
Abstract
Background
Metformin hydrochloride (MFH) is a biguanide class anti-diabetic drug used to treat type-2 diabetes mellitus. Its reaction with two charge-transfer complexing agents, p-chloranilic acid (PCA) and 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) in acetonitrile medium to yield coloured products measurable at wavelengths of maxima 530.0 and 460.0 nm, respectively, was conveniently used to develop two spectrophotometric methods for analyses of bulk sample and tablets.
Results
The effect of solvent, reagent concentration and reaction time to form charge-transfer (CT) complexes was meticulously studied and optimized. Under optimised conditions, the absorbance at the respective wavelength of maximum versus concentration of MFH was in linear correlation for the range from 8.0 to 320.0 and from 1.6 to 64.0 μg mL-1 in PCA and DDQ methods, respectively, and correspondingly, the values of molar absorptivity of 0.733 × 103 and 0.257 × 104 L mol-1 cm-1 and Sandell sensitivity of 0.3620 and 0.0644 μg cm-2. The quantification (QL) and detection (DL) limits were 2.67 and 0.88 μg mL-1 for PCA method, and 0.33 and 0.11 μg mL-1 for DDQ method.
Conclusion
The new methods were emerged as repeatable and reproducible, with replicate measurements for intra- and inter-day variations as showed by obtained RSD values of < 2%. Within a day and between day relative errors were ≤ 2.18%. Methods were also validated for robustness, ruggedness and selectivity and agreeing results were produced. The methods were used to analyse MFH-containing tablets very accurately and precisely as reflected by the mean recovery value close to 100% and lower RSD values, respectively. Analysis of spiked human urine yielded excellent mean recoveries, indicating the absence of interference from endogenous substances.
Similar content being viewed by others

Background
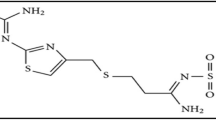
Metformin hydrochloride (MFH), has the IUPAC name of 1,1-dimethylbiguanide monohydrochloride (Fig. 1), is an anti-diabetic drug belonging to biguanide class [1]. MFH is the anti-hyperglycemic compound and used to treat type 2 diabetic patients [2]. The drug lowers the glucose level of plasma.

Structure of MFH
MFH is official in European (EP) [3] and the United States Pharmacopeia (USP) [4]. Both describe titrimetry to determine MFH in pharmaceuticals; in the former, drug solution in anhydrous formic acid-acetonitrile mixture is titrated with perchloric acid with potentiometric end-point detection and the latter involves the titration of MFH in anhydrous mixture of formic and acetic acids versus perchloric acid, determining the endpoint visually, with crystal violet indicator.
Different analytical techniques have been employed by different workers for MFH determination in real pharmaceutical samples and they are liquid chromatography (LC) [5,6,7,8,9,10,11,12,13,14,15], high performance-thin layer chromatography (HPTLC) [16,17,18,19], gas chromatography (GC) [20], capillary electrophoresis (CE) [21], uv-spectrophotometry [22,23,24,25,26,27,28], spectrofluorimetry [25], potentiometry [29,30,31,32] and conductometry [33]. MFH was determined simultaneously along with other therapeutic drugs in pharmaceuticals by ultra-violet spectrometry [34], LC [35,36,37] and CE [38]. Fourier transform infra-red spectrometric technique has also been utilised to determine chemical stability of the drug MFH [15] in pharmaceutical samples. These methods, no doubt, are selective and sensitive, but lack the simplicity and ease of operation, which are desirable for rapid routine analytical application. Visible spectrophotometry meets these criteria, but has been sparsely used for quantifying MFH.
A direct charge-transfer (C-T) complex formation reaction of MFH with iodine in acetonitrile medium has been reported [22]. The linear correlation exists between absorbance and concentration of the range 1.66–72.86 μg mL-1 MFH. A 2–12 μg mL-1 MFH quantification is possible in the method of iodine-drug C-T complex measurement at 295 nm, as reported by El-Bardicy et al. [24]. A complexation reaction between MFH and Cu(II) in cyclohexylamine medium to yield purple coloured product (λmax 540 nm) was found described [25]. The MFH-ninhydrin complex, measurable at 570 nm, is reported by Mubeen and Noor [39]. Vandana et al. [40] described the assay procedure of reaction between MFH and ninhydrin-molybdate mixture to form Ruhemann’s purple product with 570 nm as wavelength of maximum. Quantitative reaction was found to occur for 10 min at 90 ± 1 °C. In this ninhydrin-molybdate mixture method, MFH can be determined over 10–30 μg mL-1 concentration range. Pignard [41] has reported a method for MFH with NaOCl using NaOH and ZnSO4 to obtain and measure yellow coloured product. Methods were also developed to determine MFH with triacetyl and 1-naphthol in alkaline ethandiol [42] and bromothymolblue in phosphate buffer [43], and these are applicable exclusively for urine samples.
