
Abstract
The gut microbiota is emerging as a prominent player in maintaining health through several metabolic and immune pathways. Dysregulation of gut microbiota composition, also known as dysbiosis, is involved in the clinical outcome of diabetes, inflammatory bowel diseases, cancer, aging and HIV infection. Gut dysbiosis and inflammation persist in people living with HIV (PLWH) despite receiving antiretroviral therapy, further contributing to non-AIDS comorbidities. Metformin, a widely used antidiabetic agent, has been found to benefit microbiota composition, promote gut barrier integrity and reduce inflammation in human and animal models of diabetes. Inspired by the effect of metformin on diabetes-related gut dysbiosis, we herein critically review the relevance of metformin to control inflammation in PLWH. Metformin may improve gut microbiota composition, in turn reducing inflammation and risk of non-AIDS comorbidities. This review will pave the way towards innovative strategies to counteract dysregulated microbiota and improve the lives of PLWH.
Similar content being viewed by others

Introduction
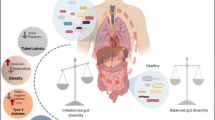
The total human body hosts over 1014 microbes, of which around 99% are present in the gastrointestinal (GI) tract [1]. The GI microbiota encompasses thousands of bacteria, fungi, archaea, viruses and eukaryotic microbes. Bacteria make up the greatest proportion of microbes in the GI tract and are therefore most frequently studied. In addition to supporting nutrient absorption, the GI microbiota has an important role in homeostasis by preventing pathogens from entering the mucosa. Accordingly, a breakdown in the balance between “protective” versus “harmful” intestinal bacteria, a concept termed dysbiosis [2], can lead to barrier dysfunction and intestinal homeostasis disruption through translocation of microbial products leading to inflammation [3]. Increasing evidence has put a spotlight on the contribution of gut dysbiosis and its related inflammation in diabetes, inflammatory bowel diseases, cancer, aging and HIV infection [4,5,6,7]. Furthermore, people with type 2 diabetes mellitus (DM2) or HIV infection share comorbidities such as dyslipidemia, cardiovascular disease, depression and cancer in part through gut microbiome-mediated inflammation [8, 9].
HIV infection is characterized by a rapid decline in mucosal CD4+ T cell count, epithelial gut damage, translocation of microbial products into the systemic circulation and immune activation [10]. By suppressing host immune function, HIV leads to microbial dysbiosis and translocation, further contributing to chronic inflammation and immune activation [10]. Antiretroviral therapy (ART) has transformed care, leading to major improvements in the health of people living with HIV (PLWH). However, despite controlling viral load and CD4+ T-cell count, long-term ART reduces but does not normalize inflammation and immune activation compared to healthy people [11]. Gut barrier dysfunction persists, allowing microbial products to enter the circulation [12]. This heightened inflammation has been associated with non-AIDS comorbidities including dyslipidemia, cardiovascular disease, depression and cancer [13]. Given the close interaction between the intestinal microbiota and HIV-related inflammation, improving gut health by targeted therapies may reduce comorbidities and constitutes the topic of this review.
Isolated in the 1920s from French lilac, metformin (dimethylbiguanide) is the most commonly used drug to treat DM2. This drug acts as an anti-diabetic agent that promotes euglycemia without inducing hypoglycaemia and has few side effects. Compared with other classes of anti-diabetic drugs such as sulfonylureas or insulin, metformin use might have an anti-inflammatory effect as its use is associated with a lower risk of cardiovascular disease [14, 15]. More recently, metformin has been shown to be also beneficial in non-diabetic subjects, by reducing inflammation and aging biomarkers [16]. Metformin was reported to extend lifespan in some animal models, acting as a diet mimetic agent [17, 18]. In women with polycystic ovary syndrome, metformin decreased infertility rate while lowering markers of inflammation such as IL-6, TNF-α and intracellular adhesion molecule-1 (ICAM-1) [19]. Remarkably, Arrieta et al. showed that metformin, when combined with epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) therapy, improved survival in a randomized study for patients with advanced lung adenocarcinoma compared to EGFR-TKIs alone [20]. Aside from cancer (recently reviewed by Klil-Drori et al. [21]), multiple clinical trials are ongoing in non-diabetic individuals with different conditions using metformin as an immunometabolic drug (Table 1).
Gut dysbiosis, increased gut permeability, chronic inflammation and systemic immune activation are common features of PLWH or DM2 [22,23,24]. Common microbiota composition changes such as decreased abundance of Bifidobacterium, Bacteroides and Akkermansia were found in DM2 and PLWH [7, 25, 26]. In some studies, metformin has been shown to positively influence GI microbiota composition and promote GI barrier integrity, resulting in reduced inflammation [27,28,29,30,31,32,33]. Given the benefits of metformin use in non-diabetic subjects and its well-documented effect on the composition of gut microbiota in DM2, we hypothesize that metformin lowers risk of non-AIDS comorbidities in ART-treated PLWH. Herein, we review and discuss advances in understanding the effects of metformin on gut dysbiosis and its potential applications in management of HIV-related inflammation, to reduce the risk of inflammatory non-AIDS comorbidities.
