
Abstract
Background
Four rapid, accurate, and validated stability-indicating spectrophotometric methods have been described in the present work for the analysis of trimebutine maleate (TM) in existence of its degradation products in its authentic form and in pharmaceutical formulations excluding any separation steps.
Results
These methods were a dual-wavelength (DW) method which allows the determination of TM in existence of its degradation products at 243 nm and 269 nm, second derivative (D2) method measured at peak amplitude at 268 nm, ratio difference (RD) method at 242 nm and 278 nm, and constant center coupled with spectrum subtraction (CC-SS) method at 242 nm and 278 nm versus 278 nm. By applying the suggested methods, TM could be quantified in the range of 5.0–60.0 μg/mL with percentage recoveries 99.97 ± 0.40, 100.36 ± 0.58, 99.90 ± 0.42, and 100.15 ± 0.45 for DW, D2, RD, and CC-SS methods, respectively. International Conference on Harmonization guidelines were followed for validation of the described methods, and the application of laboratory-prepared mixtures along with different pharmaceutical drugs including the target drug showed favorable results without any contribution from additives.
Conclusions
Statistical comparison was used to compare the proposed and official methods, and satisfactory results for both accuracy and precision were obtained. The results confirm the applicability of the suggested methods for the determination of TM in quality control laboratories.
Similar content being viewed by others

Background
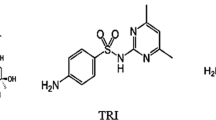
Chemically, trimebutine maleate (TM) identified as (2RS)-2-(dimethylamino)-2-phenylbutyl 3,4,5-trimethoxybenzoate (Z)-butenedioate (Fig. 1) is an antispasmodic drug and is effective in the remediation of irritable bowel syndrome which is the well-known usage of TM over a lot of previous years [1]. It is a noncompetitive spasmolytic agent with mild opiate receptor affinity and remarkable anti-serotonin activity. Over the last decade, it reduces abnormal intestinal activity but does not alter normal GI motility and is indicated for the remediation and repose of spastic colon symptoms. Moreover, it is also used to cure patients having postoperative paralytic ileus following an abdominal surgery [2, 3].

Chemical structure of trimebutine maleate (TM)
Trimebutine maleate (TM) is an official drug presented in the British Pharmacopoeia [1]. Literature review encountered that several analytical methods were published for the quantitative analysis of TM in pharmaceutical drugs and in physiological fluids. These methods include few simple and direct UV-spectrophotometric methods for the resolution of trimebutine maleate in existence of its degradation products through the application of first derivative and first derivative of ratio spectra spectrophotometric methods [4]; different visible spectrophotometric methods were reported for the determination of TM in mixture with other pharmaceutical drugs using different coloring agents in different reaction conditions [5,6,7,8,9], high performance liquid chromatographic methods [4, 10,11,12,13,14,15,16,17,18,19], and electrochemical methods [20,21,22,23].
Stability is considered to be one of the most important criteria in quality control related to pharmaceuticals. Only stable pharmaceutical drugs would guarantee accurate delivery of the drug to the patients. Expiration dating on any drug formulation depends on scientific studies at ordinary and stressed conditions. The International Conference on Harmonization (ICH) [24] approved a stability test which suggests that it is important to investigate the inherent stability properties of the drug product through certain stress studies, for instance, hydrolysis study; therefore, this will lead to the identification and determination of drug products which will support the developed stability-indicating analytical method.
Degradation of trimebutine maleate (TM) is considered an easy technique as it is an ester-type antispasmodic drug. It was reported that it was subjected to hydrolysis under alkaline and acidic conditions to yield the same degradation products, namely 2-(dimethyl amino)-2-phenylbutanol and 3,4,5-trimethoxy benzoic acid [4]. However, the degradation products used in this work were prepared using the alkaline hydrolysis as mentioned in [4] where its condition was 100 mL 0.1 M NaOH at 100 °C for only 30 min that was enough for a complete degradation, whereas the conditions for acid hydrolysis was 100 mL 1 M HCl at 100 °C for 12 h which was enough for complete degradation even though it produces the same degradation products as that produced by the alkaline hydrolysis. That is why it is time-saving to stick to the alkaline hydrolysis that also produces the same degradation products as that obtained from acidic hydrolysis. Therefore, the proposed methods could be applied for the analysis of TM in the presence of either alkali or acid-induced degradation products.
