
Abstract
Background
Malignant rhabdoid tumor of the kidney is the most aggressive childhood renal tumor. A preoperative diagnosis is critical in order to correctly establish a therapeutic strategy and a full metastatic workup.
Case presentation
We report on a 3-month-old case with fever, diarrhea, and abdominal distension treated surgically with adjuvant chemotherapy. The diagnosis was confirmed postoperatively. Relapse was quick, and the child died 5 months after surgical resection.
Conclusions
Rhabdoid renal tumor in young age is associated with a high mortality rate even with invasive strategies. Case reports and research are critical for evaluating existing protocols and improving prognosis. Diverse clinical trials are being conducted in the hopes of improving the prognosis of rhabdoid renal tumors.
Similar content being viewed by others

Background
The malignant rhabdoid renal tumor is an extremely rare and aggressive childhood renal tumor. In children, it accounts for 0.9 to 2% of kidney tumors [1]. As it arises from primitive medullar cells and invades both the hilum and the collecting system, imagery can be characteristic [1]. A full metastatic workup with a specific therapeutic strategy could improve its outcomes if a correct preoperative diagnosis is made. However, it represents a low overall survival rate ranging between 22 and 2% [2]. Giving its rarity, case reports and in vitro studies are critical in improving its prognosis. Herein, we present a case of an aggressive renal tumor affecting a 3-month-old infant that resulted in poor outcomes.
Case presentation
A previously fit and healthy 3-month-old girl presented to an outside emergency department with a 2-day history of relentless fever of 39 °C, diarrhea, and abdominal distension. Upon examination, the infant was dehydrated and lethargic and had a palpable mass in the right hypochondria. Laboratory tests revealed low hemoglobin and hematocrit as well as an elevated C-reactive protein value at 280. There was no hematuria, and the rest of the tests was normal, particularly renal function and ionized calcium levels.
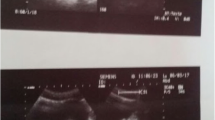
Abdominal ultrasound was initially performed revealing a 7 cm heterogeneous spherical mass arising from the right kidney with multiple hemorrhagic areas and fines calcifications and no concomitant adenomegaly (Fig. 1). Given the lack of necessary equipment in the outside hospital, she was subsequently referred to our Department of Pediatric Surgery in Children’s Hospital of Tunis which is a well-established institution in Tunisia with a multidisciplinary team.

Ultrasound picture: revealing a 7 cm heterogeneous spherical mass arising from the apical pole of the right kidney with multiple hemorrhagic areas and fine calcifications
An abdominal CT was performed revealing a 7 cm heterogeneous spherical mass arising from the apical pole of the right kidney with multiple hemorrhagic areas and fine calcifications with no concomitant adenomegaly. There were no subscapular fluid accumulations or vascular thrombosis. There was an important uretero-pyelo-calicielle dilation (Fig. 2).

A CT scan: revealing an important uretero-pyelo-calicielle dilation with fine calcifications
There was no sign of metastatic disease to the chest. At this point, a collaborative discussion between radiologists, pediatric oncologists, and pediatric surgeons took place, and two diagnoses were proposed: rhabdoid tumor or mesoblastic nephroma. Given her young age, it was decided not to begin with preoperative chemotherapy and instead start with a primary surgery.
Peroperatively, the tumor was 10 cm long and purplish-red in color. It had a solid-cystic consistency that had developed at the expense of the posterior and medial renal lip. It did not infiltrate the psoas, or adrenal gland, or the renal vein with multiple reactional adenomegaly. The patient underwent a radical primary ureteronephrectomy with regional lymphadenectomy.
The histological examination confirmed the diagnosis of a malignant rhabdoid tumor. It was composed of solid sheets showing an unusual proliferation of epithelioid cells with eosinophilic cytoplasm, single or multiple atypical nuclei, and easily identifiable multiple mitoses. The tumor was partially hemorrhagic and cystic with focal necrosis. Urothelium was not involved. It showed hilum and capsular invasion with no evidence of vascular or ganglion extension. The tumor was classified as a rhabdoid kidney malignant tumor at stage 3 according to COG’s staging of Wilms tumors. Postoperative extension workout included a brain magnetic resonance imaging and a bone scan and showed neither bone nor brain metastases. The course of postoperative treatment was chemotherapy and no radiation given her young age despite a negative histology evaluation. The patient was subsequently treated with vincristine, cisplatin, doxorubicin, and cyclophosphamide. Serial imaging detected a relapse with a local recurrence, cutaneous, and inferior vena cava invasion (Fig. 3). The patient was given intensive chemotherapy; however, she was later deceased 5 months after the surgery with cardiac failure.