The stable molecular system formed in solution between an electron-donating molecule having sufficiently low ionization energy, and an electron accepting molecule having high electron affinity and these molecules are C-T complexes. Principally, these complexes possess new and intense absorption bands in either uv or visible region of the electromagnetic spectrum. Absorption bands of this type are known as C-T bands since they involve electronic transition from an orbital on the donor atom to the vacant orbital on the acceptor. C-T complexes are formed due to transfer of an electron from a π–molecular orbital of donor (Lewis base) to a vacant π–molecular orbital of the acceptor (Lewis acid), i.e. π–π electronic transitions [44]. These complexes are of particular interest, and they were widely applicable for assay of many therapeutics by spectrophotometry [45,46,47,48,49,50].
In spite of wide utility of C-T complexation reactions in analysis of therapeutic agents [45,46,47,48,49,50], MFH, according to literature survey, has not been determined based on these versatile reactions. Taking into account the basic nature of metformin, it was made to get reacted with PCA and DDQ (as π–acceptors) in acetonitrile medium to yield coloured products. Thus, two highly feasible and simple quantitation procedures with spectrophotometry were proposed herein for MFH. The coloured radical anions such as MFH-PCA (measurable at 530.0 nm) and MFH-DDQ (measurable at 460.0 nm) complexes are of greater interest in this report to quantify MFH. MFH was able to be determined more selectively and satisfactorily in human urine sample spiked by drug by these methods.
Methods
Instrumentation
The absorbances were recorded with digital Systronics manufactured spectrophotometer (Ahmedabad, Gujarat, India) and quartz cells (path length: 1 cm).
Analytical grade reagents and spectroscopic grade organic solvents were used through the investigation. The pure drug of pharmaceutical grade metformin hydrochloride (MFH, 99.9% pure) was procured from Sanofi Aventis, Mumbai, India, was utilised in the analysis. Cetapin XR and Glyciphage tablets (500 mg MFH/tablet), manufactured and supplied by Franco Pharmaceuticals and Sanofi Aventis of India, respectively, were obtained from local stores and used for analyses.
Preparation of stock solutions
Solutions of 0.2% (w/v) each of p-chloranilic acid (PCA) and 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) were prepared freshly using standard compounds (both from S.D. Fine Chem. Ltd., Mumbai) in 1,4-dioxan (Merck, India). A 10% (w/v) solution of NaOH was prepared in doubly-deionised water.
Preparation of standard metformin base (MEB) solution from metformin hydrochloride (MFH)
An amount (128 mg) of standard MFH corresponding to 100 mg of its base (MEB) was dissolved in 20 mL of water. After adding a 15 mL of 10% NaOH solution, the content was extracted for 2 min with 2 × 30 mL portions of dichloromethane. Each extract was washed thoroughly with 15 mL of deionised water in another separating funnel. The washed and pooled extracts were then passed through 5 g of anhydrous Na2SO4 and collected to 100 mL beaker. The solvent was then evaporated, and the residue dissolved in and diluted to 100 mL in a volumetric flask to get 1000 μg mL-1 MEB in acetonitrile. Suitable aliquots were then diluted to get 400 and finally to 80 μg mL-1 solutions in equivalent to MEB and analysed by PCA and DDQ methods, respectively.
General procedures
Procedure for bulk drug
Construction of calibration line
PCA method
A clean micro-buret was filled with 400.0 μg mL-1 MEB solution and transferred 0.1, 0.25, 0.5, 1.0,....... 4.0 mL into an array of 5 mL calibrated flasks. The total volume in each flask was increased to 4.0 mL with acetonitrile, 1.0 mL of PCA was added and contents were mixed and kept standby for 10 min. Then, the absorbance was recorded in reference to the reagent blank employing 530.0 nm as absorption maximum.
DDQ method
Different aliquots of 80.0 μg mL-1 MEB solution so as to cover 1.60 to 64.0 μg mL-1 concentrations were accurately transferred to a sequence of 5 mL calibrated flasks, the volume was raised to 4 mL with the aid of acetonitrile, 1 mL of DDQ solution added and after mixing, the contents were let stand ahead for 2 min. Each solution was used and recorded the absorbance at 460.0 nm versus the reagent blank.