Microbiota dysbiosis in obesity and DM2
DM2 is an increasing public health issue arising from genetic factors, sedentary lifestyle, Western diet and excessive visceral fat. First noted in 2008, alterations of gut microbiota composition in DM2 individuals have been well studied and reviewed [4, 25, 34,35,36,37,38]. Among the commonly reported findings, the genera of Bifidobacterium, Bacteroides, Faecalibacterium, Akkermansia and Roseburia abundance were decreased in DM2, while the genera of Ruminococcus, Fusobacterium, and Blautia were increased in DM2 [25]. It still remains unclear whether the DM2-associated dysbiosis is a cause or a consequence of glucose intake and/or regulation. In DM2 individuals, dysbiosis fosters bacterial translocation through the damaged epithelial gut barrier, leading to systemic immune activation. Bacterial lipopolysaccharide (LPS) binds to TLR4 and activates monocytes/macrophages leading to pro-inflammatory IL-6 and TNF secretion, and insulin resistance by inhibiting the insulin tyrosine kinase receptor signalling [39]. Moreover, LPS-induced inflammatory response reduced insulin-receptor signaling and glucose transport in human muscle cells [40]. In addition, DM2 patients have lower levels of short chain fatty acids (SCFAs), especially propionate and butyrate, in their feces compared with non-diabetic subjects [41]. SCFAs are a subset of fatty acids produced by the gut microbiota during the fermentation of polysaccharides, among which, anti-inflammatory acetate, propionate and butyrate are the most abundant [42, 43]. As the primary energy source for colonic epithelial cells, SCFAs improve intestinal barrier function, prevent microbial translocation and further reduce inflammation [42, 44]. Therefore, SCFAs and SCFA-producing bacteria are crucial in dampening inflammation.
Based on the association between microbiota and DM2, several groups have tried to modulate gut dysbiosis with prebiotics, probiotics and fecal microbiota transplantation (FMT) to improve insulin sensitivity in animals and humans [45,46,47]. Vrieze et al. conducted two studies in 2012 showing that FMT from lean controls improved insulin sensitivity in participants with metabolic syndrome, in association with increased intestinal abundance of butyrate-producing bacteria when compared with the control group receiving autologous FMT [45, 46]. Everard et al. reported that the abundance of Akkermansia muciniphila, a gut-protective bacterium, was 3300-fold lower in obese mice than in their lean littermates. Encouragingly, a 4-week oral gavage of live A. muciniphila in mice reversed high-fat diet-induced metabolic disorders [47]. In 2019, A muciniphila supplementation in obese people improved insulin sensitivity and reduced cholesterol levels in the absence of toxicity [48]. These studies demonstrate the implication of a disturbed gut microbiota in obesity and DM2 outcomes.
More than meets the eye: metformin and gut microbiota modification in DM2
Among different anti-diabetic medications, metformin has been shown to profoundly alter the gut microbiota composition. Metformin decreases insulin resistance in DM2 via AMPK stimulation, reducing hepatic gluconeogenesis through modulation of several intracellular pathways [49]. However, growing evidence suggests that the effects of metformin are also mediated through changes in gut microbiota composition, an effect conserved from the nematode Caenorhabditis elegans to humans [17]. Metformin is predominantly concentrated in the jejunum with levels 30–300 times higher than in plasma [50]. Sum et al. showed in 1992 that intravenous administration of metformin did not improve blood glucose in contrast with oral administration in humans [51]. Moreover, depleting the microbiota using broad-spectrum antibiotics abrogated the anti-diabetic effects of metformin in high-fat diet (HFD) mice [52].
Microbiota compositional changes associated with metformin use in DM2 or healthy people are summarized in Table 2. Factors such as study population, sequencing method, dietary intake and medication may explain discrepancies between studies. However, increased A. muciniphila and Lactobacillus, and decreased Intestinibacter abundance were observed after metformin therapy in three studies [27, 29, 52]. A. muciniphila is a commensal anaerobic mucin-degrading bacterium whose abundance is positively associated with glucose regulation [52, 53]. This bacterium represents 1–5% of all intestinal bacteria in healthy individuals and has been shown to reduce insulin resistance following treatment with prebiotic polyphenols in animal models of obesity [54]. Metformin also increased abundance of A. muciniphila in HFD-fed mice [52]. Similarly, blood SCFA butyrate and propionate levels were shown to be increased in metformin-treated DM2 subjects due to microbiota modification [27]. Moreover, metformin treatment was shown to decrease the frequency of pathogenic Th17 cells and increase the frequency of regulatory T cells (Tregs), thus reducing inflammation in diabetes or IBD murine models [55, 56]. Gut dysbiosis and low SCFA production were associated with lower frequency of mucosal Tregs in mice and humans [57]. Bhaskaran et al. demonstrated that Tregs were essential in the anti-inflammatory effect of gut-derived SCFA in mice [58].