A local recurrence: cutaneous bulging appearing 5 months postoperatively and revealing a local recurrence
Discussion
Renal tumors account for 7 to 8% of all pediatric malignancies. Wilms tumor with favorable histology represents 75% of new cancers. It has been one of modern medicine’s greatest achievements. Its overall survival rate had exceeded 90% in both Children Oncology Group (COG) trials and the International Society of Pediatric Oncology (SIOP) trials [2]. High-risk renal tumors such as anaplastic Wilms tumor, metastatic renal sarcoma, malignant rhabdoid tumor, and carcinoma make up the remaining 25% [1]. In 1978, the malignant rhabdoid renal tumor was first recognized as a sarcomatoid variety of Wilms tumor, but it was later characterized as a distinct entity [3]. It represents 2% of all childhood renal tumors according to the database of the International Incidence of Childhood Cancer study [4]. According to this epidemiological study published by 2020, the incidence of the renal rhabdoid tumor had increased, while incidence of other types had decreased which could indicate a butter detection of this tumor [4]. It denotes a diverse epidemiology, therapy, and prognostic variables which make it harder to distinguish. Development in the histopathology and cytogenetic studies has made this entity well-known by pathologists. It has been by far the most lethal pediatric tumor affecting primarily infants and young children. The majority of individuals are diagnosed within the first year of life, with a peak frequency occurring between the ages of 10 and 18 months. Age has a predictive significance in this type of tumors with worst prognosis between 0 and 5 months of age [5]. Our patient was a 3-month-old infant which is associated with a prognosis of 8.8% at 4 years [5]. This age is linked to concomitant central nervous system malignancies, which can lead to an early relapse [6]. Gender represents as well a prognostic factor [5,6,7]. Although there is no clear gender bias in the literature, there has been some evidence of a slight male predominance (1.5/1) [5,6,7]. Aside from the patient’s age at the time of diagnosis and gender, tumor stage and the presence or absence of central nervous system lesions are all prognostic factors [3]. Gross hematuria, hypercalcemia due to high serum parathormone levels, abdominal distension or pain, fussiness, and fever are all common symptoms at presentation [5,6,7]. Hematuria appears to be more frequently related with renal rhabdoid tumor patients than with Wilms tumor patients. An investigation conducted by Amar et al. of 50 patients had shown gross hematuria to be a presenting symptom in 59% of renal rhabdoid patients and only 18% of Wilms tumor patients [8]. However, severe hematuria is less frequent. In rare cases, embolization [9] and urgent nephrectomy [3] may be required. In our patient, hematuria was absent. On the other hand, fever, diarrhea, and stomach distension were the most concerning symptoms. Despite the fact that our patient had no alarming clinical symptoms at the time of presentation, the tumor was in an advanced stage. It was classified as a stage 3 malignant rhabdoid tumor. Renal rhabdoid tumors are staged according to the COG’s staging of Wilms tumors after surgical resection [5]. At the time of diagnosis, more than two-thirds of patients have advanced disease, including metastases to the lungs and brain [5]. Stages 3 and 4 have typically had a lower survival probability of 15% compared to 40% survival at lower stages [5].
The treatment is multimodal following the renal tumor guidelines according to the highest risk recommendations. The gold standard is a radical surgical resection with lymph node dissection, followed by chemotherapy. However, considering the tumor’s rarity, the appropriate treatment remains unknown. No center has gained enough clinical expertise to provide a standardized protocol to disease management. Although surgical scheduling varies, chemotherapeutic drugs such as vincristine, actinomycine D, doxorubicin, carboplatin, cyclophosphamide, and etoposide have been used often in conjunction [10].
No single chemotherapeutic drug or combination of agents has been proved to be the most effective in treating this malignancy [11]. Furthermore, after numerous trials and diverse protocols with intense regimens, overall survival remains the poorest of all aggressive kidney malignancies, with just 25% survival in SIOP and NWTS studies [12]. Recently, the Eu-RHAB registry protocol in Europe had permitted the use of high-dose chemotherapy and stem cell rescue in addition to the conventional aggressive protocol recommended [11].
