
Abstract
Background
The health index of any population is directly correlated with the water quality, which in turn depends upon physicochemical characteristics and the microbiome of that aquatic source. For maintaining the water quality, knowledge of microbial diversity is a must. The present investigation attempts to evaluate the microflora of Baner. Metagenomics has been proven to be the technique for examining the genetic diversity of unculturable microbiota without using traditional culturing techniques. The microbial profile of Baner is analyzed using metagenomics for the first time to the best of our knowledge.
Results
To explore the microbial diversity of Baner, metagenomics analysis from 3 different sites was done. Data analysis identified 29 phyla, 62 classes, 131 orders, 268 families, and 741 genera. Proteobacteria was found to be the most abundant phylum in all the sampling sites, with the highest abundance at S3 sampling site (94%). Bacteroidetes phylum was found to be second abundant in S1 and S2 site, whereas Actinobacteria was second dominant in sampling site S3. Enterobacteriaceae family was dominant in site S1, whereas Comamonadaceae and Pseudomonadaceae was abundant in sites S2 and S3 respectively. The Baner possesses an abundant bacterial profile that holds great promise for developing bioremediation tactics against a variety of harmful substances.
Conclusion
Baner river’s metagenomic analysis offers the first insight into the microbial profile of this hilly stream. Proteobacteria was found to be the most abundant phylum in all the sampling sites indicating anthropogenic interference and sewage contamination. The highest abundance of proteobacteria at S3 reveals it to be the most polluted site, as it is the last sampling site downstream of the area under investigation, and falls after crossing the main city, so more human intervention and pollution were observed. Despite some pathogens, a rich profile of bacteria involved in bioremediation, xenobiotic degradation, and beneficial fish probiotics was observed, reflecting their potential applications for improving water quality and establishing a healthy aquaculture and fishery section.
Similar content being viewed by others

Background
Water covers around 75% of the earth’s crust, but nearly 3% of that water is fresh, and 99% of that 3% is trapped in glaciers, polar ice caps, or reservoirs. So, only a trace of the whole water of this earth is available as fresh water. The hydrosphere is an essential part of a sustainable environment, life originated in water, and even the origin and development of human civilization are closely related to the river. So, preserving valuable water resources and aquatic ecosystems is of utmost importance [53]. The quality of freshwater resources has significantly declined in recent years due to population growth, rapid urbanization, industrial effluents, using fertilizers and pesticides for agricultural purposes, and waste disposal. Sustainable water management of a river is quite essential to maintain stability. The effects of water pollution incidents on water quality safety and local inhabitants’ quality of life are currently receiving global attention [20, 25, 89]. In India, several major rivers have been found to exhibit excessive levels of pollution that have adversely affected the ecosystem and human population [41, 50, 88, 34, 14]. Community structure and function dynamics across contaminated rivers are essential for understanding and assessing human activities’ impacts on water ecosystems [63]. In the present scenario, there is an increasing need for regular monitoring and assessment of aquatic ecosystems so that further steps may be taken to preserve the sustainability of these resources. Comprehensive monitoring involves various techniques like physiochemical, hydrological, and biological approaches giving the exact status of the aquatic ecosystem. The latest trend in biomonitoring the ecology of aquatic systems is metagenomics through environmental DNA (e-DNA). Free DNA molecules, or “eDNA,” are those nucleic acids that exist outside of organisms, in the surrounding environment like soil, water, and snow [12]. They may be shed through skin, saliva, gametes, excreta, or corpus remains. Whole-genome shotgun sequencing (WGS based on next-generation sequencing (NGS is used to create a metagenome from eDNA which is taken straight from the environment, revealing complete genetic information of all the organisms present in that particular environment in a single stretch [87, 64]. In situations where collecting whole organisms is difficult or not feasible, or in case of cryptic, endangered, hidden, invasive species, unculturable bacterial species, the fast-expanding study of eDNA has proved a boon to identifying species and conducting genetic analyses for conserving, managing, and research through metagenomics. Microbes are significant organisms in freshwater habitats that can play a role in a variety of ecological events. Bacteria and fungi play a significant role in the conversion of biological and non-biological materials by participating in numerous biogeochemical cycles such as the nitrogen, carbon, sulfur, and phosphorus cycles, which are responsible for the ecosystem’s health and balance. Thorough understanding of the microbial variety and their functionalities found in freshwater bodies is critical for their long-term management of aquatic ecosystems [33, 76].
Bacteria are the most prevalent creatures on the planet; their composition as well their richness highly affects the ecosystem functions and stability whether they are present in host-associated colonies, soil, grassland, or seas [100, 8, 67, 91, 96, 104]. Aquatic microbiomes play crucial roles in nutrient recycling and aquatic ecology functioning. Various estimations of cell density, volume, and carbon reveal that prokaryotes of aquatic habitats are cosmopolitan, and amidst the wide variety of cell densities that have been observed till now, the average values of bacterial composition for several aquatic habitats are remarkably similar [100, 76]. The use of river water directly for drinking poses severe risks as anthropogenic activities generate environmental pollution [9]. Understanding the composition of the bacterial populations in a freshwater habitat is one of the essential steps in ensuring the health of that particular ecosystem [32]. Moreover, compared to marine pathogenic bacteria affecting human health, freshwater bacteria are less explored [30]. Kangra Valley’s residents rely heavily on the portability of Baner rivulet water [14]. The emergence of next-generation sequencing technologies and computational biology has open up the new horizons for mining the genome, transcriptome, and proteome sequence data. The bioinformatics approach for characterization and classification of genes and proteins is time-consuming, cost-effective, and less laborious and makes it possible to mine huge biological dataset [82, 84, 85]. The present investigation was an attempt made to evaluate the microflora of Baner rivulet using the metagenomics technique. The complete microbial profile of Baner is analyzed for the first time to the best of our knowledge.
