liang Y, et al. Phellinus linteus polysaccharide extract improves insulin resistance by regulating gut microbiota composition. FASEB J. 2020;34(1):1065–78. https://doi.org/10.1093/gerona/glaa056.
Lee H, Ko G. Effect of metformin on metabolic improvement and gut microbiota. Appl Environ microbiol. 2014;80(19):5935–43. https://doi.org/10.1128/AEM.01357-14.
Chelakkot C, Choi Y, Kim D-K, Park HT, Ghim J, Kwon Y, et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Mol Med. 2018;50(2): e450-e. https://doi.org/10.1038/emm.2017.282.
Huang Q-Y, Yao F, Zhou C-R, Huang X-Y, Wang Q, Long H, et al. Role of gut microbiome in regulating the effectiveness of metformin in reducing colorectal cancer in type 2 diabetes. World J Clin Cases. 2020;8(24): 6213.https://doi.org/10.12998/wjcc.v8.i24.6213.
Lee H, Kim J, An J, Lee S, Choi D, Kong H, et al. Downregulation of IL-18 expression in the gut by metformin-induced gut microbiota modulation. Immune Network. 2019;19(4).https://doi.org/10.4110/in.2019.19.e28.
Broadfield LA, Saigal A, Szamosi JC, Hammill JA, Bezverbnaya K, Wang D, et al. Metformin-induced reductions in tumor growth involves modulation of the gut microbiome. Mol Metabol. 2022;61:101498. https://doi.org/10.1016/j.molmet.2022.101498.
He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S, et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68(2):289–300. https://doi.org/10.1136/gutjnl-2018-317200.
Xiao Y, Liu F, Li S, Jiang N, Yu C, Zhu X, et al. Metformin promotes innate immunity through a conserved PMK-1/p38 MAPK pathway. Virulence. 2020;11(1):39–48.https://doi.org/10.1080/21505594.2019.1706305.
Kumar NP, Moideen K, Nancy A, Viswanathan V, Shruthi BS, Sivakumar S, et al. Systemic RAGE ligands are upregulated in tuberculosis individuals with diabetes co-morbidity and modulated by anti-tuberculosis treatment and metformin therapy. BMC Infect Dis. 2019;19(1):1–10. https://doi.org/10.1186/s12879-019-4648-1.
Volarevic V, Misirkic M, Vucicevic L, Paunovic V, Simovic Markovic B, Stojanovic M, et al. Metformin aggravates immune-mediated liver injury in mice. Arch Toxicol. 2015;89(3):437–50. https://doi.org/10.1007/s00204-014-1263-1.
Curry JM, Johnson J, Mollaee M, Tassone P, Amin D, Knops A, et al. Metformin clinical trial in HPV+ and HPV–head and neck squamous cell carcinoma: impact on cancer cell apoptosis and immune infiltrate. Front Oncol. 2018;8:436. https://doi.org/10.3389/fonc.2018.00436.
Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, et al. Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology. 2018;155(1):33–7. e6.https://doi.org/10.1053/j.gastro.2018.04.001.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews M, Karpinets T, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. https://doi.org/10.1126/science.aan4236.
Rodriguez J, Hiel S, Delzenne NM. Metformin: old friend, new ways of action–implication of the gut microbiome? Curr Opinion Clin NutrMetabol Care. 2018;21(4):294–301. https://doi.org/10.1097/MCO.0000000000000468.
Haikala HM, Anttila JM, Marques E, Raatikainen T, Ilander M, Hakanen H, et al. Pharmacological reactivation of MYC-dependent apoptosis induces susceptibility to anti-PD-1 immunotherapy. Nat Commun. 2019;10(1):1–17. https://doi.org/10.1038/s41467-019-08541-2.
Pietras R, Xu H, Hu X, Matheny C, Sandler A, Patel M. P1. 04–33 retrospective descriptive analysis of metformin with atezolizumab in advanced non-small cell lung cancer in the OAK trial. J Thoracic Oncol. 2018;13(10):S538–9. https://doi.org/10.1016/j.jtho.2018.08.748.
Kubo T, Ninomiya T, Hotta K, Kozuki T, Toyooka S, Okada H, et al. Study protocol: Phase-Ib trial of nivolumab combined with metformin for refractory/recurrent solid tumors. Clin Lung Cancer. 2018;19(6): e861-e4.https://doi.org/10.1016/j.cllc.2018.07.010.