It is critical to distinguish this tumor from other kidney cancers since it requires a different therapeutic approach, a different metastatic workup, and a different prognosis. It is still a challenge because it could be mistaken for a clear cell carcinoma on imaging [13]. They both start in the renal medulla and spread to the kidney, but the latter has extensive cystic regions with bone metastases [13]. Additional findings include a peripheral subcapsular fluid collection which is found in 70% of patients compared to 12% in other tumors [4]. There were no unique characteristics in our patient that confirmed the diagnosis. It was, nevertheless, highly suspected. In addition, extrarenal rhabdoid synchronous tumors or metastases are frequent [14]; thus, the diagnosis workup should include a chest X-ray and a thoracic computed tomography scan when a pulmonary lesion is suspected [15]. An analysis of 111 cases of rhabdoid tumors had discovered that 8% of the cases were extrarenal in origin [12]. Other typical sites include the central nervous system, soft tissue, and the liver [12]. In addition, metastases occur at an early stage, and they are rapidly progressive. They occupy especially the lungs, the liver, and the lymph nodes. In a study conducted by a collaborative trial of the Society of Pediatric Oncology and Hematology, SIOP had found a 12.3% rate of primary lung nodules [15]. These patients are associated with a limited estimate of survival at 36.4% [15]. As such, an MRI of the cerebrum and the spine must always be performed along with cancer genetic counseling as this location is common [6]. It could be synchronous or metachronous [6]. Even with no metastatic localized renal rhabdoid tumor, prognosis remains poor. The 1-year survival rate is less than 25%. This is particularly true for younger children because of the limited indications for radiation in this group. A study conducted by Gail et al. [4] compared the survival rate association with radiation. The overall survival was significantly higher in the irradiated group with a survival at 28.5% at 4 years compared to 12.2% in the other group [4]. Nonetheless, when accounting for age and stage, the relative risk of mortality was found to be similar. After comparing 25 Gy to no radiation, the apparent benefit of radiation on survival was significantly reduced and no longer statistically significant [4]. Prognosis could also be correlated with more intensive chemotherapy in older group. Consequently, the role of radiation is difficult to assess. Our patient underwent a primary radical nephroureterectomy and regional lymphadenectomy with no prior chemotherapy. She did not meet the criteria for radiation; therefore, an intensive course of postoperative chemotherapy was indicated. She received a vincristine, cisplatin, doxorubicin, and cyclophosphamide. Still she developed an early relapse and died after only 5 months. This category has a high recurrence rate, with some studies reporting a rate of 50% in 10 months [3,4,5,6,7,8,9,10,11,12,13,14,15,16]. In addition, early chemoresistance is common in rhabdoid tumors [16]. An Irish review was published concerning 25 patients with rhabdoid tumors including 8 patients having a renal localization [17]. Only 2 survived the tumor and remain relapse-free at 16 and 22 years. This is correlated with a higher age and an early stage of diagnosis with both diagnosed at stage 2 and being older than 6 months allowing an intensive chemotherapy protocol [17]. They both received a combination of vincristine, actinomycine D, doxorubicin, carboplatin, cyclophosphamide, and etoposide according to NWTS-5 protocol, and additional radiation of the flank was indicated in the older child [17].
An early stage diagnosis is vital; therefore, it could be interesting to determine a genetic predisposition to some rhabdoid tumors in young infants that help for an early detection. Clinical trials using targeting agents are being conducted in the hopes of improving the prognosis of rhabdoid renal tumors [18]. Both in vitro and in vivo, EZH2 inhibitors have exhibited anti-rhabdoid tumor properties [18]. Tazemetostat (EPZ-6438), a small chemical inhibitor of the EZH2 gene, had caused apoptosis and tumor reduction in mice and was effective in preventing the regrowth after dosing cessation [19]. Thus, the development of research efforts and critical examination of the ever-growing amount of evidence acquired by clinical trials cooperative groups offers a hopeful and optimistic future for both the children afflicted and the treating physicians.
Conclusion
The renal rhabdoid tumor is a highly aggressive embryonal tumor that has an overall survival rate of 22 to 2%. The primary therapeutic option is a radical surgical excision with lymph node dissection. Multiple trials have been conducted using extensive chemicals as adjuvant chemotherapy, but due to the rarity of this malignancy, no standardized protocol has been established. The effectiveness of radiation is not well established due to the low age of affected patients; thus, trials comparing radiation versus non-radiation therapy should be conducted in older children. Case reports and research are critical for evaluating existing protocols and improving prognosis. Clinical trials using targeting agents are being optimistic in the hopes of improving the prognosis of rhabdoid renal tumors.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Brok J, Treger TD, Gooskens SL, et al. Biology and treatment of renal tumors in childhood. Eur J Cancer. 2016;68:179–95.
Dome JS, Graf N, Geller JI, Fernandez CV, Mullen EA, Spreafico F, et al. Advances in Wilms tumor treatment and biology: progress through international collaboration. J Clin Oncol. 2015;33(27):2999–3007. https://doi.org/10.1200/JCO.2015.62.1888 Epub 2015 Aug 24. PMID: 26304882; PMCID: PMC4567702.
