
Abstract
Background
Usual interstitial pneumonia (UIP) is a fatal disease that is associated with poor prognosis and survival. Several growth factors such as IGFs (insulin-like growth factors) and IGFBPs (insulin-like growth factor binding proteins) seem to take part to this pathogenesis.
Pirfenidone is an immunosuppressant drug that is thought to have anti-inflammatory and anti-fibrotic effects both in vitro and in vivo.
Objective
To assess IGFBP1 and IGFBP2 as non-invasive biomarkers for prediction and outcomes of UIP clinical activity and therapeutic response to the anti-fibrotic pirfenidone.
Results
Serum levels of IGFBP1 and IGFBP2 were significantly higher in the UIP group than in the healthy subjects (p ≤ 0.005). After 6 months therapy, UIP patients were divided into 2 groups according to improvement in MRC dyspnea grading into clinically improved and non-improved groups. 6MWT and SPaO2 were significantly improved in the clinically improved group compared to the non-improved one with no differences as regards other parameters (p < 0.0001). Both IGFBP1 and IGFBP2 were significantly decreased in serum while only IGFBP2 was decreased in BAL of all UIP after completing 12 months therapy.
Conclusion
IGFBP1 and IGFBP2 were increased in active UIP patients and reduced after 12 months anti-fibrosing therapy. IGFBPs may be promising biomarkers and predictors of response to therapy in UIP.
Similar content being viewed by others


Background
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive fibrosing interstitial pneumonia with inevitable loss of lung function. Internationally recognized guidelines recommend the multidisciplinary evaluation of clinical, radiologic, and pathologic disease features in the diagnosis and management of ILD [1]. Radiological usual interstitial pneumonia (UIP) is characterized on high-resolution computed tomography (HRCT) by the presence of reticular opacities, often associated with traction bronchiectasis, typically in a basal, subpleural, and patchy distribution [2].
Histopathologic UIP consists of a combination of fibrotic areas with scarring and honeycomb change alternate with areas of less affected or even normal lung parenchyma [3]. The patchy interstitial fibrosis, collagen deposition, and architectural distortion characteristic of the UIP pathologic pattern, evident by surgical pathology or at the macroscale by HRCT, are generally associated with poor prognosis and survival [4].
The insulin-like growth factor (IGF) system comprises the two primary ligands, namely IGF1 and IGF2; six high-affinity fully characterized IGF-binding proteins (IGFBP1–IGFBP6); and the IGF receptors. IGF1 and IGF2 are anabolic peptides with extensive structural and functional homology with insulin. IGF1 and IGF2 affect local and systemic responses through autocrine, paracrine, and endocrine mechanisms [5].
During human fetal lung development, IGF1 and 2 transcripts are localized to cells of mesenchymal origin (pleura, interlobular septa, perivascular fibroblasts) while immunocytochemistry identifies IGF peptides in association with airway epithelium. These findings suggest that IGFs act locally in the lung in an autocrine or paracrine manner [6].
Regulation of IGF activity in the lung depends on the expression of the IGFs and IGF receptors and the modulation of IGF activity by specific IGF-binding proteins (IGFBPs). IGFBP2 is a member of a family of six insulin-like growth factor binding proteins, which has recently been identified in IPF [5].
Many clinical trials with anti-fibrotic drugs for IPF were available in the last decade, including bosentan, imatinib, and interferon (IFN)-1b. Pirfenidone, an orally administered pyridine, demonstrated combined anti-inflammatory, anti-oxidant, and anti-fibrotic actions both in vitro and in animal models of pulmonary fibrosis, consisting in the regulation of the expression of TGF-β and inhibition of fibroblast and collagen synthesis [7].
The revision of the 2011 guidelines released a conditional recommendation for the use of nintedanib and pirfenidone for the treatment of IPF. In many countries, pirfenidone and nintedanib are approved with reimbursement for the treatment of patients with mild-to-moderate disease, who therefore present with well-defined impairments of lung function [8].
Aim of the study
We aimed to assess IGFBP1 and IGFBP2 as non-invasive biomarkers for prediction and outcomes of UIP clinical activity and therapeutic response to the anti-fibrotic pirfenidone.
Methods
Study data were collected within the period from March 2016 to February 2018. According to the 2011 joint statement by the American Thoracic Society (ATS), European Respiratory Society (ERS), Latin American Thoracic Association (ALAT), and Japanese Respiratory Society (JRS) [9], the diagnosis of IPF can be secured by the presence of a UIP pattern on high-resolution computed tomography (HRCT).