The calibration lines of MEB concentration in μg mL-1 against absorbance in each method were constructed and used to calculate the MEB concentration in μg mL-1 in unknowns. Otherwise, the respective regression equation derived was also used to avail the concentrations in unknowns.
Procedure for tablets
Finely grounded tablet powder contained with 100 mg MEB was dissolved in about 20 mL of water and poured into a 125 mL separating funnel. The procedure of conversion of MFH to MEB was followed. The 400.0 and 80.0 μg mL-1 solutions in respect of MEB were obtained by dilution and correspondingly used for assay by PCA and DDQ methods. Aliquots of appropriate volumes were befitted for assay by the procedures as described under construction of calibration line.
Procedure for selectivity by analyses of placebo and synthetic mixture
A homogeneous powder of 40 mg each of acacia, hydroxyl cellulose, magnesium sodium alginate, stearate, starch, sodium citrate and talc was composed as placebo. Accurately 20 mg of placebo was used and extract prepared by following procedure described for tablets. A 2-mL aliquot of placebo extract was used for analysis following the procedures described in PCA and DDQ methods separately.
A synthetically prepared mixture of placebo and drug in the form of powder was constituted by mixing 128 mg MFH and 100 mg placebo. Metformin base (MEB) solution in acetonitrile was prepared using the procedure for preparation of placebo or tablet extract and was used for analyses by PCA and DDQ procedures. The percentage recovery of MEB was then calculated.
Procedure for spiked human urine sample
Urine (5 mL) was spiked with 128 mg of pure MFH, diluted to 10 mL with 10% NaOH and carefully taken in the separating funnel. Then, the procedure to prepare basic form of MFH was intact followed. After evaporation, the residue was constituted in acetonitrile and the resulting solution was diluted to get 400 and 80 μg mL-1 with acetonitrile, and used in PCA method or DDQ method, respectively. The drug contents recovered (in %MEB) were calculated for each case.
Results
Spectral characteristics
The reaction between MEB and PCA resulted an intense reddish violet complex with analytical wavelength of maximum (λmax) at 530.0 nm (Fig. 2). The PCA radical anion was assumed to be formed there. In resembling to the reaction of MEB with PCA, the free base of MFH, MEB also reacted with DDQ to produce an intense orange-red colored complex with highly intense band at 460.0 nm. Two other bands (at 510.0 and 570.0 nm) (Fig. 2) were also seen in the spectra, and the choice of 460.0 nm was made because of sensitivity of the reaction product and low blank absorbance.

Absorption spectra of MEB-PCA and MEB-DDQ C-T complexes
Method development
Optimization of reaction conditions
Optimum conditions were established by measuring absorbance of either MEB-PCA or MEB-DDQ C-T complexes, by varying parameters separately and individually at a time while others maintained intact or unvaried.
Effect of volume of PCA and DDQ
Figure 3 illustrates the impact of PCA and DDQ concentrations on their respective complexes with MEB. MEB was allowed to react with either 0.5–3 mL of 0.2% PCA or 0.5–3 mL of 0.2% DDQ. Maximum and minimum absorbance values were obtained for sample and blank, respectively, when 1 mL of PCA or DDQ used in PCA or DDQ method.

Effect of PCA and DDQ concentrations on absorbance of respective C-T complexes
Effect of dissolution or reaction solvent
MEB was reacted with either PCA or DDQ in acetonitrile, chloroform, ethanol, methanol, acetone, 1,4-dioxane, dichloromethane and dimethylformamide. Maximum sample absorbance and minimum blank absorbance were observed in acetonitrile. The respective results, come across during the fixing up of solvent are presented in Table 1 below, indicated that acetonitrile as a suitable solvent.
Effect of time and stability of C-T complexes
The times required to form a stable C-T complexes were fixed by measuring absorption of the respective complex formed upon adding the PCA or DDQ solution to MEB solution at different intervals of times. The color formation was complete in 10 min in PCA, and 2 min in DDQ methods were stable for 2.5 and 3 h of time in PCA and DDQ methods, respectively.
Investigation of stoichiometry of CT complexes
Principle of Job’s continuous variations method [51] was invaded to evaluate the reaction stoichiometry between MEB and either PCA or DDQ. In this experiment, equi-molar solutions of drug and reagent were mixed out of 5.0 mL total volume. The plots of absorbance at wavelength of maximum versus mole ratio of MEB in each method were prepared (Fig. 4). The maximum absorbance of both MEB-PCA and MEB-DDQ C-T complexes at mole ratio of 0.5 indicated that the drug in base form (MEB) and reagent (PCA or DDQ) followed a 1:1 reaction stoichiometry.