Chang CJ, Yeh ML, Chen CC. Rhabdoid tumor of the kidney with spontaneous rupture: case report and review of literature. Pediatr Surg Int. 2008;24:451–45.
Tomlinson GE, Breslow NE, Dome J, Guthrie KA, NorkooL P, Li S, et al. Rhabdoid tumor of the kidney in National Wilms Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol. 2005;23:7641–5. https://doi.org/10.1200/JCO.2004.00.8110 PMID: 16234525.
Van den Heuvel-Eibrink MM, van Tinteren H, Rehorst H, et al. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP renal tumour study group. Pediatr Blood Cancer. 2011;56:733–7.
Dome JS, Graf N, Geller JI, Fernandez CV, Mullen EA, Spreafico F, Van den Heuvel-Eibrink M, Pritchard-Jones K. Advances in Wilms Tumor Treatment and Biology: Progress Through International Collaboration. J Clin Oncol. 2015;33(27):2999-3007. https://doi.org/10.1200/JCO.2015.62.1888. Epub 2015 Aug 24.
Prasad SR, Humphrey PA, Menias CO, et al. Neoplasms of the renal medulla: radiologic-pathologic correlation. Radiographics. 2005;25:369–80.
Amar AM, Tomlinson G, Green DM, et al. Clinical presentation of rhabdoid tumors of the kidney. J Pediatr Hematol Oncol. 2001;23(2):105–108 5.
Sharma R, Kitchen BJ, Mody R, Chamdin A, Bruch S, Jasty R. A report of renal artery embolization for hematuria facilitating neoadjuvant chemotherapy in an unresectable malignant renal rhabdoid tumor. Pediatr Surg Int. 2013;29(5):533–5. https://doi.org/10.1007/s00383-013-3260-5 Epub 2013 Jan 24. PMID: 23344150.
Perlman EJ. Pediatric renal tumors: practical updates for the pathologist. Pediatr Dev Pathol. 2005;8:320–38 5 Raney RB, Palmer N, Sutow WW, et al. Renal cell.
Kerl K, Holsten T, Fruhwald MC. Rhabdoid tumors: clinical approaches and molecular targets for innovative therapy. Pediatr Hematol Oncol. 2013;30:587–604.
Weeks DA, Beckwith JB, Mierau GW. Rhabdoid tumor:an entity or a phenotype? Arch Pathol Lab Med. 1989;113:113–4.24.
Agrons GA, Kingsman KD, Wagner BJ, et al. Rhabdoid tumor of the kidney in children: a comparative study of 21 cases. Am J Roentgenol. 1997;168:447–51.
Warmann SW, Nourkami N, Frühwald M, Leuschner I, Schenk JP, Fuchs J, et al. Primary lung metastases in pediatric malignant non-Wilms renal tumors: data from SIOP 93-01/GPOH and SIOP 2001/GPOH. Klin Padiatr. 2012;224(3):148–52. https://doi.org/10.1055/s-0032-1304600 Epub 2012 Apr 18. PMID: 22513793.
Shukla D, Pradhan A, Bhardwaj M, Malhotra V. Malignant rhabdoid tumor of kidney and brain in an infant. Indian Pediatr. 2015;52(1):65–6. https://doi.org/10.1007/s13312-015-0570-9 PMID: 25638191.
Fanburg-Smith JC, Hengge M, Hengge UR, et al. Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immu immunohistochemical study of 18 cases. Ann Diagn Pathol. 1998;2:351e362.
Uwineza A, Gill H, Buckley P, Owens C, Capra M, O'Sullivan C, et al. Rhabdoid tumor: the Irish experience 1986-2013. Cancer Genet. 2014;207(9):398–402. https://doi.org/10.1016/j.cancergen.2014.05.015 Epub 2014 Jun 11. PMID: 25085603.
Walz AL, Fernandez CV, Geller JI. Novel therapy for pediatric and adolescent kidney cancer. Cancer Metastasis Rev. 2019;38(4):643–55. https://doi.org/10.1007/s10555-019-09822-4 PMID: 31811552.
Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7.
Acknowledgements
Not applicable
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
GH participated in investigation, writing the original draft, reviewing, and editing. SS participated in investigation, editing, and rewriting the draft. SBA participated in investigation. RJ participated in the project administration, supervision, and visualization. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Approved
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Habachi, G., Sahli, S., Ammar, S.B. et al. Rhabdoid renal tumor: an aggressive embryonal tumor in an infant — a case report. Ann Pediatr Surg 18, 63 (2022). https://doi.org/10.1186/s43159-022-00200-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43159-022-00200-4