Methods
Study area
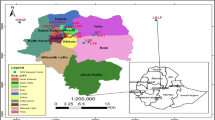
Baner rivulet in Kangra, Himachal Pradesh, the site of the current investigation, is an essential perennial tributary of river Beas, which is one of the major rivers of the Kangra district. The Baner rivulet is one of the main sources of drinking water for the people residing in district Kangra. Baner, with an area of 668 km2, originates in the southern slopes of snow-capped Dhouladhar Mountains near Aadi Himani Chamunda Temple, Palampur, Himachal Pradesh, India (Table 1; Fig. 1). It drains the central part of Kangra district and fans in a south-westward direction before merging with Pong Dam close to Mahora Village in district Kangra [84, 85]. The area of the investigation for Baner rivulet was divided into three sites. The sampling sites which were selected downstream were S1 (Jia), S2 (Chamunda Shaktipeeth), and S3 in Ranital near the confluence of Bathu rivulet and Baner.

Location map showing three sampling sites of Baner
Sample collection, analysis and sequencing
The 250 ml of surface water sample was collected in sterile polyethylene bottles from all three sampling sites, i.e., S1 (Jia), S2 (Chamunda Shaktipeeth), and S3 (Ranital). The sample was carried to the laboratory at 4 °C and stored at – 20 °C till further analysis. The debris and coarse particles were removed by filtering the sample through 1.2 μm pore size membrane. Furthermore, the sample was passed by 0.2 μm pore membrane for collecting prokaryotes cell in sample. Metagenomic DNA was isolated from the samples from each site utilizing the DNeasy PowerWater kit (QIAGEN) as per the manufacturer’s protocol. All laboratory procedures were performed following strict protocols. Enzymatic DNA fragmentation was done using KAPA fragkit to produce small double-stranded DNA segments. The library preparation was started with 200 ng g-DNA. KAPA Hyper Prep kit was used for Library Preparation from these dsDNA fragments. End repair and A tailing, i.e., the addition of A at 3′ end to the dsDNA, was followed by adapter ligation. The adapters’ sequences complement the dual-barcoded libraries in a flow cell for sequencing, enable PCR amplification of adapter-ligated fragments, and bind the conventional Illumina sequencing primers. The library amplification of adapter-ligated libraries was done using KAPA HiFi Hot Start Ready Mix and KAPA Library Quantification Kit. Sequencing was done by Novaseq 6000 Illumina to generate raw data in the form of fastq reads.
Bioinformatics analysis of Baner metagenome
Data generation and quality check
The raw fastq data generated by sequencing were checked for noise and processed by computational tools. The data was pre-processed. The reads were trimmed two from the front and three from the tail and filtered on the parameter of < Q20. The data quality was improved after trimming and filtering, and high-quality reads were used for downstream analysis.
De-novo whole gene assembly
The metaSPAdes-St. Peterburg genome assembler (Megahit genome assembler) was used for the de novo assembly of microbial genomes. MetaSPAdes tool combines the latest algorithmic ideas with already proven solutions from the SPAdes toolkit to address different challenges of metagenomic assembly [65]. For the complete assessment of organisms in the sample, CCmetagen 1.3 tool was used to analyze the diversity from high-throughput sequencing of DNA [59]. After the de novo assembly of sequence data, contigs in FASTA format were obtained for every sample. Following assembly, the contigs were uploaded to Prodigal to anticipate open reading frames (ORFs) [40]. The coding regions have been recognized and separated from noncoding DNA using the gene discovery tool Prodigal.
Taxonomic profiling and annotations of scaffolds
Taxonomic profiling was carried out on all the metagenomics samples using National Centre for Biotechnology Information (NCBI) taxonomy datasets. Each sequencing read was assigned to a taxon in the NCBI taxonomy compared to a reference database with nucleotide sequences. It uses the set of available complete non-redundant Nucleotide databases (nr Database) that includes fungi and microbial eukaryotes genomes. The similarity search of reads was performed at NCBI using BLAST with default parameters [61]. Furthermore, the taxonomy file is used for Krona plot generation using the Krona tool [66]. Functional annotations of all the contigs were performed using SEED classification. Each contig’s function was assigned using the MGA software. The protein functions of each contig having maximum alignment score from MGA results selected for the functional assignment. Pathway analysis was performed using the kofam software at KEGG Database [44].
Results
Metagenomics analysis revealed the identification of 29 phyla, 62 classes, 131orders, 268 families, and 741 genera. Krona graphs (Fig. 2) have been used to show the entire taxonomy, from kingdom up to species level [66]. The detailed description of the data has been given as a comparative account at different taxonomical levels (Tables 2, 3, 4, 5, and 6). The microbiome of Baner showed phylum-wise dominance of Proteobacteria (23,655, 85%), followed by Bacteroidetes (2815, 10%), Actinobacteria (367, 1%), and Verrucomicrobia (307, 1%), at S1. In contrast, at S2, it was Proteobacteria (3270, 82%), Bacteroidetes (272, 7%), Actinobacteria (165, 4%), and Firmicutes (40, 1%), which is the same pattern as that of S1, while at S3, phylum level analysis revealed a dominance of Proteobacteria (11,517, 94%), Actinobacteria (245, 2%), Verrucomicrobia (188, 1.9%), and Bacteroidetes (128, 1%) (Table 2).

Krona graph showing taxonomy at species level at sampling sites. a S1. b S2. c S1
At the class level, the order of dominance in decreasing order at S1 is Gammaproteobacteria (11,099, 40%), Betaproteobacteria (11,049, 40%), Alphaproteobacteria (797, 3%), and Actinomycetia (316, 1%). At S2, the order was as Betaproteobacteria (1487, 37%), Alphaproteobacteria (1257, 31%), Gammaproteobacteria (328, 8%), and Actinomycetia (143, 4%). At S3, it was Alphaproteobacteria (4520, 37%), Gammaproteobacteria (4508, 36%), Betaproteobacteria (1323, 11%), and Actinomycetia (237, 2%) (Table 3; Fig. 3a).

Comparative graphs showing relative taxonomic abundances at different taxonomic hierarchy levels at S1 (HP_B), S2 (HP_C), and S3 HP_D). a Class. b Family. c Phylum
The order level analysis at S1 resulted in the dominance of Enterobacterales (8171, 29%), Burkholderiales (7716, 28%), Rhodocyclales (1988, 7%), Pseudomonadales (1470, 5%), and Nitrosomonadales (756, 2%). At S2, the dominance order was Burkholderiales (1227, 31%), Hyphomicrobiales (487, 12%), Rhodobacterales (298, 7%) Nitrosomonadales (180, 4%), and Sphingomonadales (146, 3%). At S3, Pseudomonadales (4104, 33%), Hyphomicrobiales (3301, 27%), Burkholderiales(1127, 9%), and Sphingomonadales (1063, 9%) are shown in Table 4.