Inclusion criteria
Patients with IPF and aged 40–80 years with diagnostic criteria conforming to the current guideline based on ATS/ERS guidelines and the dose of pirfenidone ≥ 1800 mg daily were included in this analysis [9].
Exclusion criteria
Patients who had other causes of UIP except IPF (such as collagen vascular diseases and history of exposure to drugs, radiation, and asbestosis) and incomplete data collection were excluded.
Subjects
Prior to initiating pirfenidone therapy, all patients were subjected to the following:
-
1.
Thorough history taking and careful clinical examination including age, sex, body mass index (BMI), smoking habits, and associated co-morbidity.
-
2.
Laboratory tests including complete blood count (CBC), and renal and liver panels were performed before administration of the compound during the period of diagnosis, as well as 6 and 12 months post-treatment initiation.
-
3.
Pulmonary function testing using computerized spirometry with a SensorMedics Vmax 229 (SensorMedics, Yorba Linda, CA, USA).
-
4.
Chest X-ray and HRCT (high-resolution CT) chest.
-
5.
Arterial blood gasses.
-
6.
O2 saturation by pulse oximetry in follow-up of the patients and 12 months after medication.
-
7.
6MWT (6 min walking test) for all patients before and 12 months after medication.
-
8.
Bronchoscopy and bronchoalveolar lavage (BAL): for all patients before and 12 months after medication.
-
9.
Measure IGFBP1 and IGFBP2 in blood and BAL by ELISA for all patients before and 12 months after medication.
Pirfenidone treatment
Pirfenidone is available as 267 mg capsules. Initial dosing is one capsule three times daily with meals on days 1 through 7. On days 8 through 14, the dose is advanced to two capsules three times daily with meals. The daily dose should be titrated to the full dosage of nine capsules per day (2403 mg/day) or three capsules three times a day with meals onward starting on day 15. Consider temporary dosage reduction, treatment interruption, or discontinuation for management of adverse reactions. For those patients who have treatment interruption of 14 or more days, therapy should be reinitiated by undergoing the initial 2-week titration regimen to the maintenance dosage [10].
Patients were informed for known adverse events of pirfenidone and were instructed to avoid exposure to sunlight and alcohol consumption.
Dyspnea was assessed using the MRC dyspnea scale at presentation and after completed pirfenidone therapy.
Statistical analysis
Analysis using SPSS version 12 was performed with respect to the main study aim. Descriptive characteristics for participants are expressed as means and standard deviation (SD) for continuous variables, and number and percent for categorical variables. We used the independent sample test to show the significant difference between the continuous variables and chi-square test for the categorical variables. The level of significance was accepted at p ≤ 0.05.
Results
Subject demographic and functional characteristics
This study was carried out on 23 UIP patients (16 males and 7 females, with a mean age of 65.09 ± 6.01 years) and 25 healthy patients (15 males and 10 females, with a mean age of 62.87 ± 3.99 years) with no significant statistical difference between both groups as regards age, sex, or smoking habits.
Serum growth factors before start therapy
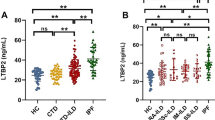
There were significant increases in serum IGFBP1 and IGFBP2 of the UIP group compared to the healthy one (p ≤ 0.005) (Table 1).
The UIP patients were divided into 2 groups according to clinical improvement in MRC dyspnea scale into improved or non-improved after completing the pirfenidone course. Comparing both groups, there were significant improvements in 6MWT and SPaO2 in the clinically improved group compared to the non-improved one with no differences as regards other parameters (Table 2).
Serum and BALF growth factors after completing 12 months therapy
On comparing all the UIP patients before and after completing their pirfenidone course, there were significant decreases in serum levels of both IGFBP1and IGFBP2 with significant improvement in the SPaO2 and 6MWT after 12 months therapy. By contrast, there was no significant difference between the groups regarding the BALF levels of IGFBP1 (Table 3, Fig. 1).

Mean and SD of biomarkers before and after treatment
Relationship between growth factors and other parameters
Moreover, our study showed a direct relationship between circulating levels of IGFBP2 and BALF levels, while we did not find any correlation between IGFBP1 in serum and BALF or between both growth factors and any other biomarkers assessed in our study (Table 4).
Of great interest, there was an inverse relationship between MRC dyspnea score and 6MWT and also with SPaO2 in UIP patients after completing 12 months treatment (Tables 4 and 5).