The ultimate target of this study is to achieve and validate stability-indicating UV-spectrophotometric methods that are simple, rapid, and selective with less cost and time for the analysis of trimebutine maleate in existence of its degradation products in its authentic form and in market samples excluding any separation steps through various manipulating pathways so as to obtain satisfying results that are characterized by high accuracy and precision and are more sensitive for the substance in charge.
Experimental
Apparatus and software
The following are the apparatus and software used:
A double beam UV/VIS spectrophotometer (Shimadzu, Japan) UV/VIS model UV-1800 PC with 1-cm path length quartz cell. The width of the spectral band was 2 nm, and the scanning speed was 2800 nm/min.
An IR spectrophotometer (Shimadzu 435, Kyoto, Japan).
A mass spectrophotometer: MS-QB 1000 EX, Finnigan Nat (USA).
Thin-layer chromatography (TLC) plates (20 cm × 20 cm) coated with silica gel 60 F254 (Merck, Germany).
Sonicator (Elmasonic – S30H), Germany.
UV lamp with short wavelength 254 nm (USA).
Magnetic stirrer, Bandelin Sonorox, Rx5l0S (Budapest, Hungarian).
Materials and solvents
Pure sample
Trimebutine maleate (TM) pure samples were kindly provided from the National Organization for Drug Control and Research (NODCAR), Egypt. Their purity was checked to be 99.73 ± 0.86 in reference to the official HPLC method [1].
Pharmaceutical formulations
Gast-reg® 100 mg tablets (Batch No. 144695), 200 mg tablets (Batch No. 162912), ampoules 50 mg/5 mL (Batch No. 154534), and suspension 24 mg/5 mL (Batch No. 163650) were manufactured by Amoun Pharmaceuticals Co., Egypt.
Solvents
The following are the chemicals of analytical grade used in this study:
Concentrated hydrochloric acid solution (Adwic, Egypt) used to prepare 1 M HCl
Sodium hydroxide pellets (Adwic, Egypt) used to prepare 0.1 M NaOH
Isopropanol (SDFCL, India)
Concentrated ammonia solution (Teba, Egypt)
De-ionized water (Egypt Otsuka Pharmaceutical Co., SAE, Egypt)
Methanol (Adwic, Egypt)
Chloroform (Adwic, Egypt)
Standard solutions
Standard stock solutions
Preparation of TM standard stock solutions (1.0 mg/mL) along with its degradation products (derived from 1.0 mg/mL) was accomplished in 100-mL measuring flasks by dissolving 100 mg of each of them in methanol. Following this step, the volumes were increased until it reaches the mark using the same solvent and then kept in the refrigerator.
Standard working solutions
The working solutions were prepared freshly from the stock solutions by dilution using methanol to achieve a concentration of 100.0 μg/mL for each component.
Procedures
Preparation of degradation products of TM (derived from 1 mg/mL)
The preparation of TM degradation products was accomplished by refluxing 0.1 g of TM with 10 mL 0.1 M sodium hydroxide at 100 °C following the reported method [4]. Complete degradation was done by refluxing for exactly 30 min that was confirmed by TLC development through the invisibility of the drug spot by the use of UV lamp at 254 nm using isopropanol-water-ammonia (14:4:2, by volumes) as a developing system. Potentiometer was used to adjust the pH of the solution to be 4.5 ± 0.02 using several values of 1.0 M hydrochloric acid and that pH was found to be adequate for precipitating the degradation products. Subsequently, filtration of the precipitate was done then dried under vacuum by the aid of separating funnel. Consequently, analysis of that dried precipitate was conducted by UV spectroscopy, IR, and mass spectrum and was confirmed that the precipitate is DEG 1. The filtrate was washed for several times, each time using 10.0 mL chloroform. The aqueous extract that was just washed was evaporated then dried under vacuum. The residue was analyzed by IR and mass spectrum and was confirmed that the filtrate is DEG 2. Then, the preparation of 1 mg/mL stock solution of the degradation products (derived from intact) was done by dissolving all the content of the solution after neutralization in 100 mL methanol.