Job’s plots in a. PCA method (3.27 × 10-4 M MEB and PCA) and b. DDQ method (4.09 × 10-4 M MEB and DDQ)
Method validation
The optimised experimental conditions have been invaded and the final procedures for PCA and DDQ methods have been described. The validity of both the analytical methods has been assessed for linearity, accuracy, precision, robustness and ruggedness on par with the ICH guidelines [52, 53]. The details and consequent assessment results from validation are presented in sections below.
Evaluation of linearity with calibration and sensitivity parameters
A linear relationship was originated between absorbance at λmax and concentration of MEB (Fig. 5) in the ranges of concentrations mentioned below (Table 2). The method of least squares was followed and the slopes (m), intercepts (b) and correlation coefficients ® in PCA and DDQ methods were calculated and are also reported (Table 2). The calibration or linear range, molar absorption coefficient, Sandell’s sensitivity and ranges of PCA and DDQ methods are also tabulated (Table 2). The detection (DL) and quantitation limits (QL) have been calculated using the slope of the calibration curve and the standard deviation of the blank absorbance readings of respective method as per the ICH Guidelines [53]. The formulae used to calculate DL and QL are given below:

Calibration curves for a. PCA method and b. DDQ method
where SD is the standard deviation of absorbance of set of blanks and m is the slope of the calibration curve of respective method. The values of DL and QL for PCA and DDQ methods have been reported in Table 2.
Accuracy, repeatability and intermediate variation
The assay procedures were repeated seven times within the day and five times on different days to evaluate repeatability and intermediate variation, respectively. The accuracies have also been reported in the mean time for proposed PCA and DDQ methods. Solutions of different concentrations of MFH equivalent to MEB were assayed at three levels. The results, expressed as %RSD, were ≤ 1.13 in intra-day and ≤ 1.75 (Table 3) in inter-day variations indicating high precision with respect to repeatability and less deviations in the results of assay in the study of intermediate variations of both the methods. Accuracy, expressed as %RE, between the mean/average concentrations and true or taken concentrations of MEB. The %RE values in Table 3 were calculated for the studied concentrations of MEB, were ≤ 2.18% demonstrated the high accuracies of presented PCA and DDQ methods.
Evaluation of robustness and ruggedness
Small incremental changes were made in the volume of reagent and time. The effects of the changes were studied on the absorption of complex systems for study of robustness. In ruggedness study, assays were done by three analysts, and also by a single analyst performing analysis on three different instruments in the same laboratory. The intermediate precision values, expressed as %RSD, were from 0.47 to 2.98 indicating acceptable robustness and ruggedness. Table 4 below is dedicated to highlight the overall results of these studies.
Selectivity
After subjecting the placebo extract for analysis the resembling results with reagent blank confirmed inactive role of substances in placebo. The selectivity was also ensured by analysing synthetic mixture extract. The analysis yielded a percentage recovery values of MFH from 97.89 to 101.70 with a standard deviation below 2%. This demonstrated the non-interference of additives in a synthetic mixture in MFH quantification.
Application to tablets
Cetapin XR 500 mg (product X) and Glyciphage 500 mg (Product Y) tablets were subjected to analysis by general quantification procedures. The mean recoveries were calculated and compared with official method [3] statistically. The results of statistical tests as revealed by the Student’s t and variance ratio F values [52] were in very good agreement. The mean recovery values of MFH from PCA, DDQ and official methods with standard deviation, t and F values are indicated below in Table 5.
Accuracy by recovery study
The accuracy and selectivity were further confirmed by performing a recovery test. To a fixed quantity of the pre-analyzed tablet extract, pure MFH solutions were spiked at three levels of different concentrations and total MFH was determined by following proposed PCA and DDQ methods. Three replicates from each level were assayed; the recoveries of MFH (pure) were calculated and compiled in Table 6. These results reveal that commonly added adjuvants did not show interference. Further, the results also outlined the selectivity of analytical methods with accurate determination.
Application to spiked human urine
The proposed analytical procedures were employed to determine MFH in extract of human urine spiked by pure drug so that the usefulness of them for physiotherapeutic administration of drug to be made possible. The obtained results of spiked human urine analysis are presented below in Table 7.