Nevertheless, metformin increased Escherichia abundance which is associated with bloating and diarrhea, contributing to discontinuation of metformin in up to 30% of diabetic people [27, 29, 59, 60]. Toxicity including gastrointestinal upset, hyperlactatemia and metabolic acidosis, occurs infrequently when metformin accumulates due renal insufficiency or overdose [61]. Some cases of lactic acidosis and ketoacidosis have been reported in metformin-treated diabetic PLWH receiving stavudine (d4T) and didanosine (ddI) nucleoside analogs, no longer used in current practice [62,63,64]. Thus, use of metformin may contribute to risks including gastrointestinal distress and drug interactions in certain antiretroviral therapies, however, the benefits outweigh the risks.
Microbiota, gut permeability and inflammation in HIV infection
During acute HIV infection, the virus rapidly disseminates while establishing a pool of latently infected cells [67]. The GI tract is critical for the pathogenesis of HIV infection and serves as a major site of viral replication [68]. Up to 70% of GI and 20% of peripheral blood CD4+ T-cells express CCR5, a chemokine receptor that serves as co-receptor allowing for the entry of HIV [67]. Thus, intestinal CD4+ T-cells are a preferential target of the virus and are massively depleted during early infection. In simian immunodeficiency virus (SIV) infected macaques and HIV infected humanized mice, damages to the intestinal epithelium were linked to microbial translocation [69,70,71]. In PLWH, the disruption in gut homeostasis also results in increased permeability of the gut and translocation of microbial products such as LPS, bacterial DNA, and fungal β-d-Glucan into the circulatory system, promoting chronic immune activation and disease progression [10, 72].
In parallel, bacterial communities found in the intestine of HIV-infected individuals have been shown to differ from those of individuals not infected with HIV independently of age, sex and sexual practice, recently reviewed by Vujkovic-Cvijin et al. [7]. Vujkovic-Cvijin et al. [73] used high-resolution bacterial community profiling and identified a dysbiotic mucosal-adherent community in HIV-infected subjects with high Proteobacteria and low Bacteroidia associated with markers of mucosal immune disruption, T-cell activation, and chronic inflammation. Rocafort et al. [74] found that ART-naïve HIV-1-infected subjects were significantly depleted in Akkermansia, Anaerovibrio, Bifidobacterium, and Clostridium, compared to HIV negative individuals. ART exposure was not associated with changes in abundance of such genera, compared with ART-naïve. In SIV infected macaques, gut dysbiosis was also observed and strongly correlated with cytokine gene expression in the gut‐draining mesenteric lymph nodes including IL-10 and IL-6 [75]. Probiotic/prebiotic supplementation improves gastrointestinal immune function, increases reconstitution and decreases inflammation in ART-treated SIV-infected macaques [76].
Accumulating evidence has shown that dysregulation of the gut microbiota metabolism plays a role in HIV disease progression. The activity of the indoleamine-2,3-dioxygenase-1 (IDO-1), an enzyme catabolising the essential amino-acid tryptophan into immunosuppressive kynurenines, has been recognized as a key factor of HIV immune dysfunction and damage to the gut mucosa [73, 77]. The activity of IDO-1 correlates with Th17 cell loss, Tregs elevation, gut and systemic inflammation, reservoir size and disease progression in HIV-infected subjects [78, 79]. Furthermore, gut dysbiosis correlated with plasma kynurenine levels in ART-treated PLWH [73]. In addition, a decreased abundance of bacteria producing the gut epithelial protector butyrate, including Roseburia, Coprococcus, Faecalibacterium, and Eubacterium, was observed in both HIV-treated and ART-naïve individuals, in association with altered SCFAs profiles [80, 81]. Finally, HIV infection is associated with increased risk of coronary heart disease beyond that explained by traditional risk factors, and altered gut microbiota has been proposed as a key contributing determinant [82]. Higher activity of the kynurenine pathway and higher trimethylamine N-oxide (TMAO) plasma levels were also associated with an increased risk of cardiovascular disease [83, 84]. TMAO is converted in the liver from trimethylamine (TMA) which is an organic compound synthesized exclusively by the gut microbiota from dietary nutrients. Haissman et al. [85] reported that microbiota-dependent TMAO levels are also associated with monocyte activation in untreated PLWH. By comparing PLWH with and without coronary heart disease, Kehrmann et al. [86] showed that high circulating TMAO was a marker of coronary heart disease in association with the fecal abundance of Phascolarctobacterium, Desulfovibrio, Sutterella, and Faecalibacterium.