At the family level, the dominance at S1 was as follows (Table 5): Enterobacteriaceae (8023, 29%), Flavobacteriaceae (2431, 9%), Comamonadaceae (2251, 8%), and Pseudomonadaceae (1463, 5%). At S2, it was Comamonadaceae (430, 11%), Rhodobacteraceae (213, 5%), Nitrobacteraceae (180, 4%), Burkholderiaceae (163, 4%), and Nitrosomonadaceae (163, 4%). At S3, it was Pseudomonadaceae (4097, 33%), Rhizobiaceae (2996, 24%), Sphingomonadaceae (1022, 8%), and Acrobacteriaceae (882, 7%), (Fig. 3b).
The dominance pattern at the genus level included (Table 6) Enterobacter (3819, 14%), Paucibacter (3756, 14%), Leclercia (2144, 8%), Citrobacter (1448, 5%), and Pseudomonas (1443, 5%), at S1, while at S2, it was Nitrosomonas (152, 4%), Bradyrhizobium (117, 3%), Aeromonas (104, 3%), Variovorax (95, 2%), Rubrivivax (76, 1%), and Methylibium (76, 1%), respectively. At S3, it was Pseudomonas (4092, 33%), Rhizobium (2401, 20%), Sphingobium (918, 8%), and Pseudoarcobacter (780, 7%).
At the species level, metagenomic analysis revealed the order of dominance at S1 (Table 7, Fig. 4a) as Paucibacter sp. KCTC42545 (3756, 14%), Enterobacter cloacae complex sp. (2648, 10%), Dechloromonas aromatica (844, 3%), and Methylophilus sp. TWE2 (589, 2%). At S2, as shown in Fig. 4b, it was as Nitrosomonas species (91, 2%), Methylibium petroleiphilum PM1 (76, 2%), Rubrivivax gelatinous IL144 (76, 2%), and Caulobacteraceae bacterium (37, 0.9%), and at S 3, presented in Fig. 4c, the order was Pseudomonas putida (1918, 16%), Pseudomonas mosselii (871, 7%), Sphingobium yanoikuyae (855, 7%), and Pseudoarcobacter suis (697, 6%).

Sankey diagrams showings composition of microbial diversity at division, phylum, family, genus, and species levels. a S1. b S2. c S3
Discussion
In general, freshwater habitats have microbial taxa that are different from those found in marine and terrestrial ecosystems. Various estimations of cell density, volume, and carbon reveal that prokaryotes of freshwater habitats are cosmopolitan, and amidst the wide variety of cell densities that have been observed till now, the average values of bacterial composition for several aquatic habitats are remarkably similar [100]. All freshwater forms are more likely to include Betaproteobacteria, Actinobacteria, Bacteroidetes, Verrucomicrobia, and Alphaproteobacteria [30, 51]. Similar findings were observed during the present study. Proteobacteria were found to be the most prevalent and dominating phylum in all three sampling sites (Fig. 3c), indicating anthropogenic interference and municipal disposal [102]. They were most abundant at S3, revealing it to be the most polluted site, as it is the last sampling site downstream, of the area under investigation, which shows more human intervention and pollution, as this area falls after crossing the main city. A similar pattern was observed by the researchers in earlier studies [42, 56]. Proteobacteria are mostly fast-growing copiotrophs that can thrive in environments with abundant nutrients, and they are thought to be crucial for nitrogen cycling, linking iron-carbon biogeochemistry, carbon sequestration, nutrient flux, and other biochemical phenomena [52]. The Baner rivulet’s metagenomic analysis initially showed a diverse microbial makeup revealing a complex microbial composition of both helpful and unfriendly species, detailed in the following section.
Beneficial microbiota of Baner
Bio-remedial bacterial profile
Several prokaryotic and eukaryotic species have evolved defense systems against toxic metals, making them benign. With the help of their genes, microbes typically biosorb metals and sequester these in their cell walls [17, 79]. They adopt various mechanisms to respond to heavy metal stress, such as exclusion, compartmentalization, complex formation, and the creation of binding proteins like metallothioneins (MTs) [80,81,82,83]. During the present investigation, some of bio-remedial species were found in Baner that can be utilized to remove heavy metals, harmful chemicals, pesticides, etc.
Few strains of cyanobacterium Synechococcus which produce MT have been discovered. Compared to mammalian MTs, prokaryotic MT has fewer cysteine residues [57]. Synechococcus has been found at all three selected sites of Baner and can reduce copper, cadmium, and zinc from the environment [103].
Leptothrix discophora, gram-negative bacteria, showed the capability of oxidizing Mn(II) [13]. It extracts manganese from the environment, synthesizes Mn(II)-oxidizing proteins as a component of an extracellular sheath matter for the purpose of growth; defense against predation, UV light, or viral attacks; and probably also for immobilizing the toxic metal [2, 92]. The bacteria have the ability to produce two different extracellular macromolecules that catalyze the oxidation of Fe(II) and Mn(II) which makes it useful for bioremediation [21, 95]. The metal-encrusted surfaces of Mn(II)-oxidizing bacteria provide biogenic Mn(II) oxidation which can be used for the removal of Mn(II), Fe(II), and As(III) from potable groundwater. The role of Leptothrix sp. and Gallionella sp. in treating the groundwater for Mn(II) and Fe(II) removal has been suggested by Katsoyiannis and Zouboulis [46, 47]. Raw water is treated biologically by Mn(II)-oxidizing bacteria, including Leptothrix cholodnii, in biological filters extensively [46, 47]. Both Gallionella and Leptothrix have been found at all three sampling sites (S1, S2, and S3).