Discussion
Various methods were reported by researchers for MFH determination. The articles describing the analytical procedures using chromatographic [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20], electrophoretic [21] and spectrofluorimetric [25] techniques were described as selective methods for MFH. Unfortunately, the need for sophisticated instruments and highly skilful operator to execute the assay plan made them disadvantageous. On the other end, the reported ultra-violet spectrophotometric methods [22,23,24,25,26,27,28] are also suffering from limitations such as shorter analytical wavelength where the possible photometric error arises, less sensitivity, narrow linear range or additional extraction steps with organic solvents. The potentiometric methods [29,30,31,32] are usable only with the fabricated electrodes or sensors which have not been thoroughly validated and moreover with shorter life time. Requirement of larger sample size per titration is the major drawback of conductometric method [33]. The methods developed for MFH using spectrophotometry [34], liquid chromatography [35,36,37] and capillary electrophoresis [38] are applicable for simultaneous determination of MFH in the presence of other therapeutics. There are few visible spectrophotometric methods were also developed and published. The details, performance characteristics and limitations of reported visible spectrophotometric methods are summarised below in Table 8.
The limitations or disadvantages of reported spectrophotometric methods have been showed in the above table (Table 8), and thus, a clear footstep was taken to develop simple analytical methods with complete validation to quantify MFH in pharmaceuticals and urine.
Metformin base (MEB) as n-donor, reacts with PCA and DDQ, the π-acceptors, to form C-T complexes, which dissociates into radical anions in solvents such as acetonitrile. The e- transfer process from the donor to the acceptor mostly occurs, and consequently, the intensely colored radical anions observed. The probable pathway is indicated below:

The reaction between π-acceptors and the hydrochloride salts of amines is infeasible because of inability of N of amine in contributing e- [50]. Since MFH contains HCl, it was converted to base form, i.e. MEB, extracted into dichloromethane, solvent evaporated and finally the solution in acetonitrile obtained [54]. The possible reaction schemes showing the pathways for formation of MEB-CAA and MEB-DDQ C-T complexes are outlined in Scheme 1 in analogous to previous works [47, 55,56,57,58,59].

Pathway of reactions showing the MEB-PCA and MEB-DDQ C-T complex formation in acetonitrile.
The acidic hydrochloride of MFH was eliminated out by extraction and obtained the drug in basic form (MEB). The basic nitrogenous group of MEB was utilised to react with PCA and DDQ to develop two new visible spectrophotometric methods based on the measurement of absorbance of 1:1 in stoichiometric MEB-PCA and MEB-DDQ C-T complexes at 530.0 and 460.0 nm, respectively. The reaction between MEB and reagents took place in 10 and 2 min to form intense MEB-PCA and MEB-DDQ C-T complexes, respectively, and respective complexes were stable for 2.5 and 3 h. Acetonitrile was showed greater selectivity for complexes’ stability over other solvents such as methanol, ethanol, acetone, 1,4-dioxane, chloroform, dichloromethane, dichloroethane and dimethylformamide. The validation of methods was performed according to ICH Guidelines [52, 53]. The calibration curves were linear over the concentration ranges of 8.0–320.0 and 1.6–64 μg mL-1 MEB in PCA and DDQ methods, respectively, with molar absorptivity values of 7.33 × 102 and 2.57 × 103 L mol-1 cm-1. The regression coefficient values close to unity indicated the closed correlation between concentration of MEB and absorbance at respective analytical λmax in both the methods. The values of Sandell sensitivity, DL and QL of both the methods revealed the acceptable sensitivity. The values of RSD of 0.54 to 1.75% and RE of 0.62 to 2.18% declared the excellent precision and accuracies of proposed methods to the assay of MFH in the form of MEB. The methods were also complied for robust nature, as per the results expressed as %RSD obtained from deliberate variations of reagents and reaction time. Besides the results in %RSD from study of intermediate variation also revealed the excellent ruggedness of the methods. The statistical tests between the results of proposed (PCA and DDQ methods) and official methods yielded a satisfactory outcome of non-significant differences between performances of proposed and official methods with respect to accuracy and precision. The procedures were checked for selectivity by applying to the analysis of placebo and synthetic mixture. The inactive role of compounds in placebo and synthetic mixture was observed. The absorbance of placebo extract was same as blank and the recovery of MEB from synthetic mixture was close to 100%. The assessment of accuracy and selectivity by analysis of tablets using standard addition procedure was produced the mean recovery of MEB as 100.86 and 99.34% in PCA and DDQ methods, respectively. The values very close to 100% declared greater selectivity of PCA and DDQ methods. The methods’ applicability to determine MFH in the form of MEB was also proved by spiked human urine analysis and in which the percent recovery of MEB was 97.87 to 98.72.
Conclusion
This is the first article utilising substituted p-benzoquinones to rapidly and reliably quantify metformin hydrochloride (MFH) in original/pure sample and tablets by spectrophotometry. The hallmarks of the presented methods are simplicity and ease of performance without the need for pH adjustment, heating, extraction and/or multireaction step as found in methods of already reported articles. The methods involve simple mixing of the reactants and absorbance measurement almost immediately. The advantageous factors of proposed methods are wide linear dynamic ranges, acceptable accuracies, precisions, robustness and ruggedness. The selectivity studies produced confident results, and they permitted the MFH determination without the need for time consuming sample preparation steps. However, the methods require the drug to be present in its base form, which is obtained by an extraction step.