Spectrophotometric methods
Spectral characteristics
The absorption spectra of 30.0 μg/mL of both TM and its degradation products in methanol were recorded all over the UV range of 200–400 nm using methanol as a blank.
Construction of calibration graphs
Aliquots equal to 50.0–600.0 μg TM were accurately transferred into a series of 10-mL measuring flasks from its corresponding working solution (100.0 μg/mL) following that the volume was completed to the mark using methanol to achieve solutions of final concentrations (5.0–60.0 μg/mL). Scanning of the absorption spectra of the resulting solutions from 200 to 400 nm was done and kept in the computer. Average of the three experiments were used to construct the calibration graphs of each method, and the regression equations were calculated.
Manipulating steps on zero-order absorption spectra
Dual-wavelength (DW) method
The absorbances of the stored spectra were recorded at 243 nm and 269 nm. The calibration curve was done gathering the differences between the recorded absorbance and the corresponding concentrations using the average of the three experiments, and the regression equation was calculated.
Second derivative method
The stored second derivative (D2) spectra of TM were measured through the range of 240–300 nm against solvent blank using Δλ = 8 nm and scaling factor of 100. Peak amplitude at 268 nm (zero-crossing of the degradation products) was used for constructing the calibration curve against the corresponding concentrations of TM, and the regression equation was calculated.
Manipulating steps on ratio spectra
Ratio difference (RD) method
In this method, ratio spectra were obtained and recorded by dividing the absorption spectra of the zero-order of TM (5.0–60.0 μg/mL) over the absorption spectrum of 30.0 μg/mL degradation products used as a divisor. The difference between the amplitudes of stored ratio spectra at 242 nm and 278 nm against the corresponding concentrations of TM was used for constructing the calibration curve, and then the regression equation was calculated.
Constant center coupled with spectrum subtraction (CC-SS) method
This method seems to look like the ratio difference method only in the beginning of the manipulating steps as the same ratio spectra were used with the same divisor 30.0 μg/mL of the degradation products. Subsequently, two calibration curves were obtained, one of them relating the absorption spectra of the zero-order of TM at 266 nm against its corresponding concentrations and the other one relating the difference between the amplitudes of the ratio spectra at 242 nm and 278 nm against the amplitudes at 278 nm, and therefore, two regression equations were calculated.
Analysis of laboratory-prepared mixtures
Different laboratory-prepared mixtures having different ratios of the target drug and its degradation products (5.0–85.0%) were prepared using different aliquots of TM working solutions (100.0 μg/mL) and its degradation products (derived from 100.0 μg/mL). These mixtures were added into a series of 10-mL measuring flasks, then the volumes were made up to the mark with methanol while mixing well of the solutions, measuring those prepared laboratory-prepared mixtures throughout the UV range. Then, follow the instructions as described in each spectrophotometric method. Consequently, the concentration of TM in each mixture was obtained from each corresponding regression equation.
Application on pharmaceutical formulations
For Gast-reg® (100 mg and 200 mg) tablets
Twenty tablets of each of Gast-reg® 100 mg and 200 mg tablets were weighed separately with high accuracy then finely powdered. An amount exactly equals to 100.0 mg of powder was added to a 100-mL beaker with 50 mL methanol. The solution was shaken in an ultrasonic bath for about 5 min then filtered into a 100-mL measuring flask, and the volume was made up to the mark with water in order to get a concentration of 1.0 mg/mL.
For Gast-reg® ampoules (50 mg/5 mL)
The content of five Gast-reg ampoules was mixed; 5.0 mL from the content of five ampoules was transferred accurately to a 50-mL measuring flask and made up to the mark with methanol to attain a final concentration of 1.0 mg/mL. Then, further dilution was made by transferring 2.5 mL in a 25-mL measuring flask to attain a working solution of concentration 100.0 μg/mL.