Acidovorax sp. is a mesophilic gram-negative bacterium decomposing nitroarene compounds to use 2-nitrotoluene (2NT) as the only source of carbon and energy [58]. Nitroarene compounds are a class of poisonous chemicals predominantly man-made and known to contaminate soil and groundwater, including chemical manufacturing units and explosive factories, grenades, and detonation sites [36]. Pseudomonas putida, the most abundant species at S3, was genetically engineered, increasing its inherent cadmium binding capacity threefold. Similarly, the metal binding capacity of Burkholderia, Alcaligenes, and Ralstonia can also be enhanced so that further they may be utilized to eliminate heavy metal pollution from sewage and industrially polluted water bodies [81, 94]. All these species were abundant in Baner rivulet.
Arthrobacter is known to possess growth-promoting activity in plants; these bacteria also possess genes for uptaking heavy metals, to decompose complex organic and inorganic compounds. Their respective genes for diverse metal degradation can be utilized for synthesizing transgene, which can then be utilized to create artificial bacteria that can degrade various xenobiotics [71]. The role of Arthrobacter in bioremediation has been proved multiple times through the removal of pesticides and herbicides, pollutants like 4 chlorophenol, pentachloronitrobenzene cypermethrin, cyhalothrin, dichlorobiphenyls, trichloroethylene, p-nitrophenol, and atrazine or removing heavy metal like chromium and iron [11, 43, 62, 69, 98, 101].
Rare earth metals cerium and neodymium are bioaccumulated by Bacillus cereus [17]. The involvement of Pseudomonas in iron and uranium removal has been proved earlier [39]. Methylibium petroleiphilum plays an essential role in bioremediation by eliminating MTBE ( methyl tert-butyl ether), a component of gasoline [35].
Paucibacter is good at remediation of Microcystis algal blooms and microcystin; microcystin is a carcinogen and hepatotoxin for humans and causes mortalities in fish and livestock [54, 70]. Leptothrix and Dechloromonas are sulfur oxidation bacteria converting hydrogen sulfide to sulfur element or sulfate and removing toxic compounds with foul-smell like H2S [19, 106]. Dechloromonas aromatica plays important role in the biodegradation of benzene [73]. Sphingobium sp. degrades phenanthrene (component of plastics), pesticides, explosives, and drugs. Aromatic molecules, glycan polymers, metal ions, xenobiotics, and resistive substances are all catabolized by the Variovorax paradoxus [74]. Acinetobacter sp., Klebsiella sp., and Elizabethkingia sp. efficiently biodegrade ciprofloxacin and levofloxacin [77]. Rhizobacter gummiphilus, found at all three sites, participates in the degradation of rubber, a component of tyres and surgical gloves [45]. Rhodobacter sphaeroides have an excellent potential for heavy metals bioremediation [5, 15]. Stenotrophomonas maltophilia has been found at both S2 and S3 which has been proven to transform As(V) to As(III) and nontoxic arsenobetaine in the fish gut [90].
Potential probiotics for fish
During the present study, several probiotic bacteria were found in Baner which are helpful for humans as well as aquatic organisms. Probiotics are microorganisms or derivatives from them that provide good health and growth of the host that are used in aquaculture to manage the disease, complement immunity, and in some circumstances even substitute the use of antibacterial agents like antibiotics. Fish are benefited in many ways through these probiotic bacteria, including growth promotion, pathogen colonization inhibition, improved digestion, water quality, stress tolerance, and fertility improvement [23]. The findings of the studies on fish probiotics were reported earlier [16] which showed that the bacteria present in the aquatic systems impacted the gut microbial diversity and vice versa. Species that can survive and proliferate in the digestive system often appear to be those from the surroundings or the food.
The probiotics can offer a promising approach for controlling the bacterial, fungal and parasitic infections in fishes [93]. The role of Enterococcus faecium as a probiotic for improving the health of Labeo rohita has been proved previously [31]. The Bacillus sp. has also been recognized a good fish probiotic involved in overall development [37]. Bacillus subtilis and B. circulans enhanced the fish growth and eliminated the anti-nutritional factor mimosine [6, 7]. Lactobacillus sp. also shows good probiotic potential, inhibiting activities against harmful fish pathogen Aeromonas hydrophila and boosting fish health [4, 27, 75]. Micrococcus and Pseudomonas are good as probiotics for enhancing fish growth [1]. The probiotic activity of Roseobacter against Vibrio anguillarum infection has been proved efficient earlier [22, 38, 68]. Rubrivivax gelatinosus is a probiotic as a bacterium, and its biomass and by-products boost the immune system of fish to combat pathogens and enhance growth [28, 29]. Sharifuzzaman and Austin [78] observed that the cellular components of Rhodococcus improved rainbow trout resistance against Vibrio anguillarum. The probiotic activity of Alteromonas sp., Phaeobacter gallaeciensis, and Pseudomonas damselae against Vibrio splendidus and V. coralliilyticus has been reported [48, 49]. Shewanella putrefaciens and Shewanella baltica were examined for their probiotic potential in fishes which heightened immune response and increased resistance to Photobacterium damselae [24]. All abovementioned probiotic bacteria have been found in the microbial profile of Baner.
Pathogens
Several disease-causing pathogens have been found during the metagenomics study of the river during the present investigation. The species of Aeromonas (Aeromonas veronii) was reported, which acts as a fish and opportunistic pathogen to humans [55]. Vibrio sp. causes diseases in fish, but being zoonotic also infects birds and animals; consumption of undercooked infected fish causes gastrointestinal problems in humans [10, 18]. Spiroplasma has been observed as a severe pathogen of crayfish [99], Nocardia is being found in multiple hosts including fish and human [26, 60] and acts as a fish pathogen [3]. Flavobacterium sp., Citrobacter sp., Proteus sp., Klebsiella sp., Comamonas, Plesiomonas, Ralstonia, Chryseobacterium, vibrio, and Edwardsiella have been recognized as fish pathogens [97]. Comamonas, Ralstonia sp., Ochrobactrum sp., Pseudomonas sp., Sphingomonas sp., and Brevundimonas sp. are also opportunistic pathogens [72]. Leclercia adecarboxylata is also a human opportunistic pathogen affecting immunocompromised hosts [105]. All these pathogenic microbiota were also reported at the sites of Baner rivulet. Furthermore, more deep studies are required to carry on the Baner regarding control of these types of pathogenic microorganisms.