Verdura S, Cuyàs E, Martin-Castillo B, Menendez JA. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. Oncoimmunology. 2019;8(10):e1633235. https://doi.org/10.1080/2162402X.2019.1633235.
Clements SJ, R. Carding S. Diet, the intestinal microbiota, and immune health in aging. Critical Rev Food Sci Nutr. 2018;58(4):651–61.https://doi.org/10.1080/10408398.2016.1211086.
Peniche AG, Spinler JK, Boonma P, Savidge TC, Dann SM. Aging impairs protective host defenses against Clostridioides (Clostridium) difficile infection in mice by suppressing neutrophil and IL-22 mediated immunity. Anaerobe. 2018;54:83–91. https://doi.org/10.1016/j.anaerobe.2018.07.011.
Kim S, Jazwinski SM. The gut microbiota and healthy aging: a mini-review. Gerontology. 2018;64(6):513–20. https://doi.org/10.1159/000490615.
Smith P, Willemsen D, Popkes M, Metge F, Gandiwa E, Reichard M, et al. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. elife. 2017;6:e27014. https://doi.org/10.7554/eLife.27014.
Durand A, Audemard-Verger A, Guichard V, Mattiuz R, Delpoux A, Hamon P, et al. Profiling the lymphoid-resident T cell pool reveals modulation by age and microbiota. Nat Commun. 2018;9(1):1–12. https://doi.org/10.1038/s41467-017-02458-4.
Ouyang J, Isnard S, Lin J, Fombuena B, Marette A, Routy B, et al. Metformin effect on gut microbiota: insights for HIV-related inflammation. AIDS Res Ther. 2020;17(1):1–9. https://doi.org/10.1186/s12981-020-00267-2.
Jenabian M-A, Patel M, Kema I, Kanagaratham C, Radzioch D, Thebault P, et al. Distinct tryptophan catabolism and Th17/Treg balance in HIV progressors and elite controllers. PLoS One. 2013;8(10):e78146. https://doi.org/10.1371/annotation/11698dd2-0bc1-4fe0-ad92-b161b5594e81.
Muzik O, Burghardt P, Yi Z, Kumar A, Seyoum B. Successful metformin treatment of insulin resistance is associated with down-regulation of the kynurenine pathway. Biochem Biophys Res Commun. 2017;488(1):29–32. https://doi.org/10.1016/j.bbrc.2017.04.155.
Mottillo EP, Desjardins EM, Fritzen AM, Zou VZ, Crane JD, Yabut JM, et al. FGF21 does not require adipocyte AMP-activated protein kinase (AMPK) or the phosphorylation of acetyl-CoA carboxylase (ACC) to mediate improvements in whole-body glucose homeostasis. Mol Metabol. 2017;6(6):471–81. https://doi.org/10.1016/j.molmet.2017.04.001.
Song YM, Lee Y-H, Kim J-W, Ham D-S, Kang E-S, Cha BS, et al. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. 2015;11(1):46–59. https://doi.org/10.4161/15548627.2014.984271.
Xu W, Deng Y-Y, Yang L, Zhao S, Liu J, Zhao Z, et al. Metformin ameliorates the proinflammatory state in patients with carotid artery atherosclerosis through sirtuin 1 induction. Transl Res. 2015;166(5):451–8. https://doi.org/10.1016/j.trsl.2015.06.002.
Raj V, Natarajan S, Marimuthu C, Chatterjee S, Ramasamy M, Ramanujam GM, et al. Cholecalciferol and metformin protect against lipopolysaccharide-induced endothelial dysfunction and senescence by modulating sirtuin-1 and protein arginine methyltransferase-1. Eur J Pharmacol. 2021;912:174531. https://doi.org/10.1016/j.ejphar.2021.174531.
Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LML, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci. 2014;111(4):E435–44. https://doi.org/10.1073/pnas.1311121111.
Pryor R, Cabreiro F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochemical Journal. 2015;471(3):307–22. https://doi.org/10.1042/BJ20150497.
Maruthur NM, Tseng E, Hutfless S, Wilson LM, Suarez-Cuervo C, Berger Z, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. 2016;164(11):740–51. https://doi.org/10.7326/M15-2650.
Davidson JA, Scheen AJ, Howlett H. Tolerability Profile of Metformin/Glibenclamide Combination Tablets (Glucovance®). Drug Saf. 2004;27(15):1205–16. https://doi.org/10.2165/00002018-200427150-00004.