Insights on the use of metformin in non-diabetic PLWH
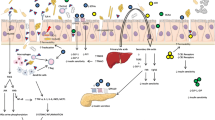
Treatment with metformin in PLWH has been shown to decrease lipodystrophy syndrome, hyperlipidemia and insulin sensitivity [87,88,89,90]. Moreover, Fitch et al. reported that metformin prevented progression of coronary artery calcification (CAC) and calcified plaque volume in PLWH with metabolic syndrome [90]. Shikuma et al. recently reported that metformin reduced CD4+ T‐cell exhaustion in non-diabetic ART‐treated PLWH [91]. Our team is currently carrying out a pilot study to determine the effect of metformin in non-diabetic ART-treated PLWH (NTC02659306) [92]. Metformin might be a promising treatment to control inflammation in non-diabetic PLWH through multiple pathways illustrated in Fig. 1.

Potential effects of metformin in non-diabetic PLWH
As several studies showed that metformin increased A. muciniphila abundance in diet-induced obese mice and DM2 patients [27, 29, 52, 53, 93], metformin may also increase A. muciniphila abundance in ART-treated PLWH to promote epithelial barrier integrity and decrease inflammation. Indeed, Reunanen et al. showed that A. muciniphila improved gut barrier integrity by binding to enterocytes in Caco-2 and HT-29 human colonic cell models [94]. Moreover, A. muciniphila administration reduced translocation of bacterial LPS and adipose tissue inflammation in an obese insulin-resistant mouse model [47].
In PLWH, there is a lower abundance of butyrate-producing bacteria in the gut microbiota [80, 81]. Interestingly, metformin was shown to increase the abundance of butyrate producing bacteria in both diabetic and healthy individuals [27, 60]. We therefore suggest that metformin, through increasing butyrate production, may decrease inflammation in ART-treated PLWH by enhancing intestinal epithelial barrier function, preventing microbial translocation and increasing mucosal Treg frequency [44, 58, 81].
Tryptophan catabolism and the kynurenine pathway were also associated with disease progression and HIV reservoir size in ART-treated PLWH [78]. Moreover, dysbiosis was associated with the kynurenine pathway in PLWH [73]. As Muzik et al. reported that metformin treatment of insulin resistant diabetic subjects was associated with down-regulation of the kynurenine pathway [95], metformin might also decrease tryptophan catabolism in non-diabetic ART-treated PLWH by altering microbiota composition.
Metformin may also reduce HIV-related inflammation independently of microbiota modification by modulating several signalling pathways: (1) suppressing nuclear factor κB activation, which enhances HIV transcription and induces the expression of various pro-inflammatory genes; (2) indirectly reducing secretion of proinflammatory cytokines such as tumour necrosis factor-alpha (TNF-α) and interleukin-1-β (IL-1β), which remain a high level in PLWH; (3) inhibiting mTOR, through an AMPK-dependent mechanism, reducing CD4 T cell activation, in turn reducing inflammation; (4) indirectly blocking mTOR signalling by inhibiting Rag GTPases [96,97,98].
Conclusions
Gut dysbiosis has been associated with DM2- and HIV-related gut permeability, microbial translocation and inflammation. Metformin has been shown to modulate gut microbiota composition in diabetic and non-diabetic people, in association with reduction of gut damage and inflammation. However, the efficacy and safety of metformin to control inflammation and reduce risk of inflammatory comorbidities in non-diabetic PLWH are still unknown. Direct evidence is needed to verify and endorse the beneficial effects of metformin as a possible modulator of HIV-related inflammation. Following our pilot study, larger randomized placebo-controlled studies will be needed to assess the independent effect of metformin on gut dysbiosis and inflammation in non-diabetic PLWH. Collaborative effort encompassing microbiology, clinical care, epidemiology and artificial intelligence will define the dose and duration to obtain the optimal benefit of metformin as an immune modulator in ART-treated PLWH.