Conclusion
The first-time metagenomic study of Baner rivulet revealed Proteobacteria to be the most dominating phylum at all the sampling sites indicating municipal wastes, human excreta, and anthropogenic pollution. Proportionately, more presence of Proteobacteria at S3 indicates fecal pollution and more anthropogenic interference at this site. S3 is the last point of investigation and falls downstream the river after crossing the main city, so more anthropogenic impact has been observed. As several human, fish, and zoonotic pathogens were found in the water, it is advised to be watchful, periodically disinfect, and use appropriate bio-remedial techniques as the water of Baner has been lifted by the Himachal Pradesh Irrigation and Public Health Department both for drinking and irrigation purposes. However, the Baner also possesses an abundant bacterial profile that holds great promise for developing bioremediation tactics against a variety of harmful substances, such as heavy metals, pesticides, xenobiotic, nitrogen and sulfur, phenol, and toluene metabolism, and also has good potential for probiotics concerned with fish health. The genes of bio-remedial and probiotic bacteria can be identified and exploited through genetic engineering or transformation for healthy aquaculture and fishery sector. As the water is used for drinking and agriculture, it is good that it is being purified of heavy metals and pollutants by natural mechanisms. The findings of the present research may also be used as a basis or case study in the future for supporting further freshwater microbiome research. Additionally, it is possible to recover and clone the expected gene pool for xenobiotic degradation to create new bioremediation techniques in the future.
Availability of data and materials
The data will be provided on reasonable request.
Abbreviations
- BLAST:
-
Basic local alignment search tool
- DNA:
-
Deoxyribonucleic acid
- dsDNA:
-
Double-stranded DNA
- e-DNA:
-
Environmental DNA
- FASTA:
-
Fast-All
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MGA:
-
Multiple Genome Aligner
- MT:
-
Metallothionein
- NCBI:
-
National Centre for Biotechnology Information
- NGS:
-
Next-generation sequencing
- ORF:
-
Open reading frames
- PCR:
-
Polymerase chain reaction
- WGS:
-
Whole-genome shotgun sequencing
References
Abd El-Rhman, A.M., Khattab, Y.A.E. and Shalaby, A.M.E. (2009) ‘Micrococcus luteus and Pseudomonas species as probiotics for promoting the growth performance and health of Nile tilapia, Oreochromis niloticus’, Fish and Shellfish Immunology, 27(2), pp. 175–180. https://doi.org/10.1016/j.fsi.2009.03.020
Adams, L.F. and Ghiorse, W.C. (1985) ‘Influence of manganese on growth of a sheathless strain of Leptothrix discophora’, Applied and Environmental Microbiology, 49(3), pp. 556–562. https://doi.org/10.1128/aem.49.3.556-562.1985
Alderman D.J, Feist S.W, Polglase J.L (1986) Possible nocardiosis of crayfish, Austropotamobius pallipes. J Fish Dis 9(4):347–345
Arndt RE, Wagner EJ (2007) Enriched Artemia and probiotic diets improve survival of Colorado River cutthroat trout larvae and fry. N Am J Aquac 69(2):190–196
Asztalos E et al (2010) Early detection of mercury contamination by fluorescence induction of photosynthetic bacteria. Photochem Photobiol Sci 9(9):1218–1223
Bagheri T et al (2008) Growth, survival and gut microbial load of rainbow trout (Onchorhynchus mykiss) fry given diet supplemented with probiotic during the two months of first feeding. Turk J Fish Aquat Sci 48(1):43–48
Bairagi, A. et al. (2004) ‘Evaluation of the nutritive value of Leucaena leucocephala leaf meal, inoculated with fish intestinal bacteria Bacillus subtilis and Bacillus circulans in formulated diets for rohu, Labeo rohita (Hamilton) fingerlings’, Aquaculture Research, 35(5), pp. 436–446. https://doi.org/10.1111/j.1365-2109.2004.01028.x
Barberán A, Casamayor EO (2010) Global phylogenetic community structure and β-diversity patterns in surface bacterioplankton metacommunities. Aquat Microb Ecol 59(1):1–10
Barik RM, Patel RK (2004) Seasonal variation of water quality of Attharabanki River near Paradeep. J Environ Protect 24(3):161–166
Bean NH et al (1998) Crayfish: a newly recognized vehicle for Vibrio infections. Epidemiol Infect 121(2):269–273
Bjerketorp J et al (2018) Formulation and stabilization of an Arthrobacter strain with good storage stability and 4-chlorophenol-degradation activity for bioremediation. Appl Microbiol Biotechnol 102(4):2031–2040
Bohmann, K. et al. (2014) ‘Environmental DNA for wildlife biology and biodiversity monitoring’, Trends in Ecology and Evolution, 29(6), pp. 358–367. https://doi.org/10.1016/j.tree.2014.04.003
Boogerd, F.C. and De Vrind, J.P.M. (1987) ‘Manganese oxidation by Leptothrix discophora’, Journal of Bacteriology, 169(2), pp. 489–494. https://doi.org/10.1128/jb.169.2.489-494.1987
Brar, B. et al. (2023) ‘Mapping water quality parameters in Baner and Gaj rivulets: insights into the potential impact on river Beas in Himachal Pradesh using ArcGIS’, World Water Policy, pp. 1–41. https://doi.org/10.1002/wwp2.12122
Buccolieri A et al (2006) Testing the photosynthetic bacterium Rhodobacter sphaeroides as heavy metal removal tool. Annali di Chimica: J Analytical Environ Cultural Heritage Chemistry 96(3–4):195–203
Cahill MM (1990) Bacterial flora of fishes: a review. Microb Ecol 19:21–41
Challaraj Emmanuel ES et al (2011) Bioaccumulation of cerium and neodymium by Bacillus cereus isolated from rare earth environments of Chavara and Manavalakurichi, India. Indian J Microbiol 51(4):488–495