Blonde L, Rosenstock J, Mooradian A, Piper BA, Henry D. Glyburide/metformin combination product is safe and efficacious in patients with type 2 diabetes failing sulphonylurea therapy. Diabetes Obes Metab. 2002;4(6):368–75. https://doi.org/10.1046/j.1463-1326.2002.00229.x.
Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med. 1998;338(13):867–73. https://doi.org/10.1056/NEJM199803263381303.
Wulffelé MG, Kooy A, Lehert P, Bets D, Ogterop JC, Borger van der Burg B, et al. Combination of insulin and metformin in the treatment of type 2 diabetes. Diabetes Care. 2002;25(12):2133–40. https://doi.org/10.2337/diacare.25.12.2133.
Group UPDS. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352(9131):854–65. https://doi.org/10.1016/S0140-6736(98)07037-8.
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577–89. https://doi.org/10.1056/NEJMoa0806470.
Hong J, Zhang Y, Lai S, Lv A, Su Q, Dong Y, et al. Effects of metformin versus glipizide on cardiovascular outcomes in patients with type 2 diabetes and coronary artery disease. Diabetes Care. 2013;36(5):1304–11. https://doi.org/10.2337/dc12-0719.
Larsen AH, Jessen N, Nørrelund H, Tolbod LP, Harms HJ, Feddersen S, et al. A randomised, double-blind, placebo-controlled trial of metformin on myocardial efficiency in insulin-resistant chronic heart failure patients without diabetes. Eur J Heart Fail. 2020;22(9):1628–37. https://doi.org/10.1002/ejhf.1656.
Wiggers H, Køber L, Gislason G, Schou M, Poulsen MK, Vraa S, et al. The DANish randomized, double-blind, placebo-controlled trial in patients with chronic HEART failure (DANHEART): A 2× 2 factorial trial of hydralazine-isosorbide dinitrate in patients with chronic heart failure (H-HeFT) and metformin in patients with chronic heart failure and diabetes or prediabetes (Met-HeFT). Am Heart J. 2021;231:137–46. https://doi.org/10.1016/j.ahj.2020.09.020.
Zhou J-B, Tang X, Han M, Yang J, Simó R. Impact of antidiabetic agents on dementia risk: a Bayesian network meta-analysis. Metabolism. 2020;109:154265. https://doi.org/10.1016/j.metabol.2020.154265.
Samaras K, Makkar S, Crawford JD, Kochan NA, Wen W, Draper B, et al. Metformin use is associated with slowed cognitive decline and reduced incident dementia in older adults with type 2 diabetes: the Sydney Memory and Ageing Study. Diabetes Care. 2020;43(11):2691–701. https://doi.org/10.2337/dc20-0892.
Løvvik TS, Carlsen SM, Salvesen Ø, Steffensen B, Bixo M, Gómez-Real F, et al. Use of metformin to treat pregnant women with polycystic ovary syndrome (PregMet2): a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019;7(4):256–66. https://doi.org/10.1016/s2213-8587(19)30002-6.
Jacob S, Brewer C, Tang T, Picton H, Barth J, Balen A. A short course of metformin does not reduce OHSS in a GnRH antagonist cycle for women with PCOS undergoing IVF: a randomised placebo-controlled trial. Hum Reprod. 2016:1–9. https://doi.org/10.1093/humrep/dew268.
Nascimento IBd, Sales WB, Dienstmann G, Souza MLRd, Fleig R, Silva JC. Metformin for prevention of cesarean delivery and large-for-gestational-age newborns in non-diabetic obese pregnant women: a randomized clinical trial. Arch Endocrinol Metabol. 2020;64:290–7. https://doi.org/10.20945/2359-3997000000251.
Cluver CA, Hiscock R, Decloedt EH, Hall DR, Schell S, Mol BW, et al. Use of metformin to prolong gestation in preterm pre-eclampsia: randomised, double blind, placebo-controlled trial. bmj. 2021;374. https://doi.org/10.1136/bmj.n2103.
Saif MW, Rajagopal S, Caplain J, Grimm E, Serebrennikova O, Das M, et al. A phase I delayed-start, randomized and pharmacodynamic study of metformin and chemotherapy in patients with solid tumors. Cancer Chemother Pharmacol. 2019;84(6):1323–31. https://doi.org/10.1007/s00280-019-03967-3.
Pimentel I, Lohmann AE, Ennis M, Dowling RJ, Cescon D, Elser C, et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast. 2019;48:17–23. https://doi.org/10.1016/j.breast.2019.08.003.