Availability of data and materials
Not applicable.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- APC:
-
Antigen presenting cell
- ART:
-
Antiretroviral therapy
- cAMP:
-
Cyclic adenosine monophosphate
- EGFR-TKI:
-
Epidermal growth factor receptor-tyrosine kinase inhibitor
- FMT:
-
Fecal microbiota transplantation
- GI:
-
Gastrointestinal
- HFD:
-
High-fat diet
- IDO-1:
-
Indoleamine-2,3-dioxygenase-1
- ICAM-1:
-
Intracellular adhesion molecule-1
- LPS:
-
Lipopolysaccharide
- MGWAS:
-
Metagenome-wide association study
- NAFLD:
-
Nonalcoholic fatty liver disease
- PLWH:
-
People living with HIV
- PKA:
-
Protein kinase A
- Tregs:
-
Regulatory T cells
- SCFA:
-
Short chain fatty acid
- SIV:
-
Simian immunodeficiency virus
- TLR-4:
-
Toll-like receptor
- TMA:
-
Trimethylamine
- TMAO:
-
Trimethylamine N-oxide
- IL-1β:
-
Tumour necrosis factor-alpha (TNF-α) and interleukin-1-β
- DM2:
-
Type 2 diabetes mellitus
References
D’Angelo C, Reale M, Costantini E. Microbiota and probiotics in health and HIV infection. Nutrients. 2017;9:615.
Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53:1–4.
Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3:858–76.
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE. 2010;5:e9085.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Choi J, Hur TY, Hong Y. Influence of altered gut microbiota composition on aging and aging-related diseases. J Lifestyle Med. 2018;8:1–7.
Vujkovic-Cvijin I, Somsouk M. HIV and the gut microbiota: composition, consequences, and avenues for amelioration. Curr HIV/AIDS Rep. 2019;16:204–13.
Iglay K, Hannachi H, Joseph Howie P, Xu J, Li X, Engel SS, et al. Prevalence and co-prevalence of comorbidities among patients with type 2 diabetes mellitus. Curr Med Res Opin. 2016;32:1243–52.
Gallant J, Hsue PY, Shreay S, Meyer N. Comorbidities among us patients with prevalent HIV infection-a trend analysis. J Infect Dis. 2017;216:1525–33.
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71.
Zicari S, Sessa L, Cotugno N, Ruggiero A, Morrocchi E, Concato C, et al. Immune activation, inflammation, and non-AIDS co-morbidities in HIV-infected patients under long-term ART. Viruses. 2019;11:200.
Ramendra R, Isnard S, Lin J, Fombuena B, Ouyang J, Mehraj V, et al. CMV seropositivity is associated with increased microbial translocation in people living with HIV and uninfected controls. Clin Infect Dis. 2019. https://doi.org/10.1093/cid/ciz1001.
Hsu DC, Sereti I. Serious non-AIDS events: therapeutic targets of immune activation and chronic inflammation in HIV infection. Drugs. 2016;76:533–49.
Roumie CL, Chipman J, Min JY, Hackstadt AJ, Hung AM, Greevy RA Jr, et al. Association of treatment with metformin vs sulfonylurea with major adverse cardiovascular events among patients with diabetes and reduced kidney function. JAMA. 2019;322:1167–77.
Roumie CL, Greevy RA, Grijalva CG, Hung AM, Liu X, Murff HJ, et al. Association between intensification of metformin treatment with insulin vs sulfonylureas and cardiovascular events and all-cause mortality among patients with diabetes. JAMA. 2014;311:2288–96.
Pollak M. The effects of metformin on gut microbiota and the immune system as research frontiers. Diabetologia. 2017;60:1662–7.
Pryor R, Norvaisas P, Marinos G, Best L, Thingholm LB, Quintaneiro LM, et al. Host-microbe-drug-nutrient screen identifies bacterial effectors of metformin therapy. Cell. 2019;178(1299–312):e29.
Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192.
Victor VM, Rovira-Llopis S, Banuls C, Diaz-Morales N, Lopez-Domenech S, Escribano-Lopez I, et al. Metformin modulates human leukocyte/endothelial cell interactions and proinflammatory cytokines in polycystic ovary syndrome patients. Atherosclerosis. 2015;242:167–73.
Arrieta O, Barron F, Padilla MS, Aviles-Salas A, Ramirez-Tirado LA, Arguelles Jimenez MJ, et al. Effect of metformin plus tyrosine kinase inhibitors compared with tyrosine kinase inhibitors alone in patients with epidermal growth factor receptor-mutated lung adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5:e192553.
Klil-Drori AJ, Azoulay L, Pollak MN. Cancer, obesity, diabetes, and antidiabetic drugs: is the fog clearing? Nat Rev Clin Oncol. 2017;14:85–99.
Estrada V, Gonzalez N. Gut microbiota in diabetes and HIV: inflammation is the link. EBioMedicine. 2018;38:17–8.
Hoel H, Hove-Skovsgaard M, Hov JR, Gaardbo JC, Holm K, Kummen M, et al. Impact of HIV and type 2 diabetes on gut microbiota diversity, tryptophan catabolism and endothelial dysfunction. Sci Rep. 2018;8:6725.
Moon JY, Zolnik CP, Wang Z, Qiu Y, Usyk M, Wang T, et al. Gut microbiota and plasma metabolites associated with diabetes in women with, or at high risk for HIV infection. EBioMedicine. 2018;37:392–400.