Availability of data and materials
Data and material are available upon request.
Abbreviations
- MFH:
-
Metformin hydrochloride
- PCA:
-
p-Chloranilic acid
- DDQ:
-
2,3-Dichloro-5,6-dicyano-p-benzoquinone
- DL:
-
Detection limit
- QL:
-
Quantitation limit
- RSD:
-
Relative standard deviation
- RE:
-
Relative error
- CT:
-
Charge transfer
- MEB:
-
Metformin base
- CAA:
-
p-Chloranilic acid
References
British Pharmacopoeia (2007), British Pharmacopeia Commission, Her Majesty’s Stationery Office, London.
Bailey CJ, Path MRC, Turner RC (1996) Metformin. N Engl J Med 334:574–579
European Pharmacopoeia, Strasbourg, Council of Europe, 2007;6.0: 2370.
The United States Pharmacopoeia, XXXII Revision, the National Formulary XXVII, Rockville, USP Convention, 2009; 2905.
Al-Rimawi F (2009) Development and validation of an analytical method for metformin hydrochloride and its related compound (1-cyanoguanidine) in tablet formulations by HPLC-UV. Talanta 79:1368–1371
Kar M, Choudhury P (2009) HPLC Method for estimation of metformin hydrochloride in formulated microspheres and tablet dosage form. Indian J Pharm Sci 71:318–320
Lad NR, Bhoir SI, Bhoir IC, Sundaresan M (2003) Concurrent assay of Metformin and Glimepiride in tablet using RP-HPLC with wavelength programming. Indian J Pharm Sci 65(6):650–653
Gindy AE, Nassar MW, Abasawy NME, Attia KAS, Shabrawi MA (2010) Optimization and validation of an RP-HPLC method for direct determination of metformin hydrochloride in human urine and in a dosage form. J AOAC Intl 93:1821–1828
Devendra K, Saurabh S, Rachumallu R, Rakesh KD (2014) Development and validation RP-HPLC-PDA method for the determination of metformin in bulk and dosage form. World J Pharm Pharm Sci 3:745–757
Gomes P, Sippel J, Jablonski A, Steppe M (2004) Determination of rosiglitazone in coated tablets by MEKC and HPLC methods. J Pharm Biomed Anal 36:909–913
Radhakrishna J, Satyanarayana J, Satyanarayana A (2002) LC Determination of rosiglitazone in bulk and pharmaceutical formulation. J Pharm Biomed Anal 29:873–880
Vasudevan M, Ravi J, Ravisankar S, Suresh B (2001) Ion-pair liquid chromatography technique for the estimation of metformin in its multicomponent dosage forms. J Pharm Biomed Anal 25:77–84
Khanolkar DH, Shindhe VM (1999) RP/HPLC method for the estimation of gliben clamide and metformin HCl from combined dosage form. Indian Drugs 36:739–742
Arayne MS, Sultana N, Zuberi MH (2006) Development and validation of RP-HPLC method for the analysis of metformin. Pakistan J Pham Sci 19:231–235
Anna G, Anna B-R, Tomasz M, Krzysztof W (2019) Determination of chemical stability of two oral antidiabetics, metformin and repaglinide in the solid state and solutions using LC-UV, LC-MS, and FT-IR methods. Molecules 24:4430
Gayathri S, Shantha A, Vaidyalingam V (2003) Simultaneous HPTLC determination of gliclazide and rosiglitazone in tablets. Indian J Pharm Sci 65:663–665
Havele SS, Dhaneshwar SR (2011) Simultaneous determination of metformin hydrochloride in its multicomponent dosage forms with sulfonyl ureas like gliclazide and glimepiride using HPTLC. J Liq Chromatogr Rel Tech 34:966–980
Sane RT, Francis M, Moghe A, Khedkar S, Anerao A (2002) High-performance thin-layer chromatographic determination of rosiglitazone in its dosage form. J Planar Chromatogr Mod TLC 15:192–195
Afnan EA, Hadir MM, Nourah ZA (2020) HPTLC method for the determination of metformin hydrochloride, saxagliptin hydrochloride, and dapagliflozin in pharmaceuticals. Current Anal Chem 16:609–619
Sane RT, Banavalikar VJ, Bhate VR, Nayak VG (1989) Gas-chromatographic determination of metformin hydrochloride from pharmaceutical preparations. Indian Drugs 26:647–648
Handom II, Bani AKJ, Abushoffa AM (2010) Development and validation of a stability indicating capillary electrophoresis method for the determination of Metformin -hydrochloride in tablets. J Pharm Biomed Anal 53:1254–1257
Ashour S, Kabbani R (2003) Direct spectrophotometric determination of metformin hydrochloride in pure form and in drug formulations. Anal Lett 36:361–370