Wang S, Lin Y, Xiong X, Wang L, Guo Y, Chen Y, et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical TrialLow-Dose Metformin Turns TIME against Cancer. Clin Cancer Res. 2020;26(18):4921–32. https://doi.org/10.1158/1078-0432.CCR-20-0113.
Ciccarese C, Iacovelli R, Buti S, Primi F, Astore S, Massari F, et al. Concurrent Nivolumab and Metformin in Diabetic Cancer Patients: Is It Safe and More Active?. Anticancer Res. 2022;42(3):1487–93. https://doi.org/10.21873/anticanres.15620.
Lalau J-D, Al-Salameh A, Hadjadj S, Goronflot T, Wiernsperger N, Pichelin M, et al. Metformin use is associated with a reduced risk of mortality in patients with diabetes hospitalised for COVID-19. Diabetes Metabol. 2021;47(5):101216. https://doi.org/10.1016/j.diabet.2020.101216.
Ojeda-Fernández L, Foresta A, Macaluso G, Colacioppo P, Tettamanti M, Zambon A, et al. Metformin use is associated with a decrease in the risk of hospitalization and mortality in COVID-19 patients with diabetes: A population-based study in Lombardy. Diabetes Obesity Metabol. 2022;24(5):891–8. https://doi.org/10.1111/dom.14648.
Crouse AB, Grimes T, Li P, Might M, Ovalle F, Shalev A. Metformin use is associated with reduced mortality in a diverse population with COVID-19 and diabetes. Front Endocrinol. 2021;11:600439. https://doi.org/10.3389/fendo.2020.600439.
Bramante CT, Huling JD, Tignanelli CJ, Buse JB, Liebovitz DM, Nicklas JM, et al. Randomized trial of metformin, ivermectin, and fluvoxamine for Covid-19. New England J Med. 2022;387(7):599–610. https://doi.org/10.1056/NEJMoa2201662.
Ting RZ-W, Szeto CC, Chan MH-M, Ma KK, Chow KM. Risk factors of vitamin B12 deficiency in patients receiving metformin. Arch Int Med. 2006;166(18):1975–9. https://doi.org/10.1001/archinte.166.18.1975.
Donnelly LA, Dennis JM, Coleman RL, Sattar N, Hattersley AT, Holman RR, et al. Risk of anemia with metformin use in type 2 diabetes: a MASTERMIND study. Diabetes Care. 2020;43(10):2493–9. https://doi.org/10.2337/dc20-1104.
Singh A, Kumar A, Karmakar D, Jha R. Association of B12 deficiency and clinical neuropathy with metformin use in type 2 diabetes patients. J Postgrad Med. 2013;59(4):253. https://doi.org/10.4103/0022-3859.123143.
Cheng X, Liu Y-M, Li H, Zhang X, Lei F, Qin J-J, et al. Metformin is associated with higher incidence of acidosis, but not mortality, in individuals with COVID-19 and pre-existing type 2 diabetes. Cell Metabol. 2020;32(4):537–47. e3. https://doi.org/10.1016/j.cmet.2020.08.013.
Alvarez CA, Halm EA, Pugh MJV, McGuire DK, Hennessy S, Miller RT, et al. Lactic acidosis incidence with metformin in patients with type 2 diabetes and chronic kidney disease: A retrospective nested case-control study. Endocrinol Diabetes Metabol. 2021;4(1):e00170. https://doi.org/10.1002/edm2.170.
Hanem LGE, Stridsklev S, Júlíusson PB, Salvesen Ø, Roelants M, Carlsen SM, et al. Metformin use in PCOS pregnancies increases the risk of offspring overweight at 4 years of age: follow-up of two RCTs. J Clin Endocrinol Metab. 2018;103(4):1612–21. https://doi.org/10.1210/jc.2017-02419.
Rowan JA, Rush EC, Plank LD, Lu J, Obolonkin V, Coat S, et al. Metformin in gestational diabetes: the offspring follow-up (MiG TOFU): body composition and metabolic outcomes at 7–9 years of age. BMJ Open Diabetes Res Care. 2018;6(1):e000456. https://doi.org/10.1136/bmjdrc-2017-000456.
Landi SN, Radke S, Engel SM, Boggess K, Stürmer T, Howe AS, et al. Association of long-term child growth and developmental outcomes with metformin vs insulin treatment for gestational diabetes. JAMA Pediatr. 2019;173(2):160–8. https://doi.org/10.1001/jamapediatrics.2018.4214.