Gurung M, Li Z, You H, Rodrigues R, Jump DB, Morgun A, et al. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine. 2020;51:102590.
Vazquez-Castellanos JF, Serrano-Villar S, Jimenez-Hernandez N, Soto Del Rio MD, Gayo S, Rojo D, et al. Interplay between gut microbiota metabolism and inflammation in HIV infection. ISME J. 2018;12:1964–76.
Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–6.
Mardinoglu A, Boren J, Smith U. Confounding effects of metformin on the human gut microbiome in type 2 diabetes. Cell Metab. 2016;23:10–2.
Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Manneras-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23:850–8.
Rosario D, Benfeitas R, Bidkhori G, Zhang C, Uhlen M, Shoaie S, et al. Understanding the representative gut microbiota dysbiosis in metformin-treated type 2 diabetes patients using genome-scale metabolic modeling. Front Physiol. 2018;9:775.
Xue Y, Zhang H, Sun X, Zhu MJ. Metformin improves ileal epithelial barrier function in interleukin-10 deficient mice. PLoS ONE. 2016;11:e0168670.
Koh SJ, Kim JM, Kim IK, Ko SH, Kim JS. Anti-inflammatory mechanism of metformin and its effects in intestinal inflammation and colitis-associated colon cancer. J Gastroenterol Hepatol. 2014;29:502–10.
Jin Q, Cheng J, Liu Y, Wu J, Wang X, Wei S, et al. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav Immun. 2014;40:131–42.
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–81.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60.
Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in european women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103.
Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut. 2016;65:426–36.
Cani PD. Human gut microbiome: hopes, threats and promises. Gut. 2018;67:1716–25.
Saad MJ, Santos A, Prada PO. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiology. 2016;31:283–93.
Liang H, Hussey SE, Sanchez-Avila A, Tantiwong P, Musi N. Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS ONE. 2013;8:e63983.
Adachi K, Sugiyama T, Yamaguchi Y, Tamura Y, Izawa S, Hijikata Y, et al. Gut microbiota disorders cause type 2 diabetes mellitus and homeostatic disturbances in gut-related metabolism in Japanese subjects. J Clin Biochem Nutr. 2019;64:231–8.
Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119.
Tedelind S, Westberg F, Kjerrulf M, Vidal A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J Gastroenterol. 2007;13:2826–32.
Wang HB, Wang PY, Wang X, Wan YL, Liu YC. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein claudin-1 transcription. Dig Dis Sci. 2012;57:3126–35.
Kootte RS, Levin E, Salojarvi J, Smits LP, Hartstra AV, Udayappan SD, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 2017;26(611–9):e6.
Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143(913–6):e7.
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. 2013;110:9066–71.
Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, et al. Supplementation with akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. 2019;25:1096–103.
Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat Rev Endocrinol. 2019;15:569–89.
Bailey CJ, Wilcock C, Scarpello JH. Metformin and the intestine. Diabetologia. 2008;51:1552–3.
Sum CF, Webster JM, Johnson AB, Catalano C, Cooper BG, Taylor R. The effect of intravenous metformin on glucose metabolism during hyperglycaemia in type 2 diabetes. Diab Med. 1992;9:61–5.
Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, et al. An increase in the Akkermansia spp. Population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63:727–35.
de la Cuesta-Zuluaga J, Mueller NT, Corrales-Agudelo V, Velasquez-Mejia EP, Carmona JA, Abad JM, et al. Metformin is associated with higher relative abundance of mucin-degrading Akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diab Care. 2017;40:54–62.
Anhe FF, Nachbar RT, Varin TV, Trottier J, Dudonne S, Le Barz M, et al. Treatment with camu camu (Myrciaria dubia) prevents obesity by altering the gut microbiota and increasing energy expenditure in diet-induced obese mice. Gut. 2018;68:453–64.
Duan W, Ding Y, Yu X, Ma D, Yang B, Li Y, et al. Metformin mitigates autoimmune insulitis by inhibiting Th1 and Th17 responses while promoting treg production. Am J Transl Res. 2019;11:2393–402.
Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK, Shin DY, et al. Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance. PLoS ONE. 2015;10:e0135858.
Pandiyan P, Bhaskaran N, Zou M, Schneider E, Jayaraman S, Huehn J. Microbiome dependent regulation of Tregs and Th17 cells in mucosa. Front Immunol. 2019;10:426.
Bhaskaran N, Quigley C, Paw C, Butala S, Schneider E, Pandiyan P. Role of short chain fatty acids in controlling Tregs and immunopathology during mucosal infection. Front Microbiol. 2018;9:1995.
Elbere I, Kalnina I, Silamikelis I, Konrade I, Zaharenko L, Sekace K, et al. Association of metformin administration with gut microbiome dysbiosis in healthy volunteers. PLoS ONE. 2018;13:e0204317.