For Gast-reg® suspension (24 mg/5 mL)
Gast-reg suspension was constituted in 70 mL distilled water and mixed well to attain a final concentration of 24.0 mg of drug/5 mL. Five milliliters of the resulting suspension was transferred quantitatively into a 50-mL measuring flask, and some methanol were added then filtered through a filter paper. After the complete filtration, the solution was made up to the mark with methanol to attain a final concentration claimed to be 0.48 mg/mL.
Appropriate dilutions were done for the analyzed formulations to get solutions with final concentrations claimed to be 20.0 μg/mL of TM in case of Gast-reg® tablets and ampoules while 24.0 μg/mL of TM in case of Gast-reg® suspension. Then, the solutions were analyzed using the procedure described under the proposed methods. The regression equations were used to calculate the concentrations of TM.
According to ICH, the accuracy of the proposed spectrophotometric methods for the analysis of the different pharmaceutical formulations was determined using a standard addition technique in case of Gast-reg® 200 mg tablets, ampoules, and suspension while using a comparative study with a reported method [11] in case of Gast-reg® 100 mg tablets.
Results and discussion
The International Conference on Harmonization (ICH) guideline, namely “stability testing of new drug substances and products” demands that the stress testing should be carried out to illustrate the stability characteristics of the active compound [24]. A typical stability-indicating method is the one that determines the amount of the standard drug alone and analyzes its degradation products. Trimebutine maleate (TM) undergoes hydrolysis under alkaline condition according to the previously reported study [4]. This work was extended to confirm such investigation where complete degradation using alkaline condition was conducted upon refluxing of pure TM with 0.1 M NaOH for 30 min at 100 °C (Fig. 2). TLC of the intact drug and degradation products using isopropanol-water-ammonia (14:4:2, by volume) as a developing system revealed that the potential degradation products were isolated and showed two new spots at Rf = 0.02 and Rf = 0.46, which is distinct from that of the intact drug (Rf = 0.81). The IR spectrum of intact TM reveals a stretching band at 1750 cm−1 which disappears in the IR spectrum of both degradation products, thus confirming the cleavage at ester linkage. In addition, the appearance of carbonyl group of acid at 1600 cm−1 (implies the structure of DEG 1) contains a carbonyl group. There is a broad band that appears at 3000 cm−1 which indicates the presence of alcoholic OH that is found in the structure of DEG 2 (Fig. 3). In the MS chart, the parent peak of the main drug, TM was identified at m/z 503 (Mol. wt of TM) and that of the isolated degradation products was identified at m/z 212 (Mol. wt of 3,4,5-trimethoxy benzoic acid) “DEG 1” and at m/z 193 (Mol. wt of 2-(dimethylamino)-2-phenylbutanol) “DEG 2” (Fig. 4).

Proposed scheme for preparing the alkali-induced degradation products of trimebutine maleate

IR spectrum of trimebutine maleate; 3,4,5-trimethoxy benzoic acid (DEG 1); and 2-(dimethylamino)-2-phenylbutanol (DEG 2)

MS chart of trimebutine maleate; 3,4,5-trimethoxy benzoic acid (DEG 1); and 2-(dimethylamino)-2-phenylbutanol (DEG 2)
The objective of this work is to develop different smart stability-indicating spectrophotometric methods for specific estimation of TM in existence of its degradation products in its authentic form, laboratory-prepared mixtures, and different pharmaceutical drugs using several manipulating steps for either direct measurements at absorption spectra of zero-order or manipulation using ratio or derivative spectra.
Spectrophotometric methods
UV spectra of 30.0 μg/mL of each of TM with DEG 1 and DEG 2 were scanned (Fig. 5) which reveals that trimebutine maleate has λmax at 266 nm and that of DEG 1 has λmax at 256 nm while that of DEG 2 has λmax at 200 nm with poor absorbance spectrum due to the weak conjugation in its structure. Therefore, this poor absorbance of DEG 2 confirms its hard determination by different analytical methods.