Sluggett JK, Koponen M, Bell JS, Taipale H, Tanskanen A, Tiihonen J, et al. Metformin and risk of Alzheimer’s disease among community-dwelling people with diabetes: a national case-control study. J Clin Endocrinol Metabol. 2020;105(4):e963–72. https://doi.org/10.3233/JAD-181017.10.1210/clinem/dgz234.
Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: a population-based case–control study. J Am Geriatr Soc. 2012;60(5):916–21. https://doi.org/10.1111/j.1532-5415.2012.03916.x.
Khattar D, Khaliq F, Vaney N, Madhu S. Is metformin-induced vitamin B12 deficiency responsible for cognitive decline in type 2 diabetes? Indian J Psychol Med. 2016;38(4):285–90. https://doi.org/10.4103/0253-7176.185952.
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–81. https://doi.org/10.1038/s41582-019-0244-7.
Wahlqvist ML, Lee M-S, Hsu C-C, Chuang S-Y, Lee J-T, Tsai H-N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Related Disord. 2012;18(6):753–8. https://doi.org/10.1016/j.parkreldis.2012.03.010.
Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS, et al. Polycystic ovary syndrome. Nat Rev Dis Primers. 2016;2(1):1–18. https://doi.org/10.1038/nrdp.2016.57.
Tang T, Lord JM, Norman RJ, Yasmin E, Balen AH. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst Rev. 2012;5:Cd003053. https://doi.org/10.1002/14651858.CD003053.pub5.
Aubuchon M, Kunselman AR, Schlaff WD, Diamond MP, Coutifaris C, Carson SA, et al. Metformin and/or clomiphene do not adversely affect liver or renal function in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2011;96(10):E1645–9. https://doi.org/10.1210/jc.2011-1093.
Tso LO, Costello MF, Albuquerque LET, Andriolo RB, Macedo CR. Metformin treatment before and during IVF or ICSI in women with polycystic ovary syndrome. Cochrane Database Syst Rev. 2020;12(12):Cd006105. https://doi.org/10.1002/14651858.CD006105.pub4.
Bordewijk EM, Nahuis M, Costello MF, Van der Veen F, Tso LO, Mol BW, et al. Metformin during ovulation induction with gonadotrophins followed by timed intercourse or intrauterine insemination for subfertility associated with polycystic ovary syndrome. Cochrane Database Syst Rev. 2017;1(1):Cd009090. https://doi.org/10.1002/14651858.CD009090.pub2.
Faure M, Bertoldo MJ, Khoueiry R, Bongrani A, Brion F, Giulivi C, et al. Metformin in reproductive biology. Front Endocrinol (Lausanne). 2018;9:675. https://doi.org/10.3389/fendo.2018.00675.
Campbell JM, Bellman SM, Stephenson MD, Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res Rev. 2017;40:31–44. https://doi.org/10.1016/j.arr.2017.08.003.
Kulkarni AS, Brutsaert EF, Anghel V, Zhang K, Bloomgarden N, Pollak M, et al. Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell. 2018;17(2):e12723. https://doi.org/10.1111/acel.12723.
Newman JC, Milman S, Hashmi SK, Austad SN, Kirkland JL, Halter JB, et al. Strategies and challenges in clinical trials targeting human aging. J Gerontol Series A Biomed Sci Med Sci. 2016;71(11):1424–34. https://doi.org/10.1093/gerona/glw149.
Triggle CR, Mohammed I, Bshesh K, Marei I, Ye K, Ding H, et al. Metformin: Is it a drug for all reasons and diseases?. Metabolism. 2022:155223. https://doi.org/10.1016/j.metabol.2022.155223.
Prattichizzo F, Giuliani A, Mensà E, Sabbatinelli J, De Nigris V, Rippo MR, et al. Pleiotropic effects of metformin: Shaping the microbiome to manage type 2 diabetes and postpone ageing. Ageing Res Rev. 2018;48:87–98. https://doi.org/10.1016/j.arr.2018.10.003.
Justice JN, Ferrucci L, Newman AB, Aroda VR, Bahnson JL, Divers J, et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: report from the TAME Biomarkers Workgroup. Geroscience. 2018;40(5):419–36. https://doi.org/10.1007/s11357-018-0042-y.