Discussion
Biomarkers are highly needed in IPF as tools for differential diagnostic, predictor of the progression of the disease and treatment response. Specifically in IPF, early diagnosis is important to reduce as much as possible the disease progression [11].
A key initiating event hypothesized in the pathobiology of IPF is alveolar epithelial dysfunction leading to innate immune cell activation, dysregulated epithelial-mesenchymal communication, myofibroblast differentiation and proliferation, and excessive extracellular matrix deposition [12].
Many biomarkers have been studied in BALF and in serum as potential diagnostic or prognostic tools. However, the predictors’ value of these new biomarkers still needs further studies.
IGFBP1 and IGFBP2 are members of a highly conserved family of six insulin-like growth factor (IGF) binding proteins; IGFBPs are described to be involved in cell proliferation and differentiation [13]. Therefore, we focused on measuring both biomarkers in serum and BALF of UIP patients. To the best of our knowledge, we are the first to study both IGFBP1 and IGFBP2 levels in both serum and BAL of patients who suffer from usual interstitial pneumonia (UIP) and to compare their results.
Although previous studies such as Chadelat et al. [14] and Mouhieddine et al. [15] were focused on IGFBP2 in fibrosis which showed an increase of IGFBP2 in the bronchoalveolar lavage and in the lung tissue of patients with interstitial lung disease (ILD) in vivo and in vitro, none of these studies focused on the usual interstitial pneumonia pattern.
Our study showed for the first time that UIP featured a marked increase in serum IGFBP1 and IGFBP2 and in BALF. Even the serum levels remained higher than those measured in healthy subjects. In addition, IGFBP1 and IGFBP2 were attenuated in those patients completing the 12-month therapy with anti-fibrotic treatment. Those results were in concordance with that reported by Guiot et al. [16] who showed a marked increase in serum IGFBP1 and IGFBP2, with decreased IGFBP2 levels in those patients receiving anti-fibrotic treatment.
Of great interest, the present study also found that IGFBP2 levels showed significant decrease in both serum and BALF of UIP patients after completing the 12-month therapy, thus supporting the idea that IGFBP2 can bind to the lung extracellular matrix [17] and it can favor the IGF activity by increasing its local availability, which results in an increase of cellular response to IGF [13]. Alternatively, we cannot rule out the fact that high IGFBP2 in IPF may actually reflect a protective feedback mechanism to limit the disease progression by neutralizing IGFs [18].
As regards the clinical effect of the anti-fibrotic therapy and its effect on the quality of life of the UIP patients and its relations to the measured IGFBP1 and IGFBP2 in both serum and BALF, we found a marked improvement in MRC dyspnea scale with significant improvement in 6MWT and SPaO2 and decrease in both IGFBP1 and IGFBP2 serum levels of UIP patients after completing the 12-month therapy with pirfenidone that may increase the awareness of using those biomarkers as predictors for the disease progression as well as monitoring anti-fibrotic therapy. It also supports the idea that IGFBP2 may play a role in the fibrotic process in the lung. As our patients were not treated with corticoids, we can here discard any possible impact of corticosteroids on IGFBP levels.
Conclusion
Our findings suggest that IGFBP1 and IGFBP2 biomarkers may have the potential to predict the progression of patients with UIP and could also be used to monitor the response to anti-fibrotic therapy. Further, longitudinal studies are needed to evaluate their usefulness as biomarkers in UIP.
Limitations
One of the limitations of our study is the reduced number of UIP patients who agree to complete the study to the end. As we have chosen IGFBP1 and IGFBP2 biomarkers’ detection in serum and BALF of UIP patients according to our thinking and their potential usefulness, the lack of longitudinal studies for those biomarkers constitutes another limitation of this study.
We believe that further longitudinal multicenter studies are highly needed to evaluate the clinical impact of those biomarkers in a single or multivariate analysis as diagnostic, prognostic, and monitoring tools.
Availability of data and materials
All data of the patients is available on computerized patient file in the Recording Department of King Fahd Hospital, Almadina Almonoura, Kingdom of Saudi Arabia. www.KFHM-moh.gov.sa.