Zero-order absorption spectra of 30 μg/mL trimebutine maleate, 30 μg/mL DEG 1, and 30 μg/mL DEG 2 in methanol
Manipulating steps on zero-order absorption spectra
Dual-wavelength method
The core of dual-wavelength method is that the difference in absorbance between the two selected points on the mixture spectra is directly proportional only to the concentration of component X and equals to zero for the component Y. In order to determine TM, two wavelengths (243 nm and 269 nm) were chosen so that the absorbance difference between them is directly proportional only to the concentration of TM and equals to zero for degradation products (Fig. 6). Under optimum spectrophotometric conditions, a linear relationship was achieved between the difference in absorbance at ΔA (243 nm and 269 nm) and the corresponding concentration of TM over the range of 5.0–60.0 μg/mL, and the regression equation was calculated as listed in Table 1.

Zero-order absorption spectra of 30.00 μg/mL of trimebutine maleate and 30.00 μg/mL of its alkali-induced degradation products showing DW method
This method has the advantage of being having minimum error and high accuracy as well as it can save effort and time, whereas its main drawback is the restriction in the selected wavelengths which are having constant absorbance of the interfering compound. This can lead to a critical measurement of the absorbance of the target drug and if any minute change in those selected wavelengths will result in unsatisfactory results with poor reproducibility.
Second derivative method
Derivative spectrophotometry is an unpretentious and of low-cost analytical technique of high utility for producing both qualitative and quantitative information form unresolved band spectra. This method has the advantage of producing an increase in sensitivity and selectivity, whereas its disadvantage is the critical measurement at a certain wavelength and its reliance on instrumental parameters such as scan speed and slit width. The instrumental parameters of obtaining the absorption spectrum of zero-order have a high effect on the form and intensity of its derivative generations. The selectivity is the key in this method without any contribution of degradation products and depends on measuring the peak amplitude of D2 spectrum of TM using smoothing factor and Δλ = 8 and scaling factor = 100 at 268 nm with zero-crossing of the spectrum of degradation products (Fig. 7). Under optimum spectrophotometric conditions, a linear relationship was achieved between the peak amplitude of TM at 268 nm and its corresponding concentration over the range of 5.0–60.0 μg/mL, and the regression equation was calculated as listed in Table 1.

The decond derivative spectrum of 30.00 μg/mL of trimebutine maleate and 30.00 μg/mL of its alkali-induced degradation products measured at 268 nm at the zero-crossing point
Manipulating steps on ratio spectra
Ratio difference method
In RD method, the absorption spectrum of the main drug was divided by the absorption spectrum of the substance that is needed to be canceled, and the obtained ratio spectrum represents TM/DEG + constant. The method was applied to fix the issue of overlapping spectra of TM and its degradation products where the interfering substance was canceled and therefore showed no contribution. The choice of suitable divisor and the selected two wavelengths considered to be the main steps that influence the ratio difference method. The requirements for selecting suitable divisor is that it should be adjusted between lower noise and higher sensitivity, and regarding the chosen wavelengths, they should have high absorbance difference with considerable contribution from the degradation products. Consequently, the amplitudes at 242 nm and 278 nm were chosen for the determination of TM using 30.0 μg/mL of degradation products as the divisor (Fig. 8).

Ratio spectra of 30.00 μg/mL of trimebutine maleate and its laboratory-prepared mixture using 30.00 μg/mL of its alkali-induced degradation products as a divisor
Under optimum spectrophotometric conditions, a linear relationship was attained between the amplitude differences of the ratio spectra at 242 nm and 278 nm and concentrations of TM in the range of 5.0–60.0 μg/mL, and the regression equation was calculated. Substitution in the regression equation was done to obtain the concentration of TM as listed in Table 1. Without the need for derivatization, the obtained constant will be removed in addition to any instrumental error following the subtraction of the recorded amplitudes at the selected wavelengths. Therefore, the ratio of signal to noise is reinforced. Ratio difference method was an appropriate method for the determination of TM in the existence of its degradation product because of its ease in application, minimal data used in manipulation, and ultimate reproducibility and robustness, which allow the analysis of drug and its degradation products without overlapped spectra and therefore can be applied in quality control testing with excessively pleasing results. Complete elimination of the degradation products was obtained in the form of a constant.