Hosono K, Endo H, Takahashi H, Sugiyama M, Sakai E, Uchiyama T, et al. Metformin Suppresses Colorectal Aberrant Crypt Foci in a Short-term Clinical TrialMetformin Suppresses Colorectal ACF in Humans. Cancer Prev Res. 2010;3(9):1077–83. https://doi.org/10.1158/1940-6207.capr-10-0186.
Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016;17(4):475–83. https://doi.org/10.1016/S1470-2045(15)00565-3.
Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe VV, Ford LG. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia. 2017;60(9):1639–47. https://doi.org/10.1007/s00125-017-4372-6.
Schuler KM, Rambally BS, DiFurio MJ, Sampey BP, Gehrig PA, Makowski L, et al. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015;4(2):161–73. https://doi.org/10.1002/cam4.353.
Sivalingam V, Kitson S, McVey R, Roberts C, Pemberton P, Gilmour K, et al. Measuring the biological effect of presurgical metformin treatment in endometrial cancer. Br J Cancer. 2016;114(3):281–9. https://doi.org/10.1038/bjc.2015.453.
Rebours V, Gaujoux S, d’Assignies G, Sauvanet A, Ruszniewski P, Lévy P, et al. Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN) Fatty Pancreatic Infiltration and PanIN Lesions. Clin Cancer Res. 2015;21(15):3522–8. https://doi.org/10.1158/1078-0432.ccr-14-2385.
Bethea TN, Kitahara CM, Sonderman J, Patel AV, Harvey C, Knutsen SF, et al. A Pooled Analysis of Body Mass Index and Pancreatic Cancer Mortality in African AmericansBMI and Pancreatic Cancer Mortality in African Americans. Cancer Epidemiol Biomark Prev. 2014;23(10):2119–25. https://doi.org/10.1158/1055-9965.epi-14-0422.
Eibl G, Rozengurt E. Metformin: review of epidemiology and mechanisms of action in pancreatic cancer. Cancer Metastasis Rev. 2021;40(3):865–78. https://doi.org/10.1007/s10555-021-09977-z.
Chae YK, Arya A, Malecek M-K, Shin DS, Carneiro B, Chandra S, et al. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget. 2016;7(26):40767. https://doi.org/10.18632/oncotarget.8194.
Wallace T, Matthews D. The assessment of insulin resistance in man. Diabetic Med. 2002;19(7):527–34. https://doi.org/10.1046/j.1464-5491.2002.00745.x.
Cazzaniga M, DeCensi A, Pruneri G, Puntoni M, Bottiglieri L, Varricchio C, et al. The effect of metformin on apoptosis in a breast cancer presurgical trial. Br J Cancer. 2013;109(11):2792–7. https://doi.org/10.1038/bjc.2013.657.
Hadad SM, Coates P, Jordan LB, Dowling RJ, Chang MC, Done SJ, et al. Evidence for biological effects of metformin in operable breast cancer: biomarker analysis in a pre-operative window of opportunity randomized trial. Breast Cancer Res Treat. 2015;150(1):149–55. https://doi.org/10.1007/s10549-011-1612-1.
Joshua A, Zannella V, Downes M, Bowes B, Hersey K, Koritzinsky M, et al. A pilot ‘window of opportunity’neoadjuvant study of metformin in localised prostate cancer. Prostate Cancer Prostatic Dis. 2014;17(3):252–8. https://doi.org/10.1038/pcan.2014.20.
Tan BX, Yao WX, Ge J, Peng XC, Du XB, Zhang R, et al. Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer. 2011;117(22):5103–11. https://doi.org/10.1002/cncr.26151.
Coyle C, Cafferty F, Vale C, Langley R. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol. 2016;27(12):2184–95. https://doi.org/10.1093/annonc/mdw410.
Zhang ZJ, Li S. The prognostic value of metformin for cancer patients with concurrent diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2014;16(8):707–10. https://doi.org/10.1111/dom.12267.
Lega IC, Shah PS, Margel D, Beyene J, Rochon PA, Lipscombe LL. The Effect of Metformin on Mortality Following Cancer among Patients with DiabetesMetformin and Cancer Mortality: Review and Meta-analysis. Cancer Epidemiol Biomark Prev. 2014;23(10):1974–84. https://doi.org/10.1158/1055-9965.EPI-14-0327.
Jang WI, Kim M-S, Kang SH, Jo AJ, Kim YJ, Tchoe HJ, et al. Association between metformin use and mortality in patients with type 2 diabetes mellitus and localized resectable pancreatic cancer: a nationwide population-based study in korea. Oncotarget. 2017;8(6):9587.https://doi.org/10.18632/oncotarget.14525.