Bryrup T, Thomsen CW, Kern T, Allin KH, Brandslund I, Jorgensen NR, et al. Metformin-induced changes of the gut microbiota in healthy young men: results of a non-blinded, one-armed intervention study. Diabetologia. 2019;62:1024–35.
Wang GS, Hoyte C. Review of biguanide (metformin) toxicity. J Intensive Care Med. 2018. https://doi.org/10.1177/0885066618793385.
Hughes CA, Taylor GD. Metformin in an HIV-infected patient with protease inhibitor-induced diabetic ketoacidosis. Annals Pharmacother. 2001;35:877–80.
Ortiz-Brizuela E, Perez-Patrigeon S, Recillas-Gispert C, Gomez-Perez FJ. Lactic acidosis complicating metformin and non-nucleoside reverse transcriptase inhibitor combination therapy: a smoldering threat in the post-HAART era. Rev Invest Clin. 2015;67:273–4.
Worth L, Elliott J, Anderson J, Sasadeusz J, Street A, Lewin S. A cautionary tale: fatal lactic acidosis complicating nucleoside analogue and metformin therapy. Clin Infect Dis. 2003;37:315–6.
Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24:1919–29.
Li F, Dong YZ, Zhang D, Zhang XM, Lin ZJ, Zhang B. Molecular mechanisms involved in drug-induced liver injury caused by urate-lowering chinese herbs: a network pharmacology study and biology experiments. PLoS ONE. 2019;14:e0216948.
Mehandru S, Tenner-Racz K, Racz P, Markowitz M. The gastrointestinal tract is critical to the pathogenesis of acute HIV-1 infection. J Allergy Clin Immunol. 2005;116:419–22.
Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008;1:23–30.
Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010;6:e1001052.
Crakes KR, Santos Rocha C, Grishina I, Hirao LA, Napoli E, Gaulke CA, et al. PPARα-targeted mitochondrial bioenergetics mediate repair of intestinal barriers at the host-microbe intersection during SIV infection. Proc Natl Acad Sci USA. 2019;116:24819–29.
Meng J, Banerjee S, Zhang L, Sindberg G, Moidunny S, Li B, et al. Opioids impair intestinal epithelial repair in HIV-infected humanized mice. Front Immunol. 2019;10:2999.
Mehraj V, Ramendra R, Isnard S, Dupuy FP, Ponte R, Chen J, et al. Circulating (1→3)-β-d-glucan is associated with immune activation during human immunodeficiency virus infection. Clin Infect Dis. 2020;70:232–41.
Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5:193ra91.
Rocafort M, Noguera-Julian M, Rivera J, Pastor L, Guillen Y, Langhorst J, et al. Evolution of the gut microbiome following acute HIV-1 infection. Microbiome. 2019;7:73.
Glavan TW, Gaulke CA, Hirao LA, Sankaran-Walters S, Dandekar S. SIV-infection-driven changes of pattern recognition receptor expression in mesenteric lymph nodes and gut microbiota dysbiosis. J Med Primatol. 2015;44:241–52.
Klatt NR, Canary LA, Sun X, Vinton CL, Funderburg NT, Morcock DR, et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. J Clin Invest. 2013;123:903–7.
Jenabian MA, Patel M, Kema I, Kanagaratham C, Radzioch D, Thebault P, et al. Distinct tryptophan catabolism and Th17/Treg balance in HIV progressors and elite controllers. PLoS ONE. 2013;8:e78146.
Routy JP, Mehraj V, Vyboh K, Cao W, Kema I, Jenabian MA. Clinical relevance of kynurenine pathway in HIV/AIDS: an immune checkpoint at the crossroads of metabolism and inflammation. AIDS Rev. 2015;17:96–106.
Chen J, Xun J, Yang J, Ji Y, Liu L, Qi T, et al. Plasma indoleamine 2,3-dioxygenase activity is associated with the size of the human immunodeficiency virus reservoir in patients receiving antiretroviral therapy. Clin Infect Dis. 2019;68:1274–81.
Gonzalez-Hernandez LA, Ruiz-Briseno MDR, Sanchez-Reyes K, Alvarez-Zavala M, Vega-Magana N, Lopez-Iniguez A, et al. Alterations in bacterial communities, SCFA and biomarkers in an elderly HIV-positive and HIV-negative population in western Mexico. BMC Infect Dis. 2019;19:234.
Dillon SM, Kibbie J, Lee EJ, Guo K, Santiago ML, Austin GL, et al. Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS. 2017;31:511–21.
Haissman JM, Knudsen A, Hoel H, Kjaer A, Kristoffersen US, Berge RK, et al. Microbiota-dependent marker tmao is elevated in silent ischemia but is not associated with first-time myocardial infarction in HIV infection. J Acquir Immune Defic Syndr. 2016;71:130–6.
Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–84.
Polyzos KA, Ketelhuth DF. The role of the kynurenine pathway of tryptophan metabolism in cardiovascular disease. Hamostaseologie. 2015;35:128–36.
Haissman JM, Haugaard AK, Ostrowski SR, Berge RK, Hov JR, Troseid M, et al. Microbiota-dependent metabolite and cardiovascular disease marker trimethylamine-n-oxide (TMAO) is associated with monocyte activation but not platelet function in untreated HIV infection. BMC Infect Dis. 2017;17:445.
Kehrmann J, Menzel J, Saeedghalati M, Obeid R, Schulze C, Holzendorf V, et al. Gut microbiota in human immunodeficiency virus-infected individuals linked to coronary heart disease. J Infect Dis. 2019;219:497–508.
Hadigan C, Corcoran C, Basgoz N, Davis B, Sax P, Grinspoon S. Metformin in the treatment of HIV lipodystrophy syndrome: a randomized controlled trial. JAMA. 2000;284:472–7.
Driscoll SD, Meininger GE, Lareau MT, Dolan SE, Killilea KM, Hadigan CM, et al. Effects of exercise training and metformin on body composition and cardiovascular indices in HIV-infected patients. AIDS. 2004;18:465–73.
Mulligan K, Yang Y, Wininger DA, Koletar SL, Parker RA, Alston-Smith BL, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS. 2007;21:47–57.
Fitch K, Abbara S, Lee H, Stavrou E, Sacks R, Michel T, et al. Effects of lifestyle modification and metformin on atherosclerotic indices among HIV-infected patients with the metabolic syndrome. AIDS. 2012;26:587–97.
Shikuma C, Chew GM, Kohorn L, Souza SA, Chow D, SahBandar IN, et al. Metformin reduces CD4 T-cell exhaustion in HIV-infected adults on suppressive antiretroviral therapy. AIDS Res Hum Retroviruses. 2019. https://doi.org/10.1089/aid.2019.0078.
Routy JP, Isnard S, Mehraj V, Ostrowski M, Chomont N, Ancuta P, et al. Effect of metformin on the size of the HIV reservoir in non-diabetic ART-treated individuals: single-arm non-randomised lilac pilot study protocol. BMJ Open. 2019;9:e028444.
Lee H, Ko G. Effect of metformin on metabolic improvement and gut microbiota. Appl Environ Microbiol. 2014;80:5935–43.
Reunanen J, Kainulainen V, Huuskonen L, Ottman N, Belzer C, Huhtinen H, et al. Akkermansia muciniphila adheres to enterocytes and strengthens the integrity of the epithelial cell layer. Appl Environ Microbiol. 2015;81:3655–62.
Muzik O, Burghardt P, Yi Z, Kumar A, Seyoum B. Successful metformin treatment of insulin resistance is associated with down-regulation of the kynurenine pathway. Biochem Biophys Res Commun. 2017;488:29–32.
Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits MTORC1 in a Rag GTPase-dependent manner. Cell Metab. 2010;11:390–401.
Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. Mtor regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–12.
Moyo D, Tanthuma G, Cary MS, Mushisha O, Kwadiba G, Chikuse F, et al. Cohort study of diabetes in HIV-infected adult patients: evaluating the effect of diabetes mellitus on immune reconstitution. Diab Res Clin Pract. 2014;103:e34–6.
Acknowledgements
We are highly grateful to Angie Massicotte, Josée Girouard for coordination and assistance.
Funding
Our work was funded by the Fonds de la Recherche Québec-Santé (FRQ-S): Réseau SIDA/Maladies infectieuses and Thérapie cellulaire; the Canadian Institutes of Health Research (CIHR; Grants HOP 103230 and PTJ 166049); the Vaccines & Immunotherapies Core of the CIHR Canadian HIV Trials Network (CTN; Grant CTN 257 and CTN PT027); the Canadian Foundation for AIDS Research (CANFAR; Grant 02-512); CIHR- funded Canadian HIV Cure Enterprise (CanCURE) Team Grant HB2-164064; and the Chinese National Science and Technology Major Project (No. 2018ZX10302104). André Marette holds a CIHR/Pfizer research Chair in the pathogenesis of insulin resistance and cardiovascular complications and is supported by a CIHR Foundation Grant.
Author information
Authors and Affiliations
Contributions
JO wrote the first draft of the manuscript. SI, JL, BF, AM and BR provided critical revision of the manuscript. YC and JPR conceived and designed the review. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ouyang, J., Isnard, S., Lin, J. et al. Metformin effect on gut microbiota: insights for HIV-related inflammation. AIDS Res Ther 17, 10 (2020). https://doi.org/10.1186/s12981-020-00267-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12981-020-00267-2