Constant center coupled with spectrum subtraction method
The constant center method has the advantages of being able to obtain the original spectrum of TM. In this method, ratio spectra were achieved by dividing the absorption spectra of TM over that of a certain concentration of its degradation products. The requirements for selecting a suitable divisor is that it should be adjusted between lower noise and higher sensitivity. The divisor concentration of 30.0 μg/mL of the degradation products produced preferable results concerning average recovery percent when used for the analysis of TM concentrations. The interfering substance was removed after applying ratio difference at the two chosen wavelengths of the ratio spectra of the target drug and therefore showed any contribution. The two selected wavelengths were 242 nm and 278 nm, and they should be selected on the basis of the contribution of both the target drug and its interfering substance (Fig. 8).
For the determination of TM in the laboratory-prepared mixture, scanning of the zero-order absorption spectra of the mixtures was done, and the ratio spectra of the mixtures were achieved by using 30.0 μg/mL of degradation products as a divisor where recorded amplitude at 278 nm was measured for each mixture, whereas the postulated amplitude value at 278 nm can be mathematically computed by using the equation explaining the linear relationship between the ratio difference of ratio spectra at 242 nm and 278 nm (degradation products were canceled) and the corresponding ratio amplitudes at 278 nm (Fig. 9).

Linear relationship between ∆P (278 nm and 242 nm) on the y-axis and P (278 nm) on the x-axis
where P1 and P2 are the postulated amplitudes at 242 nm and 278 nm, respectively, and r is the correlation coefficient. The constant value was calculated by observing the influence on the amplitude of the ratio spectrum of TM at 278 nm (∆Precorded − postulated), so the constant value was mathematically computed by measuring the difference between the recorded amplitude and postulated amplitude at this wavelength.
where C.V is the constant value.
Precorded is the recorded amplitude of the ratio spectrum of the laboratory-prepared mixture after being divided by 30.0 μg/mL of degradation products at 278 nm.
Ppostulated is the calculated amplitude using the exact regression equation.
The obtained constant values were multiplied by the absorption spectrum of the divisor (30 μg/mL of degradation products) to obtain the absorption spectrum of zero-order of the degradation products then be subtracted from the absorption spectrum of zero-order of the mixture to get the absorption spectrum of zero-order of TM. Under optimum spectrophotometric conditions, the concentration of TM in the mixture could be calculated by substitution in the regression equation attained by plotting the absorbance at its λmax (266 nm) and its corresponding concentration in the range of 5.0–60.0 μg/mL as listed in Table 1.
Constant center method provides many alluring features such as maximum resolution since absorption spectrum of zero-order of the target drug is achieved which can acts as a defined fingerprint and provides enhancement of sensitivity and specificity in mixture analysis.
Method validation
Method validation was accomplished according to ICH guidelines [25] for all the proposed methods as follows.
Linearity range
Linearity of the methods is examined by making different calibration curves on three consecutive days. The calibration curves are set up within concentration ranges that were picked on the basis of the expected drug concentration throughout the assay of the dosage form. Each concentration is iterated three times. All the validation parameters are listed in Table 1.
Accuracy
The accuracy of the presented methods was examined by analyzing three samples of TM standard solutions. The recovery percentages were listed in Table 1, and the results showed high accuracy of the suggested methods.
Precision
Repeatability and intermediate precision (intra-day and inter-day precision) are expressed as RSD and can be obtained by analyzing three different concentrations of the target drug in the linearity range on single day and on three following days as shown in Table 1.
Specificity
Specificity is checked by analyzing different mixtures consisting of the drug and its degradation products in several ratios within the linearity range. Favorable percentage recoveries with minimal standard deviation among the other methods are obtained as presented in Table 2. The results of the analysis of the different pharmaceutical formulations are illustrated in Table 3. According to ICH, the accuracy of pharmaceutical formulation using the proposed methods was checked in case of Gast-reg® 200 mg tablets, suspension, and ampoules through standard addition technique, which is a recovery attained when spiking the sample solution of the pharmaceutical formulation with known concentration of the intact drug as illustrated in Table 4. A comparative study with a reported method [11] was done in case of Gast-reg® 100 mg tablets as illustrated in Table 5.