Sadeghi N, Abbruzzese JL, Yeung S-CJ, Hassan M, Li D. Metformin Use Is Associated with Better Survival of Diabetic Patients with Pancreatic CancerMetformin and Pancreatic Cancer Survival. Clin Cancer Res. 2012;18(10):2905–12. https://doi.org/10.1158/1078-0432.CCR-11-2994.
Kordes S, Pollak MN, Zwinderman AH, Mathôt RA, Weterman MJ, Beeker A, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16(7):839–47. https://doi.org/10.1016/S1470-2045(15)00027-3.
Li X, Li T, Liu Z, Gou S, Wang C. The effect of metformin on survival of patients with pancreatic cancer: a meta-analysis. Sci Rep. 2017;7(1):1–8. https://doi.org/10.1038/s41598-017-06207-x.
Shi Y-Q, Zhou X-C, Du P, Yin M-Y, Xu L, Chen W-J, et al. Relationships are between metformin use and survival in pancreatic cancer patients concurrent with diabetes: A systematic review and meta-analysis. Medicine. 2020;99(37). https://doi.org/10.1097/md.0000000000021687.
Farmer RE, Ford D, Forbes HJ, Chaturvedi N, Kaplan R, Smeeth L, et al. Metformin and cancer in type 2 diabetes: a systematic review and comprehensive bias evaluation. Int J Epidemiol. 2017;46(2):728–44. https://doi.org/10.1093/ije/dyw275.
Wen Y, Liu Y, Chen C, Chi J, Zhong L, Zhao Y, et al. Metformin loaded porous particles with bio-microenvironment responsiveness for promoting tumor immunotherapy. Biomaterials Sci. 2021;9(6):2082–9. https://doi.org/10.1039/D0BM01931C.
Ganesh A, Randall MD. Does metformin affect outcomes in COVID‐19 patients with new or pre‐existing diabetes mellitus? A systematic review and meta‐analysis. Br J Clin Pharmacol. 2022. https://doi.org/10.1111/bcp.15258.
Dawed AY, Zhou K, van Leeuwen N, Mahajan A, Robertson N, Koivula R, et al. Variation in the plasma membrane monoamine transporter (PMAT)(encoded by SLC29A4) and organic cation transporter 1 (OCT1)(encoded by SLC22A1) and gastrointestinal intolerance to metformin in type 2 diabetes: an IMI DIRECT study. Diabetes Care. 2019;42(6):1027–33. https://doi.org/10.2337/dc18-2182.
Berchtold P, Bolli P, Arbenz U, Keiser G. Disturbance of intestinal absorption following metformin therapy (observations on the mode of action of biguanides. Diabetologia. 1969;5(6):405–12. https://doi.org/10.1007/BF00427979.
Tomkin G, Hadden D, Weaver J, Montgomery D. Vitamin-B12 status of patients on long-term metformin therapy. Br Med J. 1971;2(5763):685–7. https://doi.org/10.1136/bmj.2.5763.685.
Reinstatler L, Qi YP, Williamson RS, Garn JV, Oakley GP Jr. Association of biochemical B12 deficiency with metformin therapy and vitamin B12 supplements: the National Health and Nutrition Examination Survey, 1999–2006. Diabetes Care. 2012;35(2):327–33. https://doi.org/10.2337/dc11-1582.
Pflipsen MC, Oh RC, Saguil A, Seehusen DA, Seaquist D, Topolski R. The prevalence of vitamin B12 deficiency in patients with type 2 diabetes: a cross-sectional study. J Am Board Fam Med. 2009;22(5):528–34. https://doi.org/10.3122/jabfm.2009.05.090044.
DeFronzo RA, Goodman AM, Group MMS. Efficacy of metformin in patients with non-insulin-dependent diabetes mellitus. New England J Med. 1995;333(9):541-9. https://doi.org/10.1056/NEJM199508313330902.
Wulffele M, Kooy A, Lehert P, Bets D, Ogterop J, Borger Van Der Burg B, et al. Effects of short‐term treatment with metformin on serum concentrations of homocysteine, folate and vitamin B12 in type 2 diabetes mellitus: a randomized, placebo‐controlled trial. J Int Med. 2003;254(5):455–63. https://doi.org/10.1046/j.1365-2796.2003.01213.x.
Bauman WA, Shaw S, Jayatilleke E, Spungen AM, Herbert V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care. 2000;23(9):1227–31. https://doi.org/10.2337/diacare.23.9.1227.