Aruna A, Nancy K (2000) Simultaneous estimation of metformin hydrochloride and glipizide solid dosage forms by ultraviolet spectrophotometry. Indian Drugs 37:533–534
El-Bardicy MG, El-Khateeb SZ, Ahmad AKS, Assad HN (1989) Spectrophotometric determination of metformin via charge-transfer complex with iodine. Spectrosc Lett 22:1173–1181
Hassan SSM, Mahmoud WH, Elmosallamy MAF, Othman OHM (1999) Determination of metformin in pharmaceutical preparations using potentiometry, spectrofluorimetry and UV–visible spectrophotometry. Anal Chim Acta 378:299–311
Saxena PN, Ajay S, Raghuvamshi S, Jain VK, Patel A, Gupta N (2010) UV spectrophotometric method for the quatitation of metformin hyrochloride in pharmaceutical dosage form. Orient J Chem 26:1553–1553
Umapathi P, Ayyappan J, Quine SD (2012) Quantitative determination of metformin hydrochloride in tablet formulation containing croscarmellose sodium as disintegrant by HPLC and UV spectrophotometry. Trop J Pharm Res 11:107–116
Shrikrishna B, Vinod P, Smrutidevi S, Mahesh R, Deepali K (2012) Validation of uv spectrophotometric method with stress degradation study for metformin hydrochloride. Pharma Tutor:Article ID-1298
Rizk MS, Abdel-Fattah M, Issa YM, Atia EM (1993) A new metformin selective plastic membrane electrode based on metformin tetraphenylborate. Anal Lett 26:415–428
Shoukry AF, Rizk MS, Abdel-Fattah HM, Issa YM, Atia EM (1994) Construction and performance characteristics of a metformin electrode based on the metformin phosphotungstate ion-associate. J Chem Technol Biotechnol 60:217–222
Rizk MS (1994) Metformin-selective PVC membrane electrode based on metforminium phosphomolybdate ion pair. J Chem Technol Biotechnol 61:67–72
Rizk MS (1995) Metformin-selective poly(vinyl chloride) (PVC) membrane electrode based on the metforminium phosphomolybdate ion pair. Electroanalysis 7:687–691
Abou-dan M, Ashour S, Abou-dan H (2001) Conductometric titration of metformin in pure form and in pharmaceutical preparations using sodium tetraphenylborate and cetylpyridinium bromide. Asian J Chem 13:1–7
Kharbade S, Asnani A, Pratyush K (2019) Development and validation of UV spectrophotometric method for simultaneous estimation of metformin HCl and repaglinide in pharmaceutical formulation. J Drug Deliv Ther 9:344–347
Hanan AM, Nesrin KR, Sherine SD, Azza AM (2017) Chromatographic methods for the simultaneous determination of binary mixture of Saxagliptin HCl and metformin HCl. Bull Fac Pharm Cairo Univ 55:311–317
Ahmed G, Hani A-S, Crispin RD (2019) Development and validation of a new analytical HPLC method for simultaneous determination of the antidiabetic drugs, metformin and gliclazide. J Food Drug Anal 27:315–322
Nawab S, Nasreen F, Shahnaz P, Farhan AS (2019) Simultaneous determination of anti-diabetic drugs. Braz J Pharm Sci 55:e17394
Hadir MM, Afnan EA, Nourah ZA, Haya IA (2019) Stability-indicating capillary electrophoresis method for the simultaneous determination of metformin hydrochloride, saxagliptin hydrochloride, and dapagliflozin in pharmaceutical tablets. J Liq Chromatogr Rel Tech 42:161–171
Mubben G, Noor K (2009) Spectrophotometric method for analysis of metformin hydrochloride. Ind J Pharm Sci 71:100–101
Vandana PP, Subhash JD, Suvarna HK, Sudarshan TK, Pramod OK (2014) Molybdate assisted ninhydrin based spectrophotometric method for the estimation of metformin hydrochloride in bulk drugs and tablet dosage form. J Indian Chem Soc 91:171–177
Pignard P (1962) Dosage spectrophotométrique du NN dimethylbiguanide dans le sang et l’urine. Ann Biol Clin 20:325–333
Siest G, Ross F, Gabou JJ (1963) Dosage du NN dimethylbiguanide par le diacetyle en milieu alcalin. Bull Soc Pharm 58:29–38
Garret ER, Tsau J (1972) Application of ion-pair methods to drug extraction from biological fluids I: quantitative determination of biguanides in urine. J Pharm Sci 16:1404–1410