Chen, Y., Ai, X. and Yang, Y. (2022) ‘Vibrio cholerae: a pathogen shared by human and aquatic animals’, The Lancet Microbe, 3(6), p. e402. https://doi.org/10.1016/S2666-5247(22)00125-2
Cheng Y et al (2018) Use of sulfur-oxidizing bacteria enriched from sewage sludge to biologically remove H2S from biogas at an industrial-scale biogas plant. Bioresour Technol Rep 3:43–50
Choudhary, M. et al. (2023) ‘Status of fish assemblage structure in the Ganga and Indus riverine systems of the western Himalaya’, World Water Policy, pp. 1–26. https://doi.org/10.1002/wwp2.12116
Corstjens, P.L.A.M. et al. (1992) ‘Enzymatic iron oxidation by Leptothrix discophora: identification of an iron-oxidizing protein’, Applied and Environmental Microbiology, 58(2), pp. 450–454. https://doi.org/10.1128/aem.58.2.450-454.1992
D'Alvise Paul W et al (2012) Phaeobacter gallaeciensis Reduces Vibrio anguillarum in Cultures of Microalgae and Rotifers, and Prevents Vibriosis in Cod Larvae. PLoS One 7(8):e43996–e43996. https://doi.org/10.1371/journal.pone.0043996
Dawood M.A.O et al (2018) Probiotic application for sustainable aquaculture. Rev Aquac 11(3):907–924
Díaz-Rosales P et al (2009) Effects of two closely related probiotics on respiratory burst activity of Senegalese sole (Solea senegalensis, Kaup) phagocytes, and protection against Photobacterium damselae subsp. piscicida. Aquaculture 293(1–2):16–21
Ding, X. et al. (2017) ‘Early warning and forecasting system of water quality safety for drinking water source areas in Three Gorges Reservoir Area, China’, Water (Switzerland), 9(7). https://doi.org/10.3390/w9070465
Engelsma MY, Roozenburg I, Joly J-P (2008) First isolation of Nocardia crassostreae from Pacific oyster Crassostrea gigas in Europe. Dis Aquat Org 80(3):229–234
Galindo AB (2004) Lactobacillus plantarum 44A as a live feed supplement for freshwater fish. Wageningen University and Research. Ph.D thesis Wageningen University, pp 131
Gallani, S.U. et al. (2017) ‘Rubrivivax gelatinosus biomass as an immunostimulant for pacu Piaractus mesopotamicus’, Aquaculture Research, 48(9), pp. 4836–4843. https://doi.org/10.1111/are.13303
Gallani SU et al (2019) Dietary intake of Rubrivivax gelatinosus biomass enhances phagocytic cells in tropical fish Piaractus mesopotamicus infected with Aeromonas hydrophila. Aquacult Int 27(3):711–720
Ghai R et al (2011) Metagenomics of the water column in the pristine upper course of the Amazon river. PLoS ONE 6(8):e23785
Ghori I et al (2018) Geotrichum candidum enhanced the Enterococcus faecium impact in improving physiology, and health of Labeo rohita (Hamilton, 1822) by modulating gut microbiome under mimic aquaculture conditions. Turk J Fish Aquat Sci 18(11):1255–1267
Gómez GD, Balcázar JL (2008) A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol Med Microbiol 52(2):145–154
Hahn MW (2006) The microbial diversity of inland waters. Curr Opin Biotechnol 17(3):256–261
Hamner S et al (2013) Sewage pollution of the River Ganga: an ongoing case study in Varanasi, India. River Syst 20(3-4):157–167
Hanson RS, Hanson TE (1996) Methanotrophic bacteria. Microbiol Rev 60(2):439–471
Hartter, D.R. (1985) ‘The use and importance of nitroaromatic chemicals in the chemical industry’, Toxicity of nitroaromatic compounds, pp 1–13
Hassaan MS, Soltan MA, Ghonemy MMR (2014) Effect of synbiotics between Bacillus licheniformis and yeast extract on growth, hematological and biochemical indices of the Nile tilapia (Oreochromis niloticus). The Egyptian J Aquatic Res 40(2):199–208
Hjelm M et al (2004) Selection and identification of autochthonous potential probiotic bacteria from turbot larvae (Scophthalmus maximus) rearing units. Syst Appl Microbiol 27(3):360–371
Hu M et al (1996) Biosorption of uranium by Pseudomonas aeruginosa St ra in CSU: characterization and comparison studies. Biotechnol Bioeng 51(2):237–247
Hyatt D et al (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11(1):1–11
Jain CK (2004) Metal fractionation study on bed sediments of River Yamuna, India. Water Res 38(3):569–578
Jiravanichpaisal P et al (2009) A highly virulent pathogen, Aeromonas hydrophila, from the freshwater crayfish Pacifastacus leniusculus. J Invertebr Pathol 101(1):56–66
Joshi MN et al (2013) Draft genome sequence of Arthrobacter crystallopoietes strain BAB-32, revealing genes for bioremediation. Genome Announc 1(4):e00452-e513
Kanehisa M (2002) ‘The KEGG database’, in. ‘In silico’simulation of biological processes: Novartis Foundation Symposium, vol 247. Wiley, Chichester, pp 91–103
Kasai D et al (2017) Identification of natural rubber degradation gene in Rhizobacter gummiphilus NS21. Biosci Biotechnol Biochem 81(3):614–620
Katsoyiannis, I.A. and Zouboulis, A.I. (2004a) ‘Application of biological processes for the removal of arsenic from groundwaters’, Water Research, 38(1), pp. 17–26. https://doi.org/10.1016/j.watres.2003.09.011
Katsoyiannis, I.A. and Zouboulis, A.I. (2004b) ‘Biological treatment of Mn (II) and Fe (II) containing groundwater: kinetic considerations and product characterization’, Water Research, 38(7), pp. 1922–1932. https://doi.org/10.1016/j.watres.2004.01.014
Kesarcodi-Watson A et al (2010) Alteromonas macleodii 0444 and Neptunomonas sp. 0536, two novel probiotics for hatchery-reared Greenshell™ mussel larvae, Perna canaliculus. Aquaculture 309(1–4):49–55
Kesarcodi-Watson A et al (2012) Protective effect of four potential probiotics against pathogen-challenge of the larvae of three bivalves: pacific oyster (Crassostrea gigas), flat oyster (Ostrea edulis) and scallop (Pecten maximus). Aquaculture 344:29–34
Khwaja AR, Singh R, Tandon SN (2001) Monitoring of Ganga water and sediments vis-a-vis tannery pollution at Kanpur (India): a case study. Environ Monit Assess 68(1):19–35