Connelly PJ, Lonergan M, Soto-Pedre E, Donnelly L, Zhou K, Pearson ER. Acute kidney injury, plasma lactate concentrations and lactic acidosis in metformin users: A GoDarts study. Diabetes Obes Metab. 2017;19(11):1579–86. https://doi.org/10.1111/dom.12978.
Krowl L, Al-Khalisy H, Kaul P. Metformin-induced lactic acidosis (MILA): review of current diagnostic paradigm. Am J Emerg Med. 2018;36(5):908. e3-. e5.https://doi.org/10.1016/j.ajem.2018.01.097.
Kajbaf F, Lalau J-D. The prognostic value of blood pH and lactate and metformin concentrations in severe metformin-associated lactic acidosis. BMC Pharmacol Toxicol. 2013;14(1):1–5. https://doi.org/10.1186/2050-6511-14-2.
Blumenberg A, Benabbas R, Sinert R, Jeng A, Wiener SW. Do patients die with or from metformin-associated lactic acidosis (MALA)? Systematic review and meta-analysis of pH and lactate as predictors of mortality in MALA. J Med Toxicol. 2020;16(2):222–9. https://doi.org/10.1007/s13181-019-00755-6.
Flory JH, Hennessy S. Metformin use reduction in mild to moderate renal impairment: possible inappropriate curbing of use based on food and drug administration contraindications. JAMA Intern Med. 2015;175(3):458–9. https://doi.org/10.1001/jamainternmed.2014.6936.
Ekström N, Schiöler L, Svensson A-M, Eeg-Olofsson K, Jonasson JM, Zethelius B, et al. Effectiveness and safety of metformin in 51 675 patients with type 2 diabetes and different levels of renal function: a cohort study from the Swedish National Diabetes Register. BMJ Open. 2012;2(4):e001076. https://doi.org/10.1136/bmjopen-2012-001076.
Salvatore T, Pafundi PC, Marfella R, Sardu C, Rinaldi L, Monaco L, et al. Metformin lactic acidosis: should we still be afraid? Diabetes Res Clin Pract. 2019;157:107879. https://doi.org/10.1016/j.diabres.2019.107879.
Lazarus B, Wu A, Shin J-I, Sang Y, Alexander GC, Secora A, et al. Association of metformin use with risk of lactic acidosis across the range of kidney function: a community-based cohort study. JAMA Intern Med. 2018;178(7):903–10. https://doi.org/10.1001/jamainternmed.2018.0292.
Hu Y, Ding B, Shen Y, Yan R-N, Li F-F, Sun R, et al. Rapid changes in serum testosterone in men with newly diagnosed type 2 diabetes with intensive insulin and metformin. Diabetes Care. 2021;44(4):1059–61. https://doi.org/10.2337/dc20-1558.
Thangthaeng N, Rutledge M, Wong JM, Vann PH, Forster MJ, Sumien N. Metformin impairs spatial memory and visual acuity in old male mice. Aging Disease. 2017;8(1):17. https://doi.org/10.14336/AD.2016.1010.
Kuhla A, Brichmann E, Rühlmann C, Thiele R, Meuth L, Vollmar B. Metformin therapy aggravates neurodegenerative processes in ApoE–/–Mice. J Alzheimers Dis. 2019;68(4):1415–27.
Kuan Y-C, Huang K-W, Lin C-L, Hu C-J, Kao C-H. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2017;79:77–83.https://doi.org/10.1016/j.pnpbp.2017.06.002.
Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, Brodaty H, et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36(10):2981–7. https://doi.org/10.2337/dc13-0229.
Acknowledgements
Not applicable.
Code availability
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (81572380) and the Sichuan Science and Technology Department Key Research and Development Project (2019YFS0539).
Author information
Authors and Affiliations
Contributions
Ji-Yan Liu contributed to the study conception and design. Material preparation, data collection and analysis were performed by Yang Du, Ya-juan Zhu and Yi-Xin Zhou. The first draft of the manuscript was written by Yang Du and Ya-Juan Zhu. Figures were drawn by Ya-Juan Zhu. Manuscript revisions were performed by Yang Du and Jing Ding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, Y., Zhu, YJ., Zhou, YX. et al. Metformin in therapeutic applications in human diseases: its mechanism of action and clinical study. Mol Biomed 3, 41 (2022). https://doi.org/10.1186/s43556-022-00108-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-022-00108-w