Zhong-hua C, Adélia JAA, Andrew CHS, Hans L (2018) Analysis of charge transfer transitions in stacked π-electron donor–acceptor complexes. Phys Chem Chem Phys 20:26957–26967
Saleh GA (1998) Charge-transfer complexes of barbiturates and phenytoin. Talanta 46:111–121
Gouda AA (2009) Utility of certain sigma- and pi-acceptors for the spectrophotometric determination of ganciclovir in pharmaceutical formulations. Talanta 80:151–157
Khaled E (2008) Spectrophotometric determination of terfenadine in pharmaceutical preparations by charge-transfer reactions. Talanta 75:1167–1174
El-Sherif ZA, Mohamed AO, Walash MI, Tarras FM (2000) Spectrophotometric determination of loperamide hydrochloride by acid-dye and charge-transfer complexation methods in the presence of its degradation products. J Pharm Biomed Anal 22:13–23
Walash M, Sharaf-El Din M, Metwalli MES, Reda Shabana M (2004) Spectrophotometric determination of nizatidine and ranitidine through charge transfer complex formation. Arch Pharm Res 27:720–726
Raza A, Ijaz AS, Atta-ur-Rehmana, Rasheed U (2007) Spectrophotometric determination of ondansetron hydrochloride in pharmaceutical bulk and dosage forms. J Chin Chem Soc 54:223–227
Douglas AS, Donald MW (1971) Principles of instrumental analysis. Holt, Renehart, Winston, New York, p 104
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonized Tripartite Guideline, Validation of analytical procedures: text and methodology Q2(R 1), Complementary Guideline on Methodology dated 06 November 1996, incorporated in November 2005, London.
International Conference on Harmonization (ICH) Harmonized Tripartite Guideline: Validation of analytical procedures: text and methodology Q2(R1), November 2005, London.
Mostafa AA, Bebawy LI, Refaat HH (2002) A spectrophotomtric determination of clobetasol propionate, Halobetasol propionate, quinagolide hydrochloride, through Charge transfer complexation. J Pharm Biomed Anal 27:889–899
Basavaiah K, Sameer AMA (2010) Use of charge transfer complexation reaction for the spectrophotometric determination of bupropion in pharmaceuticals and spiked human urine. Thai J Pharm Sci 34:134–145
Basavaiah K, Raghu MS, Vinay KB (2012) Simple and rapid spectrophotometric assay of levocetirizine in pharmaceuticals through charge-transfer complexation using chloranilic acid and 2,3-dichloro-5,6-dicyanoquinone as π-acceptors. Bull Chem Soc Ethiop 26:319–328
Rahman N, Nasrul HM (2003) Validated spectrohotometric methods for the determination of amlodipine besylate in drug formulation using 2,3-dichloro 5,6-dicyano 1,4-benzoquinone and ascorbic acid. J Pharm Biomed Anal 31:381–392
Prashanth KN, Basavaiah K (2012) Utility of p-chloranilic acid and 2,3-dichloro-5,6-dicyano-p-benzoquinone for the spectrophotometric determination of rizatriptan benzoate. ISRN Anal Chem 2012:1–12
Vinay KB, Revanasiddappa HD, Raghu MS, Abdulrahman SAM, Rajendraprasad N (2012) Spectrophotometric determination of mycophenolate mofetil as its charge-transfer complexes with two π-acceptors. J Anal Methods Chem 2012:1–8
Acknowledgements
Authors thank Sanofi Aventis, Mumbai, India, for gifting metformin hydrochloride pure sample. The first author is grateful to the JSS Mahavidyappetha, Mysuru, and the Principal of JSS College of Arts, Commerce and Science, B N Road, Mysuru, India, for providing the facilities to pursue this work.
Funding
The work is not funded by any of the government or non-government bodies. The work is self-financed one.
Author information
Authors and Affiliations
Contributions
NR and KB contributed equally to the work and in the overall work of manuscript preparation. Both authors read and approved the manuscript for further processing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rajendraprasad, N., Basavaiah, K. Application of two charge transfer complex formation reactions for selective determination of metformin hydrochloride in pharmaceuticals and urine. Futur J Pharm Sci 6, 62 (2020). https://doi.org/10.1186/s43094-020-00082-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-020-00082-6