Statistical analysis
The suggested methods and the official HPLC method of TM are statistically compared with each other regarding the t test and F values and revealed that there was no considerable difference between the official HPLC method and the experimental values which were obtained in the pure sample analysis by the proposed methods indicating identical accuracy and precision where the results are illustrated in Table 6. The developed UV-spectrophotometric methods and the official HPLC method of TM are compared with each other using one-way ANOVA where the p value (0.315) is higher than 0.05 and the F calculated (1.247) is lower than the F tabulated (2.728) at p = 0.05, indicating that there is no critical difference between the suggested methods and the official method as illustrated in Table 7.
Conclusion
This work introduced different smart and unpretentious stability indicating UV-spectrophotometric methods for concurrent determination of TM in existence of its degradation products in its authentic form, laboratory-prepared mixtures, and different pharmaceutical drugs using several manipulating pathways for either direct measurements at absorption spectra of zero-order or manipulation using ratio or derivative spectra. The proposed methods were time-saving and economic since no toxic organic solvents were used when compared to the chromatographic methods. The developed methods do not require exhaustive treatment, additional sophisticated software, or especial computer programs, thus enable their application for resolving the complex mixtures in quality control laboratories. In addition, the developed methods are direct, simple, and do not need separation, specific detector, or pretreatment steps. Validation of the proposed methods was performed in reference to the ICH guidelines regarding the linearity, range, accuracy, precision, and specificity, and all the obtained results were within the acceptable range. The results were satisfactory compared to that of the official method [1] of pure powdered form of TM using Student’s t test, variance ratio F test at 95% confidence interval, and one-way ANOVA testing where the calculated results were lower than that of the tabulated results which indicates that there is no influential difference with regard to accuracy and precision. The results confirm the applicability of the suggested methods for the routine determination of the drug of interest TM in quality control laboratories.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANOVA:
-
Analysis of variance
- CC-SS:
-
Constant center coupled with spectrum subtraction
- CV:
-
Constant value
- D 2 :
-
Second derivative
- DEG:
-
Degradation product
- DW:
-
Dual-wavelength
- HPLC:
-
High-performance liquid chromatography
- ICH:
-
International Conference on Harmonization
- IR:
-
Infrared radiation
- NODCAR:
-
National Organization for Drug Control and Research
- RD:
-
Ratio difference
- RSD:
-
Relative standard deviation
- TLC:
-
Thin-layer chromatography
- TM:
-
Trimebutine maleate
References
British Pharmacopoeia, H.M.s.S.O., London (2015). Volume II: p., 1104-1106.
Delvaux M, Wingate D (1997) Trimebutine: mechanism of action, effects on gastrointestinal function and clinical results. J Int Med Res 25(5):225–246
Lacy CF, Armstrong LL, Goldman MP, Lance LL (2005) Drug information handbook: with Canadian and International Drug Monographs. Lexi-Comp Inc, 13th Edition: p. 1595-1596.https://www.amazon.ca/Drug-Information-Handbook-Comprehensive-Professionals/dp/B004JPHUOG/ref=sr_1_1?qid=1566257202&refinements=p_27%3ACharles+F.+Lacy%3BLora+L.+Armstrong%3BMorton+P.+Goldman%3BLeonard+L+Lance&s=books&sr=1-1, https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=L.L.A.+Charles+F.+Lacy%2C+Morton+P.+Goldman%2C+Leonard+L.+Lance+Drug+Information+Handbook%3A+with+Canadian+and+International+Drug+Monographs%2C+Lexi-Comp+Inc%2C+13th+Edition+%282005%29+1595-1596.&btnG=
El-Gindy A, Emara S, Hadad GM (2003) Spectrophotometric and liquid chromatographic determination of trimebutine maleate in the presence of its degradation products. J Pharm Biomed Anal. 33(2):231–241
Ayad M et al (2016) Spectrophotometric determination of tiemonium methyl sulfate, itopride hydrochloride and trimebutine maleate via ion pair complex formation and oxidation reaction. Indian J Adv Chem Sci 4(1):85–97