Kim S et al (2021) Cultivation of dominant freshwater bacterioplankton lineages using a high-throughput dilution-to-extinction culturing approach over a 1-year period. Front Microbiol 12:700637
Krishna, M. et al. (2020) ‘Successional trajectory of bacterial communities in soil are shaped by plant-driven changes during secondary succession’, Scientific Rep, 10(1), pp. 1–10. https://doi.org/10.1038/s41598-020-66638-x
Kumar R et al (2023) Knowledge mining for the differentiation pattern with respect to the productivity and trophic status of some selected lakes of Western Himalayas. Front Environ Sci 11:125
Van Le V et al (2022) The cyanobactericidal bacterium Paucibacter aquatile DH15 caused the decline of Microcystis and aquatic microbial community succession: a mesocosm study. Environ Pollut 311:119849
Li T et al (2020) Aeromonas veronii infection in commercial freshwater fish: a potential threat to public health. Animals 10(4):608
Liu R et al (2012) Diversity of bacteria and mycobacteria in biofilms of two urban drinking water distribution systems. Can J Microbiol 58(3):261–270
M M and Bülow L (2001) M etal-binding proteins and peptides in bioremediation and phytoremediation of heavy metals. Trends Biotechnol 19(2):67–73
Mahan KM et al (2015) Selection for growth on 3-nitrotoluene by 2-nitrotoluene-utilizing Acidovorax sp. strain JS42 identifies nitroarene dioxygenases with altered specificities. Appl Environ Microbiol 81(1):309–319
Marcelino VR et al (2020) CCMetagen: comprehensive and accurate identification of eukaryotes and prokaryotes in metagenomic data. Genome Biol 21(1):1–15
Maskow, T. and Babel, W. (2001) ‘A calorimetrically based method to convert toxic compounds into poly-3-hydroxybutyrate and to determine the efficiency and velocity of conversion’, Applied Microbiology and Biotechnology, 55(2), pp. 234–238. https://doi.org/10.1007/s002530000546
McGinnis S, Madden T.L (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32(suppl_2):W20–W25
Megharaj M, Avudainayagam S, Naidu R (2003) Toxicity of hexavalent chromium and its reduction by bacteria isolated from soil contaminated with tannery waste. Curr Microbiol 47(1):51–54
Mittal, P. et al. (2019) ‘Metagenome of a polluted river reveals a reservoir of metabolic and antibiotic resistance genes’, Environmental Microbiomes, 14(1), pp. 1–12. https://doi.org/10.1186/s40793-019-0345-3
Monchamp, M.È. et al. (2022) ‘Comparative analysis of zooplankton diversity in freshwaters: What can we gain from metagenomic analysis?’, Environmental DNA, (June), pp. 1–15. https://doi.org/10.1002/edn3.335
Nurk, S. et al. (2017) ‘MetaSPAdes: a new versatile metagenomic assembler’, Genome Research, 27(5), pp. 824–834. https://doi.org/10.1101/gr.213959.116
Ondov BD, Bergman NH, Phillippy AM (2011) Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. https://doi.org/10.1186/1471-2105-12-385
Philippot L et al (2013) Loss in microbial diversity affects nitrogen cycling in soil. ISME J 7(8):1609–1619
Planas M et al (2006) Probiotic effect in vivo of Roseobacter strain 27–4 against Vibrio (Listonella) anguillarum infections in turbot (Scophthalmus maximus L.) larvae. Aquaculture 255(1–4):323–333
Qingyan LI et al (2008) Isolation and characterization of atrazine-degrading Arthrobacter sp. AD26 and use of this strain in bioremediation of contaminated soil. J Environ Sci 20(10):1226–1230
Rapala, J. et al. (2005) ‘Paucibacter toxinivorans gen. nov., sp. nov., a bacterium that degrades cyclic cyanobacterial hepatotoxins microcystins and nodularin’, Int J Syst Evol Microbiol, 55(4), pp. 1563–1568. https://doi.org/10.1099/ijs.0.63599-0
Roy, P. and Kumar, A. (2020) ‘Arthrobacter’, Beneficial microbes in agro-ecology: bacteria and fungi, pp. 3–11. https://doi.org/10.1016/B978-0-12-823414-3.00001-0.
Ryan MP et al (2022) The emergence of the genus Comamonas as important opportunistic pathogens. Pathogens 11(9):1032
Salinero KK et al (2009) Metabolic analysis of the soil microbe Dechloromonas aromatica str. RCB: indications of a surprisingly complex life-style and cryptic anaerobic pathways for aromatic degradation. BMC Genomics 10(1):1–23
Satola B, Wübbeler JH, Steinbüchel A (2013) Metabolic characteristics of the species Variovorax paradoxus. Appl Microbiol Biotechnol 97(2):541–560
Schmalenberger, A. et al. (2008) ‘The role of Variovorax and other Comamonadaceae in sulfur transformations by microbial wheat rhizosphere communities exposed to different sulfur fertilization regimes’, Environmental Microbiology, 10(6), pp. 1486–1500. https://doi.org/10.1111/j.1462-2920.2007.01564.x
Sehnal L et al (2021) Microbiome composition and function in aquatic vertebrates: small organisms making big impacts on aquatic animal health. Front Microbiol 12:567408
Shaker RAE et al (2022) Acinetobacter baumannii, Klebsiella pneumoniae and Elizabethkingia miricola isolated from wastewater have biodegradable activity against fluoroquinolone. World J Microbiol Biotechnol 38(11):1–15
Sharifuzzaman SM, Austin B (2010) Development of protection in rainbow trout (Oncorhynchus mykiss, Walbaum) to Vibrio anguillarum following use of the probiotic Kocuria SM1. Fish Shellfish Immunol 29(2):212–216
Sharma A, Sharma D, Verma SK (2017) Proteome wide identification of iron binding proteins of Xanthomonas translucens pv. undulosa: focus on secretory virulent proteins. Biometals 30:127–141
Sharma A, Sharma D, Verma S.K (2018) In silico study of iron, zinc and copper binding proteins of Pseudomonas syringae pv. lapsa: emphasis on secreted metalloproteins. Front Microbiol 9:1838