Abbreviations
- IGFBP1:
-
Insulin-like growth factor binding protein 1
- UIP:
-
Usual interstitial pneumonia
- IGFs:
-
Insulin-like growth factors
- IGFBPs:
-
Insulin-like growth factor binding proteins
- HRCT:
-
High-resolution computed tomography
- 6MWT:
-
Six minute walking test
- BALF:
-
Bronchoalveolar fluid
References
Spagnolo P, Sverzellati N, Rossi G et al (2015) Idiopathic pulmonary fibrosis: an update. Ann Med 47:15–27
Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P (2014) Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev 23:308–319 Erratum in: Eur Respir Rev 2014; 23:537
Johannson KA, Ley B, Collard HR (2015) Models of disease behavior in idiopathic pulmonary fibrosis. BMC Med 13:165
Dhooria S, Sehgal IS, Aggarwal AN, Behera D, Agarwal R (2016) Diagnostic yield and safety of cryoprobe transbronchial lung biopsy in diffuse parenchymal lung diseases: systematic review and meta-analysis. Respir Care 61:700–712
Hill DJ, Clemmons DR, Wilson S, Han VK, Strain AJ, Milner RD (1989) Immunological distribution of one form of insulin-like growth factor (IGF)-binding protein and IGF peptides in human fetal tissues. J Mol Endocrinol 2:31–38
Price GJ, Berka JL, Edmondson SR, Werther GA, Bach LA (1995) Localization of mRNAs for insulin-like growth factor binding proteins 1 to 6 in rat kidney. Kidney Int 48:402–411. https://doi.org/10.1038/ki.1995.308
Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L (2018) Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res 19:32. https://doi.org/10.1186/s12931-018-0730-2
Collard HR, Cunningham W, Homma S, et al.; American Thoracic Society; European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association (2018) An official ATS/ERS/JRS/ALAT clinical practice Guideline. Am J Respir Crit Care Med 198 (e30-e43).
Raghu G, Remy-Jardin M, Myers JF, Richeldi L, Ryerson CJ, Lederer DJ et al (2018) Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 198:e44–e68
Esbriet® (pirfenidone) [package insert]. South San Francisco: Genentech USA, Inc.; 2016.
Prasad R, Gupta N, Singh A, Gupta P (2015) Diagnosis of idiopathic pulmonary fibrosis: current issues. Intractable Rare Dis Res 4(2):65–69
Heukels P, van Hulst JAC, van Nimwegen M, Boorsma CE, Melgert BN, van den Toorn LM et al (2018) Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir Res 19(1):90
Kelley KM, Oh Y, Gargosky SE, Gucev Z, Matsumoto T, Hwa V et al (1996) Insulin-like growth factor-binding proteins (IGFBPs) and their regulatory dynamics. Int J Biochem Cell Biol 28(6):619–637
Chadelat K, Boule M, Corroyer S, Fauroux B, Delaisi B, Tournier G et al (1998) Expression of insulin-like growth factors and their binding proteins by bronchoalveolar cells from children with and without interstitial lung disease. Eur Respir J 11(6):1329–1336
Mouhieddine OB, Cazals V, Maitre B, Le Bouc Y, Chadelat K, Clement A (1994) Insulin-like growth factor-II (IGF-II), type 2 IGF receptor, and IGF-binding protein-2 gene expression in rat lung alveolar epithelial cells: relation to proliferation. Endocrinology 135(1):83–91
Guiot J, Bondue B, Henket M, Corhay JL, Louis R (2016) Raised serum levels of IGFBP-1 and IGFBP-2 in idiopathic pulmonary brosis. BMC Pulm Med 16(1):86
Russo VC, Azar WJ, Yau SW, Sabin MA, Werther GA (2014) IGFBP-2: the dark horse in metabolism and cancer. Cytokine Growth Factor Rev 26(3):329–346
Guiot J, Henket M, Corhay JL, Moermans C, Louis R (2017) Sputum biomarkers in IPF: evidence for raised gene expression and protein level of IGFBP-2, IL-8 and MMP-7. PLoS One 12(2):e0171344
Acknowledgements
Thanks to all the co-authors for their help, support, and collaboration to the redaction of this review.
Funding
WHO, up to date, Google search
Author information
Authors and Affiliations
Contributions
HM: responsible for data collection and writing the manuscript. AF: responsible for writing the manuscript. SE: responsible for data collection and writing the manuscript. ZM: responsible for data collection and statistical analysis. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
King Fahd Hospital-Medina, Kingdom of Saudi Arabia. Number (59) at May 2018. The written consent form of the patients was taken and was available when needed.
Consent for publication
Not applicable.
Competing interests
Not present in this section.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Habeb, M., Embarak, S., Fathy, A. et al. Non-invasive IGFBP1, IGFBP2 biomarkers as predictors and outcomes of usual interstitial pneumonia (UIP) therapeutic response. Egypt J Bronchol 15, 6 (2021). https://doi.org/10.1186/s43168-020-00050-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43168-020-00050-x