Elqudaby HM, Mohamed GG, El-Din GM (2014) Analytical studies on the charge transfer complexes of loperamide hydrochloride and trimebutine drugs. Spectroscopic and thermal characterization of CT complexes. Spectrochim Acta A Mol Biomol Spectrosc 129:84–95
Shaban M (2002) Spectrophotometric determination of trimebutine through ion-pair and charge-transfer complexation reactions. Sci Pharm 70(4):341–351
El-Shiekh R, Zahran F, Gouda AAE-F (2007) Spectrophotometric determination of some anti-tussive and anti-spasmodic drugs through ion-pair complex formation with thiocyanate and cobalt (II) or molybdenum (V). Spectrochim Acta A Mol Biomol Spectrosc 66(4-5):1279–1287
Abdel-Gawad FM (1998) Ion-pair formation of Bi (III)–iodide with some nitrogenous drugs and its application to pharmaceutical preparations. J Pharmaceut Biomed 16(5):793–799
Joo E-H et al (1999) High-performance liquid chromatographic determination of trimebutine and its major metabolite, N-monodesmethyl trimebutine, in rat and human plasma. J Chromatogr B Biomed Sci Appl 723(1-2):239–246
V Appala Raju ABM, Pathi PJ, Raju NA (2013) Estimation of trimebutine maleate in tablet dosage form by RP-HPLC. J Pharm Sci Innov 3:99–101
Wang W (2003) RP-ion pair HPLC determination of trimebutine maleate tablets and its related compounds. Chin J Pharm Anal 23(2):111–113
Wang H et al (2002) Quantitative determination of trimebutine maleate and its three metabolites in human plasma by liquid chromatography-tandem mass spectrometry. J Chromatogr B. 779(2):173–187
Astier A, Deutsch A (1981) Quantitative high-performance liquid chromatographic determination of antispasmodic trimebutine in human plasma: pharmacokinetic studies after intravenous administration in humans. J Chromatogr 224(1):149–155
Lin Z-h, Song M (2001) RP-HPLC determination of related impurities in trimebutine maleate and assay of components in its preparation. Chin J Pharm Anal 21(1):25–27
Sanjiu J et al (2002) Determination of trimebutine maleate tables by HPLC. Chin J Modern Appl Pharm 5:21
Lavit M et al (2000) Determination of trimebutine and desmethyl-trimebutine in human plasma by HPLC. Arzneimittelforschung. 50(07):640–644
Yong-Yu P et al (2007) Enantiomeric separation of trimebutine maleate and ondansetron on amylose chiral stationary phase. Chin J Anal Chem 35(6):880
Zekai H et al (2011) Study on detection method of impurity 3, 4, 5-trimethoxy benzoic acid in trimebutine maleate sustained-release tablets. China Pharmaceuticals. 12:37–38
Elqudaby HM, Mohamed GG, El Din GM (2013) Utilization of phosphotungestic acid in the conductometric determination of loperamide hydrochloride and trimebutine antidiarrhea drugs. J Pharm Res 7(8):686–691
Ayad M et al (2016) Conductometric determination of tiemonium methylsulfate, alizapride hydrochloride, trimebutine maleate using rose bengal, ammonium reineckate and phosphotungstic acid. Indian J Adv Chem Sci 4(2):149–159
Adhoum N, Monser L (2005) Determination of trimebutine in pharmaceuticals by differential pulse voltammetry at a glassy carbon electrode. J Pharmaceut Biomed 38(4):619–623
Elqudaby H, Mohamed GG, El Din GM (2014) Electrochemical behaviour of trimebutine at activated glassy carbon electrode and its direct determination in urine and pharmaceutics by square wave and differential pulse voltammetry. Int J Electrochemical Sci 9:856–869
ICH (1993) Stability testing of new drug substances and products. Geneva.
ICH (1997) Q2B, Note for guidance on validation of analytical methods methodology, International Conference on Harmonization. IFPMA
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
EM prepared and analyzed the stock and working solutions of the drug of interest, trimebutine maleate (TM), and its degradation product in their lab mixtures and in pharmaceutical formulations. HL was a major contributor in designing the whole frame of work and in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lotfy, H.M., Morgan, E.M., Fayez, Y.M. et al. Different aspects in manipulating overlapped spectra used for the analysis of trimebutine maleate and structure elucidation of its degradation products. Futur J Pharm Sci 5, 7 (2019). https://doi.org/10.1186/s43094-019-0004-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-019-0004-y