Sharma A, Sharma D, Verma SK (2019) In silico identification of copper-binding proteins of Xanthomonas translucens pv. undulosa for their probable role in plant-pathogen interactions. Physiol Mol Plant Pathol 106:187–195
Sharma A, Sharma D, Verma SK (2019) Zinc binding proteome of a phytopathogen Xanthomonas translucens pv. undulosa. Royal Society Open Science 6(9):190369
Sharma D et al (2019) Bioinformatic exploration of metal-binding proteome of zoonotic pathogen Orientia tsutsugamushi. Front Genet 10:797
Sharma D, Kumar S et al (2022) Functional assignment to hypothetical proteins in Orientia tsutsugamushistrain Ikeda. Bioinformation 18(3):188
Sharma D, Sharma A et al (2022) Neglected scrub typhus: an updated review with a focus on omics technologies. Asian Pac J Trop Med 15(12):531–541
Sharma, R. et al. (2022) ‘Morphometric analysis of Baner, Neogal and Awa river basins, Himachal Pradesh, India’, Journal of the Geological Society of India, 98(1), pp. 125–132. https://doi.org/10.1007/s12594-022-1938-9
Singh B et al (2017) Metagenomic Insights into Herbivore Gut: An Application-Based Perspective. In: Kalia V, Shouche Y, Purohit H, Rahi P, editors. Mining of Microbial Wealth and MetaGenomics. Springer, Singapore, pp 201–215. https://doi.org/10.1007/978-981-10-5708-3_12
Singh KP et al (2004) Multivariate statistical techniques for the evaluation of spatial and temporal Environ Sci Pollut Res variations in water quality of Gomti River (India)—a case study. Water Res 38:3980–3992
Singh UB et al (2013) Nutritional studies of Chara corallina. Recent Res Sci Technol 5(3):10–14
Song D et al (2022) Gut microbiota promote biotransformation and bioaccumulation of arsenic in tilapia. Environ Pollut 305:119321
Strickland MS et al (2009) ‘Bradford CL Testing the functional significance of microbial community composition.’ Ecology 90:441–451
Tebo, B.M. et al. (2004) ‘Biogenic manganese oxides: properties and mechanisms of formation’, Annual Review of Earth and Planetary Sciences, 32(Goldberg 1954), pp. 287–328. https://doi.org/10.1146/annurev.earth.32.101802.120213
Thakur K et al (2023) An insight into the interaction between Argulus siamensis and Labeo rohita offers future therapeutic strategy to combat argulosis. Aquacult Int 31(3):1607–1621
Valls M, Lorenzo D, Gonzalez-duarte R (2000) Engineering outer-membrane proteins in. J Inorg Biochem 79:219–223
De Vrind-De Jong, E.W. et al. (1990) ‘Oxidation of manganese and iron by Leptothrix discophora: Use of N,N,N’,N’-tetramethyl-p-phenylenediamine as an indicator of metal oxidation’, Applied and Environmental Microbiology, 56(11), pp. 3458–3462. https://doi.org/10.1128/aem.56.11.3458-3462.1990
Wagg, C. et al. (2014) ‘Soil biodiversity and soil community composition determine ecosystem multifunctionality’, Proceedings of the National Academy of Sciences of the United States of America, 111(14), pp. 5266–5270. https://doi.org/10.1073/pnas.1320054111
Wamala, S.P. et al. (2018) ‘Occurrence and antibiotic susceptibility of fish bacteria isolated from Oreochromis niloticus (Nile tilapia) and Clarias gariepinus (African catfish) in Uganda’, Fisheries and Aquatic Sciences, 21(1), pp. 1–10. https://doi.org/10.1186/s41240-017-0080-x
Wang Q, Xie S (2012) Isolation and characterization of a high-efficiency soil atrazine-degrading Arthrobacter sp. strain. Int Biodeterior Biodegradation 71:61–66
Wang W et al (2005) A novel Spiroplasma pathogen causing systemic infection in the crayfish Procambarus clarkii (Crustacea: Decapod), in China. FEMS Microbiol Lett 249(1):131–137
Whitman, W.B., Coleman, D.C. and Wiebe, W.J. (1998) ‘Prokaryotes: the unseen majority’, Proceedings of the National Academy of Sciences of the United States of America, 95(12), pp. 6578–6583. https://doi.org/10.1073/pnas.95.12.6578
Xiao W et al (2017) Isolation and characterization of chromium (VI)-reducing Bacillus sp. FY1 and Arthrobacter sp. WZ2 and their bioremediation potential. Bioremediat J 21(2):100–108
Yadav N, Sharma S (2020) Pollution shapes the bacterial community of a river: a case study. Int J Environ Sci Technol 17(4):2003–2016
Ybarra GR, Webb R (1999) Effects of divalent metal cations and resistance mechanisms of the cyanobacterium Synechococcus sp. strain PCC 7942. J Hazardous Substance Res 2(1):1
Zavaleta ES et al (2010) Sustaining multiple ecosystem functions in grassland communities requires higher biodiversity. Proc Natl Acad Sci 107(4):1443–1446
Zayet S et al (2021) Leclercia adecarboxylata as emerging pathogen in human infections: clinical features and antimicrobial susceptibility testing. Pathogens 10(11):1399
Zhang, K. et al. (2020) ‘Fish growth enhances microbial sulfur cycling in aquaculture pond sediments’, Microbial Biotechnology, 13(5), pp. 1597–1610. https://doi.org/10.1111/1751-7915.13622
Acknowledgements
All authors duly acknowledge the Central University of Himachal Pradesh for providing facilities to carry out the work. DS acknowledges the Department of Health Research, Government of India, for Young Scientist Fellowship.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
RK—conceptualization; BB—original draft preparation; KT, DS, and AKS—reviewing and editing; BB, RK, DS, and DM helped to draft the final manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes.
Competing interests
The authors declare no completing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brar, B., Kumar, R., Sharma, D. et al. Metagenomic analysis reveals diverse microbial community and potential functional roles in Baner rivulet, India. J Genet Eng Biotechnol 21, 147 (2023). https://doi.org/10.1186/s43141-023-00601-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-023-00601-x